
Combined Transcriptome and Proteome Analysis Provides Insights into Petaloidy in Pomegranate
 and
and
Abstract
:1. Introduction
2. Results
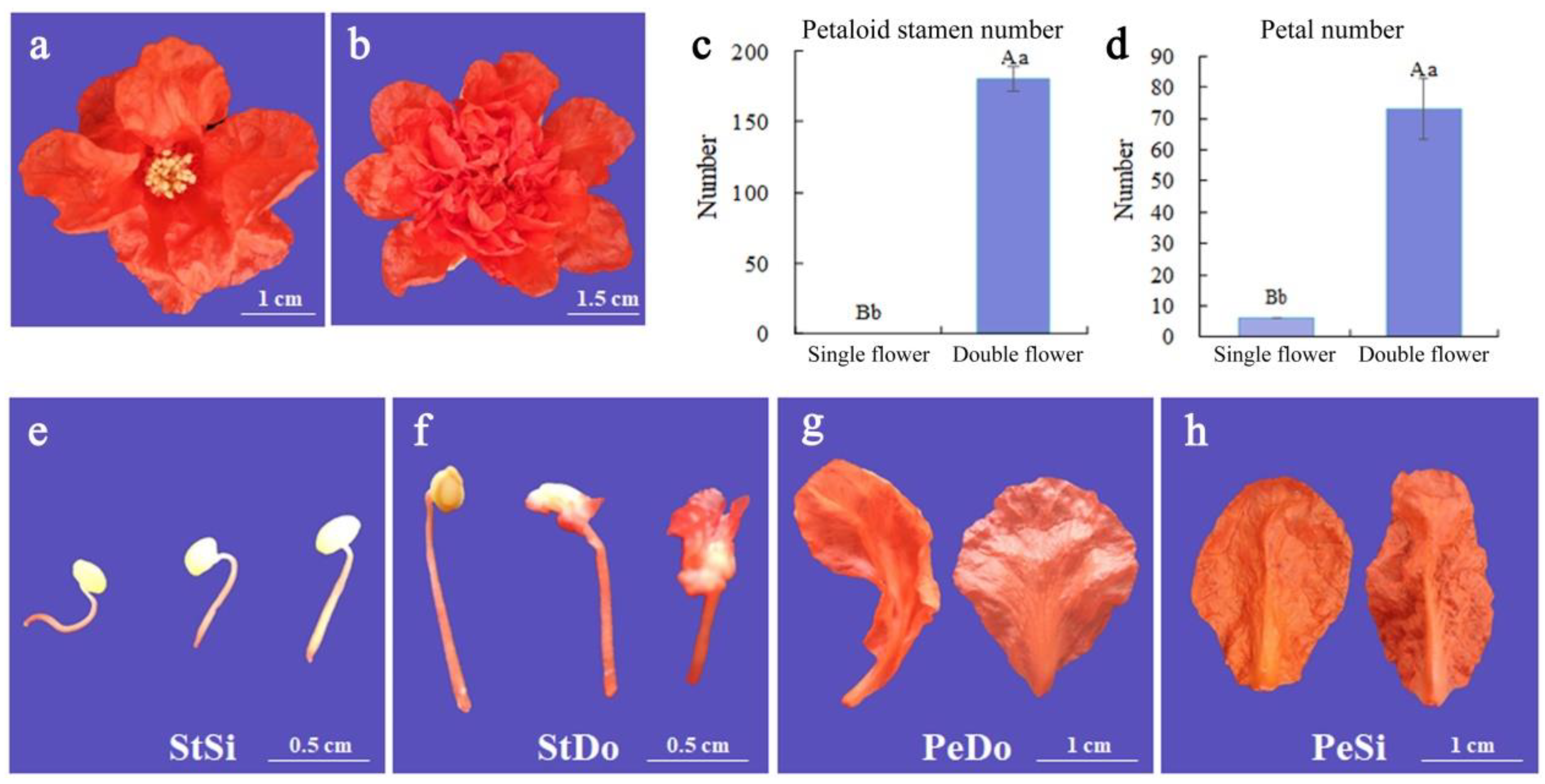
2.1. Petaloid Phenotype of Ornamental Pomegranate
2.2. Basic Information on Transcriptome Sequencing
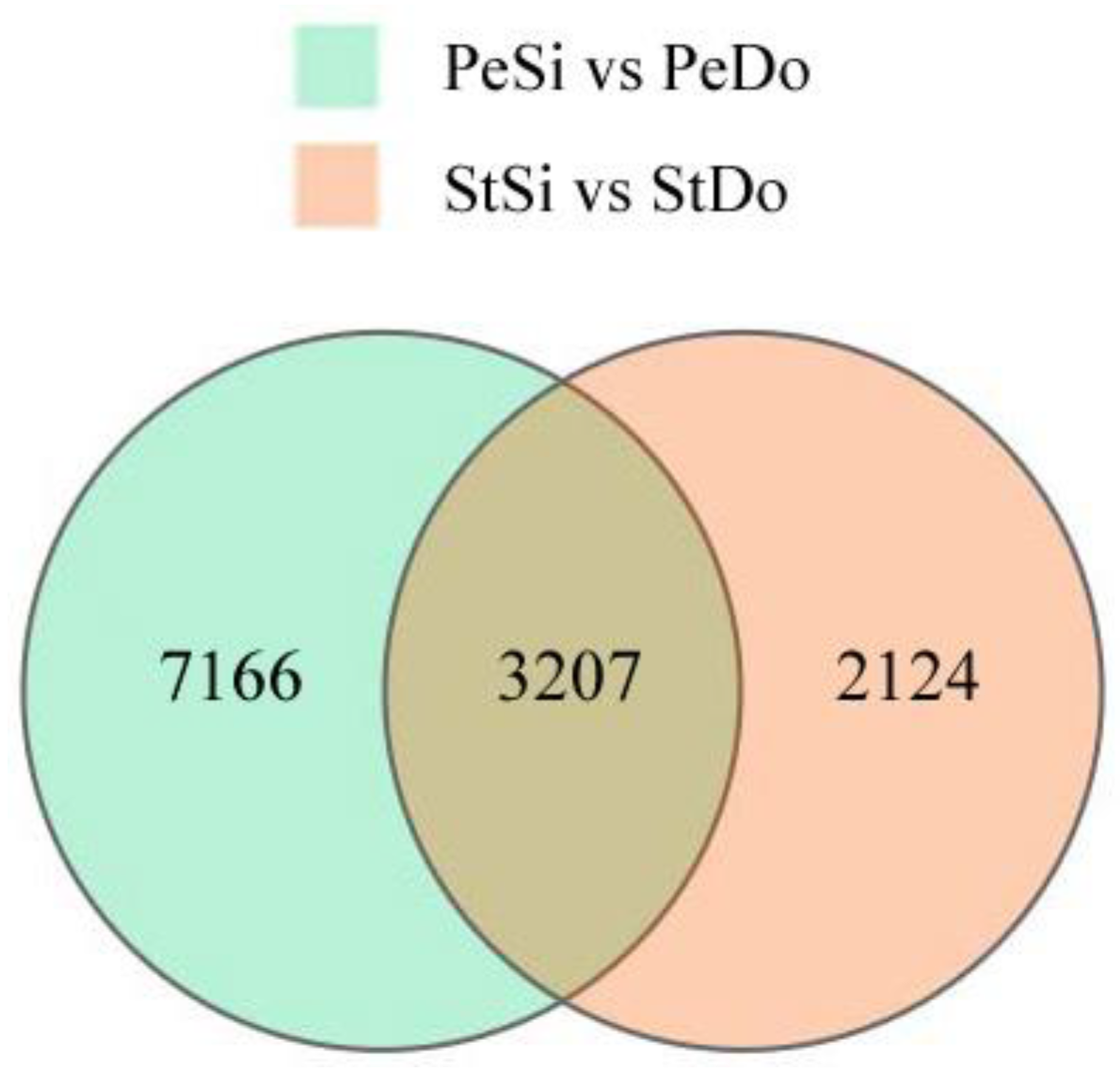
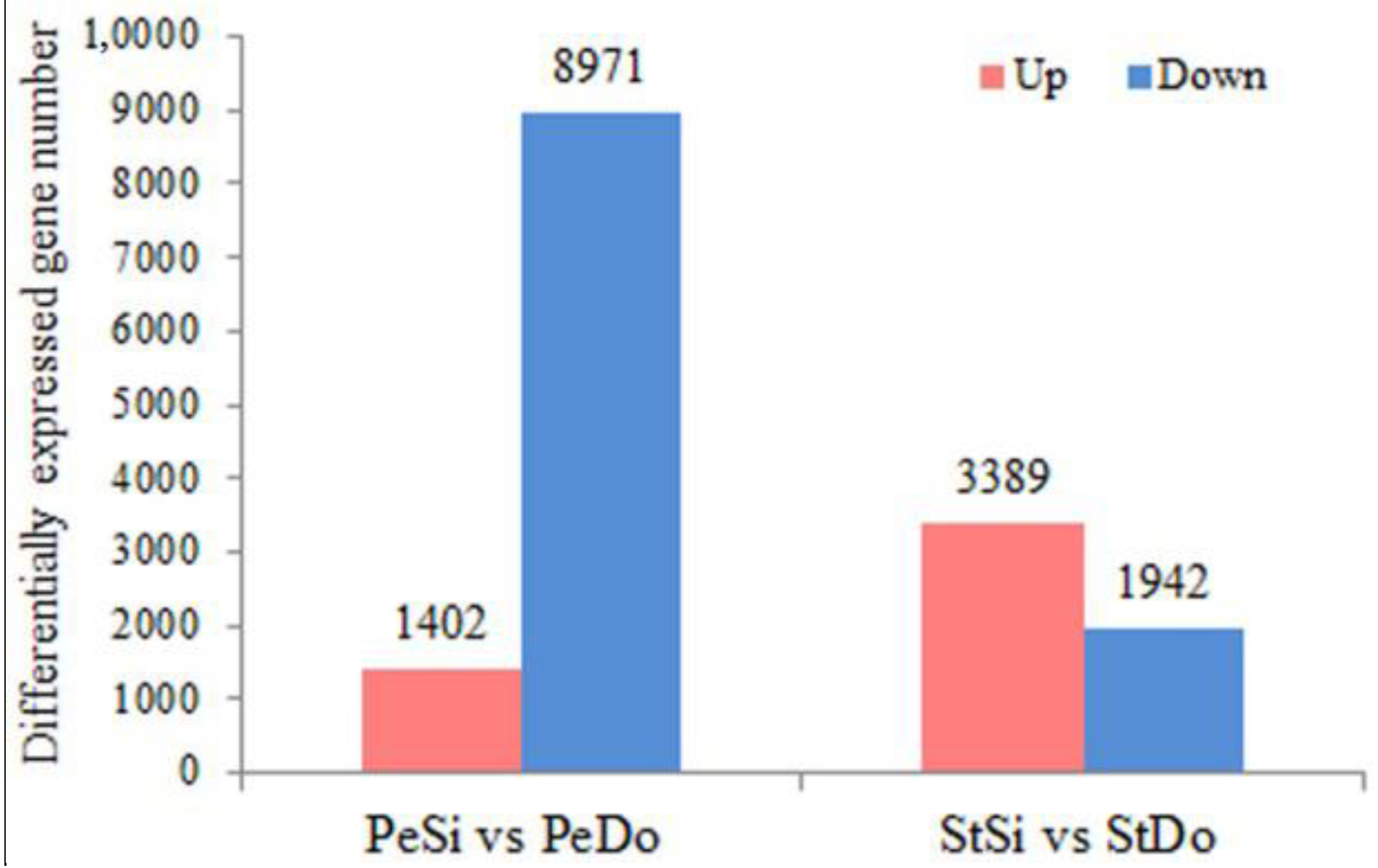
2.3. Differentially Expressed Genes among the Floral Organs
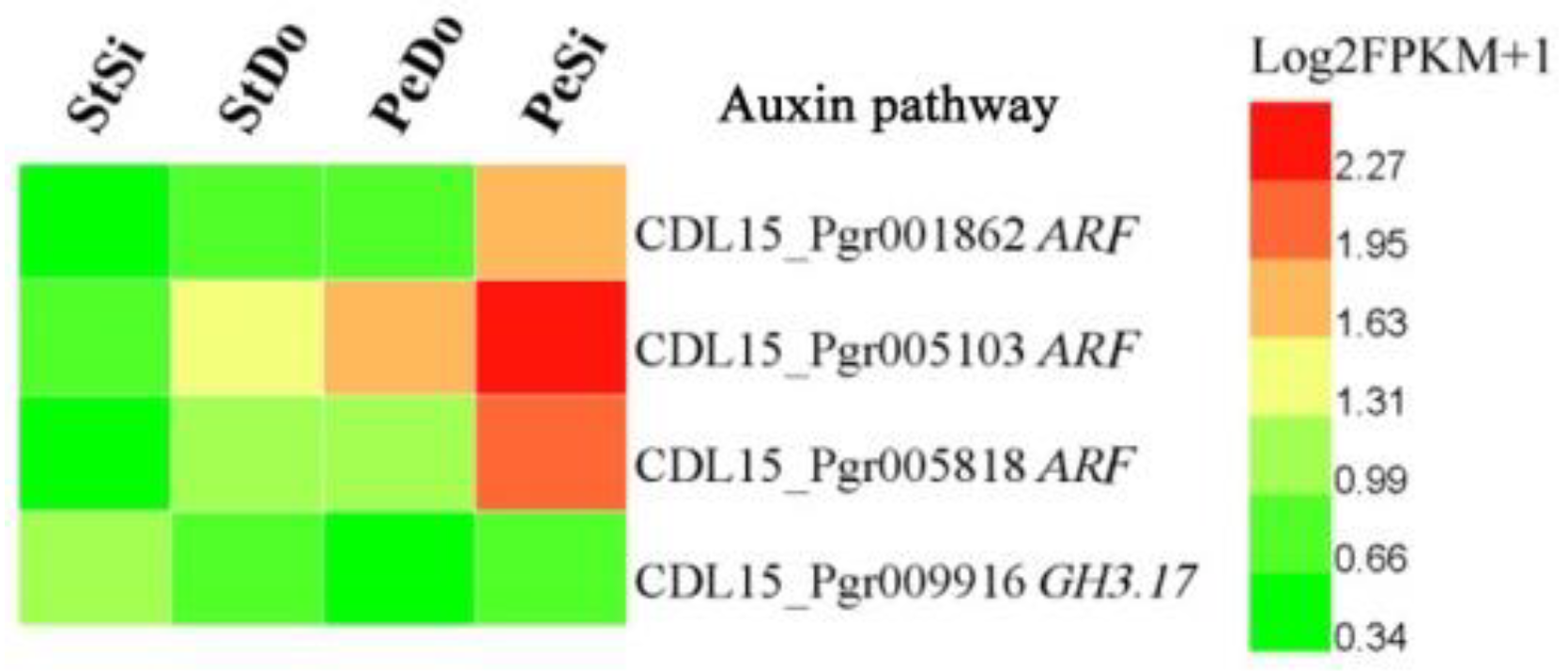
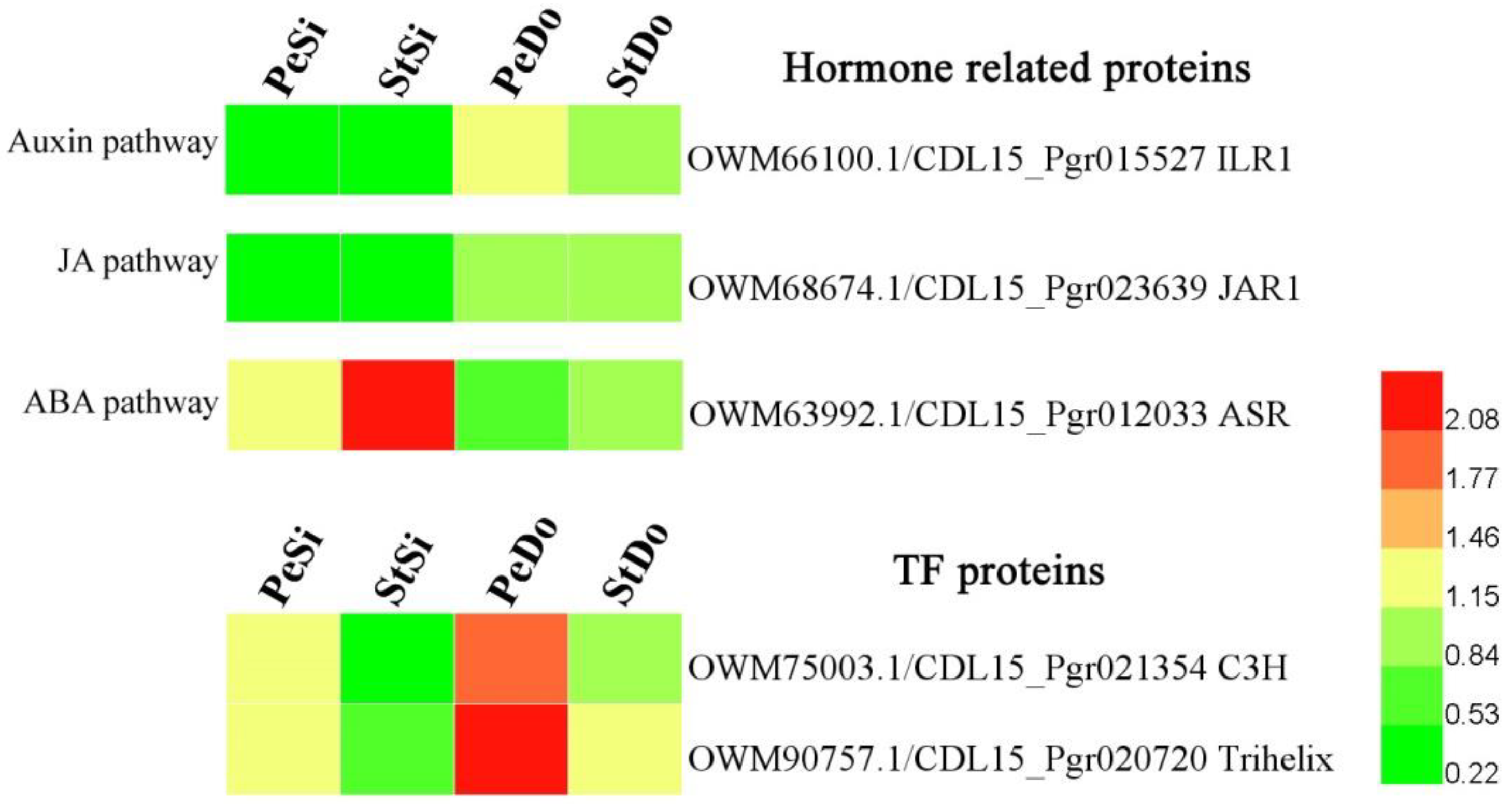
2.4. Hormone-Related and Transcription Factor Differentially Expressed Genes Involved in Petaloidy
2.5. RNA Sequencing Data Validation through Quantitative Real-Time Polymerase Chain Reaction
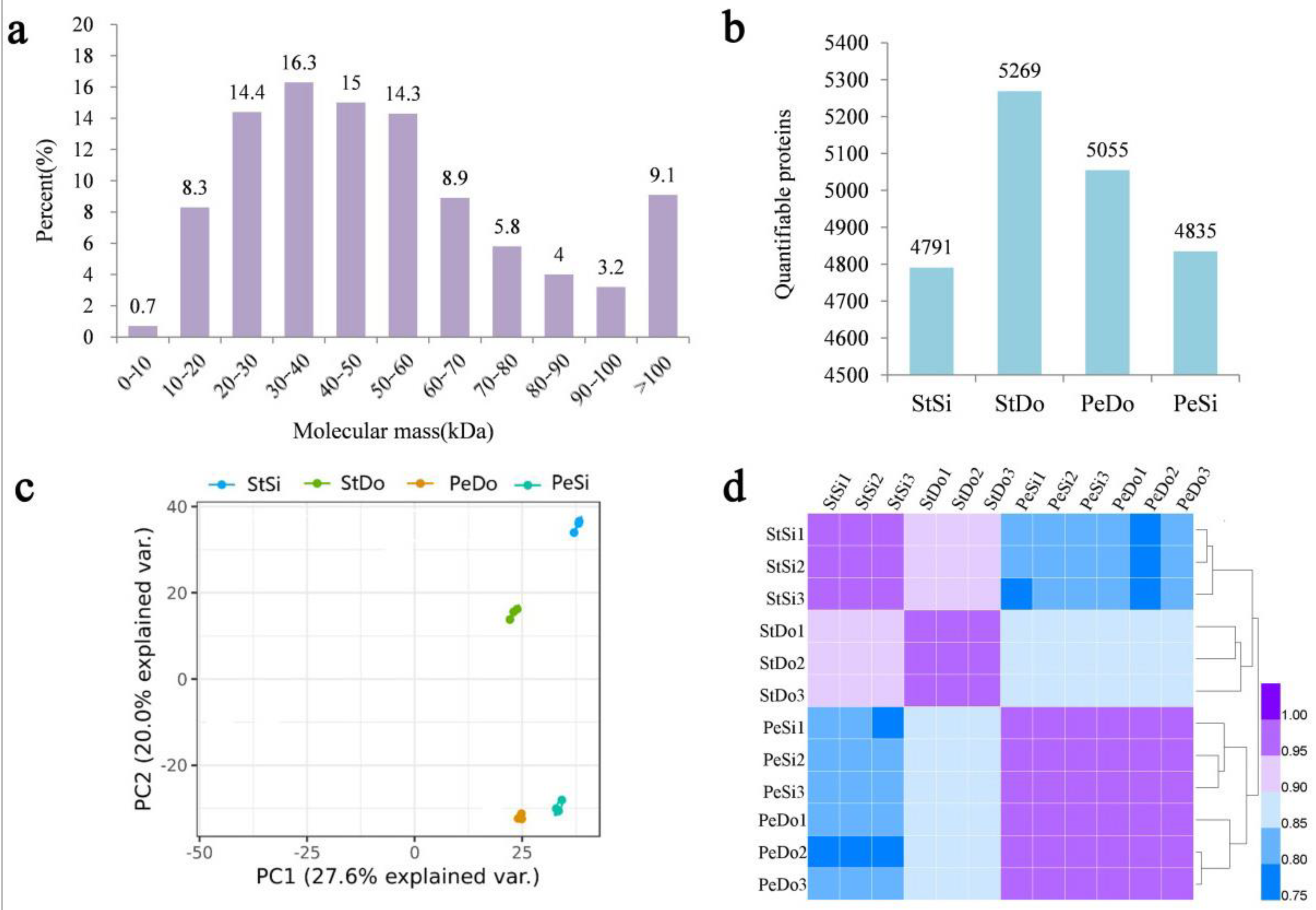
2.6. Basic Information of the Proteome Sequencing
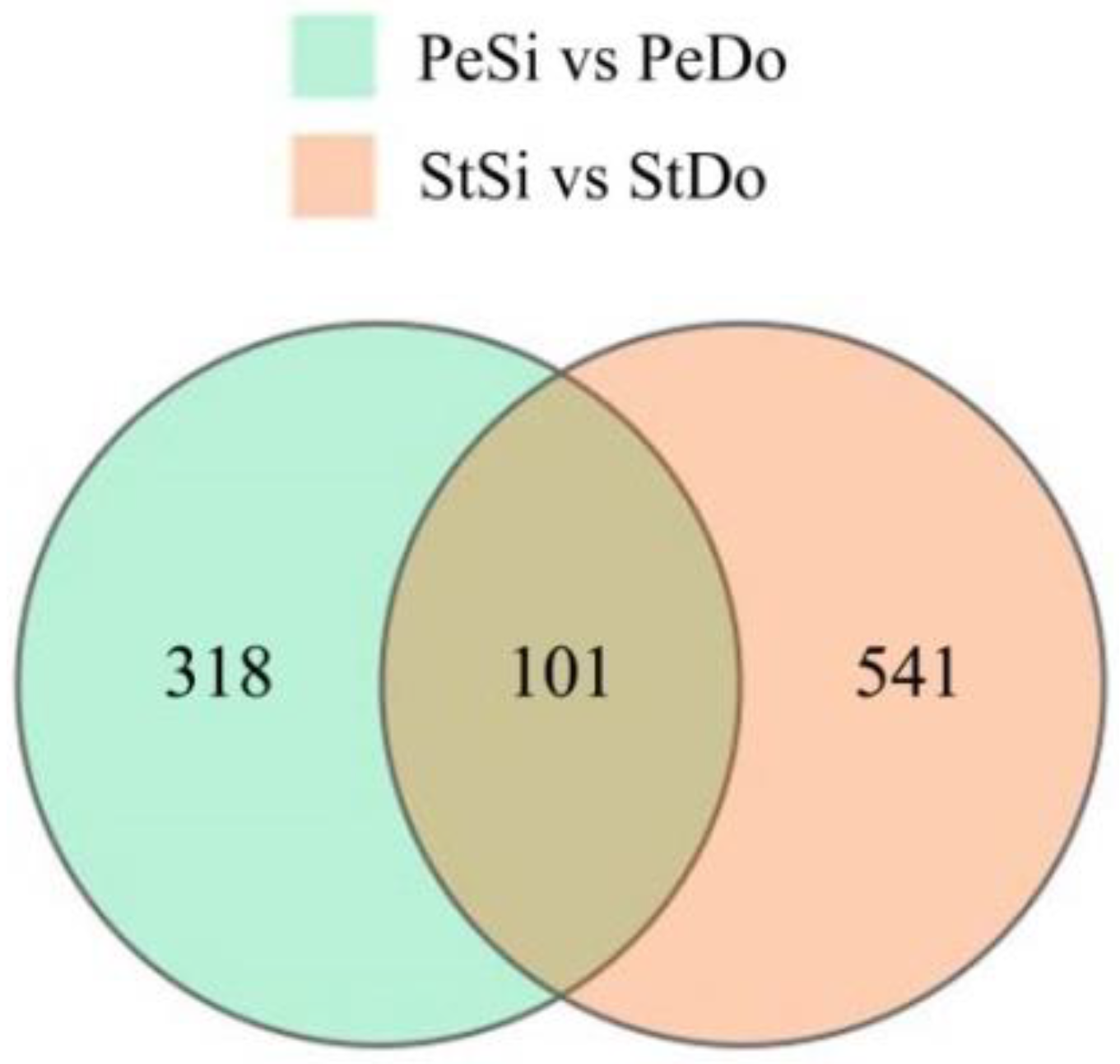
2.7. Differentially Abundant Proteins Involved in Petaloidy
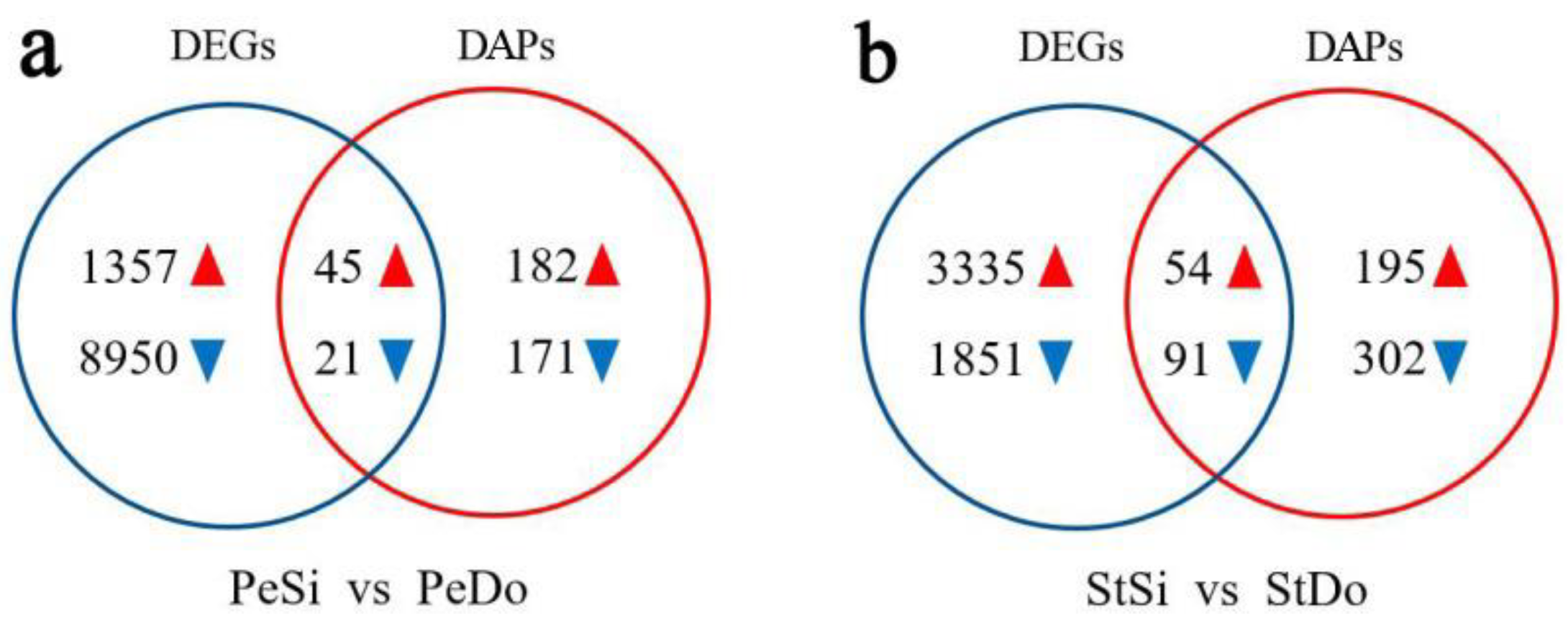
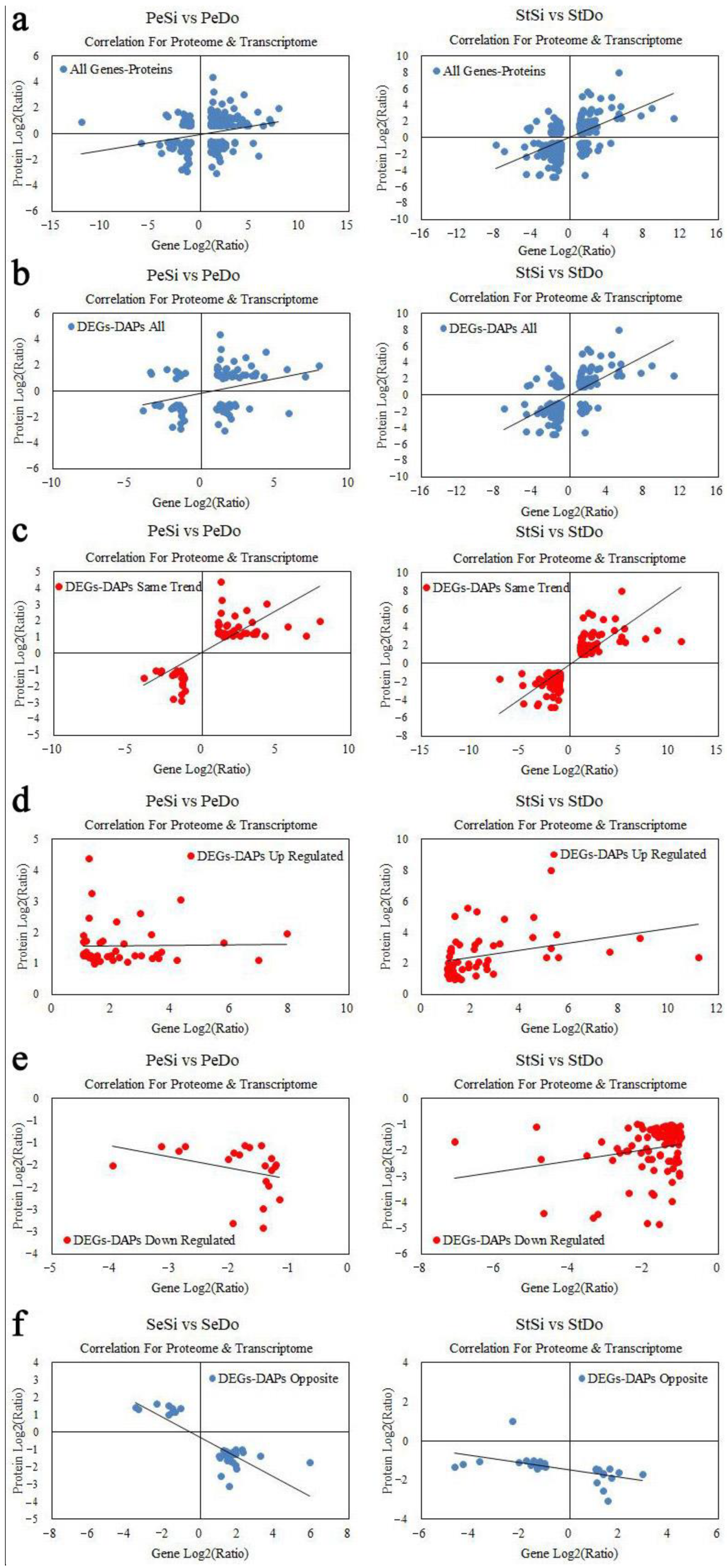
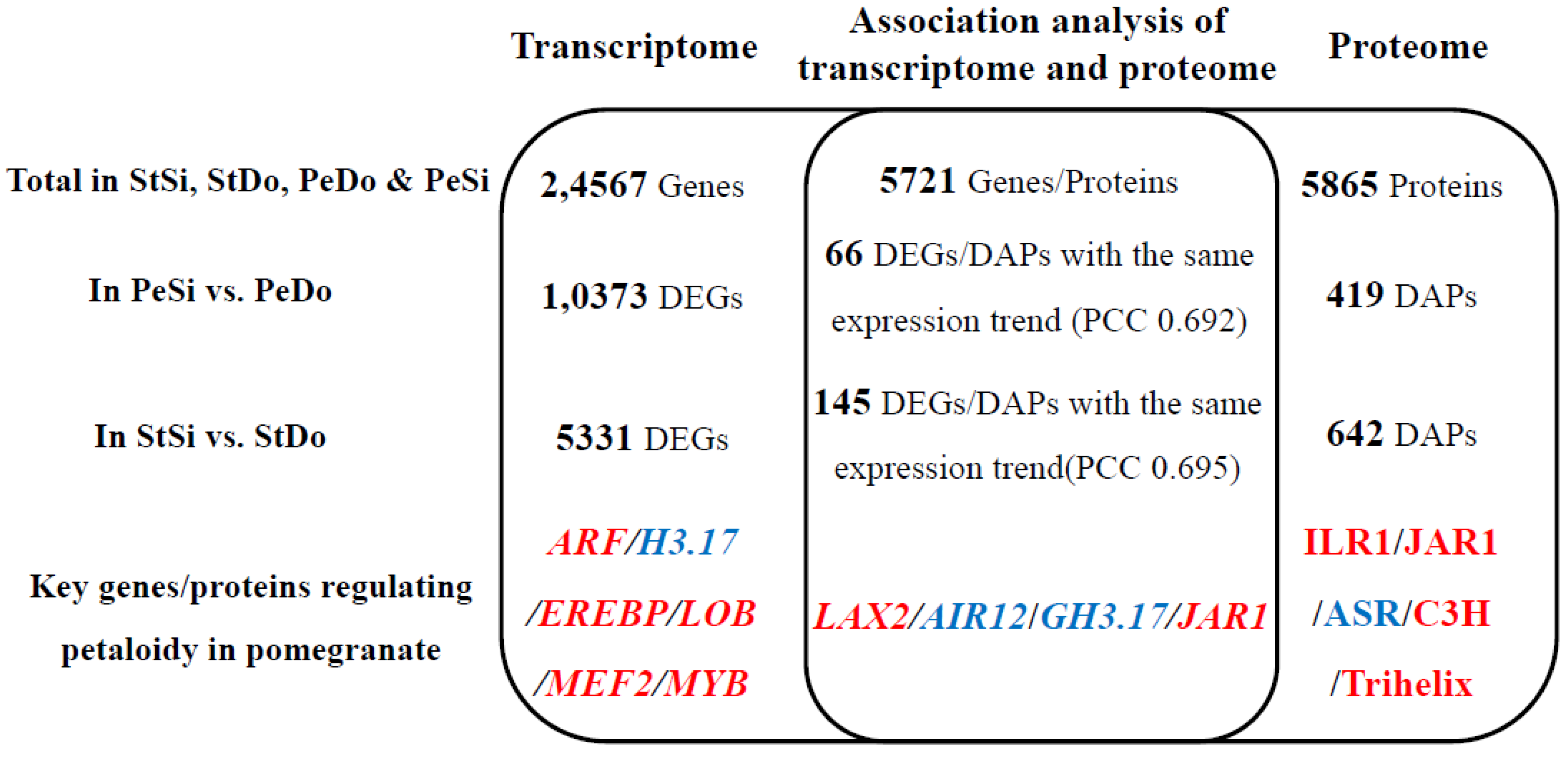
2.8. Association Analysis of the Transcriptome and Proteome
2.8.1. Quantitative Relationship between the Transcriptome and Proteome
2.8.2. Analysis of Differentially Expressed Genes/Differentially Abundant Proteins with the Same Expression Trend
3. Discussion
3.1. Overview of the Transcriptome and Proteome in Ornamental Pomegranate
3.2. Hormone Pathway Participation in Ornamental Pomegranate Petaloidy
3.3. Transcription Factor Participation in Ornamental Pomegranate Petaloidy
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Transcriptome Sequencing and Assembly and Gene Functional Annotation
4.3. RNA Sequencing Data Validation Using Quantitative Real-Time Polymerase Chain Reaction
4.4. Protein Extraction and Trypsin Digestion
4.5. Liquid Chromatography with Tandem Mass Spectrometry Analysis and Database Search
4.6. Bioinformatic Analysis of the Transcriptome and Proteome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dubois, A.; Raymond, O.; Maene, M.; Baudino, S.; Langlade, N.B.; Boltz, V.; Vergne, P.; Bendahmane, M. Tinkering with the C-function: A molecular frame for the selection of double flowers in cultivated roses. PLoS ONE 2010, 5, e9288. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Tang, A.; Wan, H.; Zhang, T.; Cheng, T.; Wang, J.; Yang, W.; Pan, H.; Zhang, Q. An APETALA2 Homolog, RcAP2, Regulates the number of rose petals derived from stamens and response to temperature fluctuations. Front. Plant Sci. 2018, 9, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, E.S.; Meyerowitz, E.M. The war of the whorls: Genetic interactions controlling flower development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koh, J.; Yoo, M.J.; Kong, H.Z.; Hu, Y.; Ma, H.; Soltis, P.S.; Soltis, D.E. Expression of floral MADS-box genes in basal angiosperms: Implications for the evolution of floral regulators. Plant J. 2005, 43, 724–744. [Google Scholar] [CrossRef] [PubMed]
- Liljegren, S.J.; Ditta, G.S.; Eshed, Y.; Savidge, B.; Bowman, J.L.; Yanofsky, M.F. SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 2000, 404, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, S.; Ditta, G.S.; Bauman, E.; Wisman, E.; Yanofsky, M.F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Fukuda, T.; Nakana, M.; Hasebe, M.; Kameya, T.; Kanno, A. The modified ABC model explains the development of the petaloid perianth of Agapanthus praecox ssp. Orientalis (Agapanthaceae) flower. Plant Mol. Biol. 2005, 58, 435–445. [Google Scholar] [CrossRef]
- Theissen, G.; Melzer, R. Molecular mechanisms underlying origin and diverdification of the angiosperm flower. Ann. Bot. 2007, 100, 603–619. [Google Scholar] [CrossRef]
- Sage-Ono, K.; Ozeki, Y.; Hiyama, S.; Higuchi, Y.; Kamada, H.; Mitsuda, N.; Ohme-Takagi, M.; Ono, M. Induction of double flowers in Pharbitis nil using a class-C MADS-box transcription factor with Chimeric REpressor gene-Silencing Technology. Plant Biotechnol. 2011, 28, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Nakatsuka, T.; Saito, M.; Yamada, E.; Fujita, K.; Yamagishi, N.; Yoshikawa, N.; Nishihara, M. Isolation and characterization of the C-class MADS-box gene involved in the formation of double flowers in Japanese gentian. BMC Plant Biol. 2015, 15, 182. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y. Isolation and Function Analysis of Class A and C genes of MADS-box family from Camellia japonica. Ph.D. Thesis, Chinese Academy of Forestry, Beijing, China, 26 June 2013. [Google Scholar]
- Almeida, A.M.; Brown, A.; Specht, C.D. Tracking the development of the petaloid fertile stamen in Canna indica: Insights into the origin of androecial petaloidy in the Zingiberale. AoB Plants 2013, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, D.; Chen, W.; Hu, R.; Zhang, Y.; Xia, Y.; Wang, S.; He, Q.; Guo, Q.; Liang, G. An integrative analysis of transcriptome, proteome and hormones reveals key differentially expressed genes and metabolic pathways involved in flower development in Loquat. Int. J. Mol. Sci. 2020, 21, 5107. [Google Scholar] [CrossRef]
- Devani, R.S.; Chirmade, T.; Sinha, S.; Bendahmane, A.; Dholakia, B.B.; Banerjee, A.K.; Banerjee, J. Flower bud proteome reveals modulation of sex-biased proteins potentially associated with sex expression and modification in dioecious Coccinia grandis. BMC Plant Biol. 2019, 19, 330. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Ng, K.H.; Lim, T.S.; Yu, H.; Meyerowitz, E.M. The homeotic protein AGAMOUS controls late stamen development by regulating a jasmonate biosynthetic gene in Arabidopsis. Plant Cell 2007, 19, 3516–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Li, Y.; Li, Y.; Wang, Y.; Jiang, C.; Choisy, P.; Xu, T.; Cai, Y.; Pei, D.; et al. AUXIN RESPONSE FACTOR 18–HISTONE DEACETYLASE 6 module regulates floral organ identity in rose (Rosa hybrida). Plant Physiol 2021, 186, 1074–1087. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Bai, S.; Lu, W.; Zhang, X. Isolation of HAG1 and its regulation by plant hormones during in vitro floral organogenesis in Hyacinthus orientalis L. Planta 2002, 215, 533–540. [Google Scholar] [CrossRef]
- Lin, Z.; Damaris, R.N.; Shi, T.; Li, J.; Yang, P. Transcriptomic analysis identifies the key genes involved in stamen petaloid in lotus (Nelumbo nucifera). BMC Genom. 2018, 19, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hera, G. Morphology of Single-Petaled/Double-Petaled Flower Traits in ‘Malus’ through Anatomical and Transcriptome analyses. Ph.D. Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 31 May 2013. [Google Scholar]
- Hu, L.; Zheng, T.; Cai, M.; Pan, H.; Wang, J.; Zhang, Q. Transcriptome analysis during floral organ development provides insights into stamen petaloidy in Lagerstroemia speciosa. Plant Physiol. Biochem. 2019, 142, 510–518. [Google Scholar] [CrossRef]
- Dong, Z.; Chen, Y. Transcriptomics: Advances and approaches. Sci. China Life Sci. 2013, 56, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, H.M. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lu, S.; Liu, K.; Wang, S.; Huang, L.; Guo, L. Proteomics: A powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, N.; Zhang, W.; Wu, C.; Jiang, Y.; Ma, J.; Li, M.; Sui, S. Integrated transcriptome and proteome analysis provides insight into chilling-induced dormancy breaking in Chimonanthus praecox. Hortic. Res. 2020, 7, 198. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, B.; Huang, R.; Suo, H.; Zhang, Z.; Chen, W.; Dai, X.; Ou, L. Transcriptome- and proteome-wide association of a recombinant inbred line population revealed twelve core QTLs for four fruit traits in pepper (Capsicum annuum L.). Hortic. Res. 2022, 9, 15. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Y.; Šmarda, P.; Bureš, P.; Guo, Q. Transcriptome and proteome associated analysis of flavonoid metabolism in haploid Ginkgo biloba. Int. J. Biol. Macromol. 2023, 224, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Yang, H.; Ding, W.; Yuan, Z.; Zhu, Z. Exploring the Relationship between Genomic Variation and Phenotype in Ornamental Pomegranate: A Study of Single and Double-Petal Varieties. Horticulturae 2023, 9, 361. [Google Scholar] [CrossRef]
- Huang, J.Z.; Lin, C.P.; Cheng, T.C.; Huang, Y.W.; Tsai, Y.J.; Cheng, S.Y.; Chen, Y.W.; Lee, C.P.; Chung, W.C.; Chang, B.C.H.; et al. The genome and transcriptome of Phalaenopsis yield insights into floral organ development and flowering regulation. PeerJ 2016, 4, e2017. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Fu, L.; Tie, W.; Yan, Y.; Wu, C.; Hu, W.; Zhang, J. Extensive post-transcriptional regulation revealed by transcriptomic and proteomic integrative analysis in Cassava under drought. J. Agric. Food Chem. 2019, 67, 3521–3534. [Google Scholar] [CrossRef]
- Lou, P.; Kang, J.G.; Zhang, G.Y.; Bonnema, G.; Fang, Z.; Wang, X. Transcript profiling of a dominant male sterile mutant (Ms-cd1) in cabbage during flower bud development. Plant Sci. 2007, 172, 111–119. [Google Scholar] [CrossRef]
- Wen, L.; Sun, X.; Ren, H.; Fan, H.; Yu, Y.; Guo, Y.; Zheng, C. Cloning and functional verification of Chrysanthemum CmXTHs genes related to petal elongation. Acta Hortic. Sinica 2017, 44, 2150–2162. [Google Scholar] [CrossRef]
- Peng, S.; Huang, S.; Liu, Z.; Feng, H. Mutation of ACX1, a jasmonic acid biosynthetic enzyme, leads to petal degeneration in Chinese cabbage (Brassica campestris ssp. pekinensis). Int. J. Mol. Sci. 2019, 20, 2310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X.; Zhang, W.; Yu, F.; Tian, J.; Li, D.; Guo, A. Functional analysis of the two Brassica AP3 genes involved in apetalous and stamen carpelloid phenotypes. PLoS ONE 2011, 6, e20930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Shahid, I.; Shi, T.; Song, J.; Ni, Z.; Gao, Z. Isolation of PmARF17 and its regulation pattern of endogenous hormones during flower development in Prunus mume. Sci. Agric. Sin. 2021, 54, 2843–2857. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Ding, L.; Song, A.; Shen, F.; Jiang, J.; Chen, S.; Chen, F. Transcriptomic and hormone analyses reveal mechanisms underlying petal elongation in Chrysanthemum morifolium ‘Jinba’. Plant Mol Biol. 2017, 6, 593–606. [Google Scholar] [CrossRef]
- Robert, H.S.; Crhak, K.L.; Mroue, S.; Benková, E. The importance of localized auxin production for morphogenesis of reproductive organs and embryos in Arabidopsis. J. Exp. Bot. 2015, 66, 5029–5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecchetti, V.; Altamura, M.M.; Falasca, G.; Costantino, P.; Cardarelli, M. Auxin regulates Arabidopsis anther dehiscence, pollen maturation, and filament elongation. Plant Cell 2008, 20, 1760–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig-Müller, J. Auxin Conjugates: Their Role for Plant Development and in the Evolution of Land Plants. J. Exp. Bot. 2011, 62, 1757–1773. [Google Scholar] [CrossRef] [Green Version]
- Swarup, K.; Benková, E.; Swarup, R.; Casimiro, I.; Péret, B.; Yang, Y.; Parry, G.; Nielsen, E.; De, S.I.; Vanneste, S.; et al. The auxin influx carrier LAX3 promotes lateral root emergence. Nat. Cell Biol. 2008, 10, 946–954. [Google Scholar] [CrossRef]
- Cardarelli, M.; Cecchetti, V. Auxin polar transport in stamen formation and development: How many actors? Front. Plant Sci. 2014, 5, 333. [Google Scholar] [CrossRef] [Green Version]
- Swarup, R.; Bhosale, R. Developmental Roles of AUX1/LAX Auxin Influx Carriers in Plants. Front. Plant Sci. 2019, 10, 1306. [Google Scholar] [CrossRef] [Green Version]
- Sessions, A.; Nemhauser, J.L.; McColl, A.; Roe, J.L.; Feldmann, K.A.; Zambryski, P.C. ETTIN patterns the Arabidopsis floral meristerm and reproductive organs. Development 1997, 134, 4481–4491. [Google Scholar] [CrossRef]
- Xu, X.F.; Wang, B.; Feng, Y.F.; Xue, J.S.; Qian, X.X.; Liu, S.Q.; Zhou, J.; Yu, Y.H.; Yang, N.Y.; Xu, P.; et al. AUXIN RESPONSE FACTOR17 directly regulates MYB108 for anther dehiscence. Plant Physiol. 2019, 181, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Sheard, L.B.; Tan, X.; Mao, H.; Withers, J.; Ben-Nissan, G.; Hinds, T.R.; Kobayashi, Y.; Hsu, F.F.; Sharon, M.; Browse, J.; et al. Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature 2010, 468, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Chen, Y.; Charnikhova, T.; Mulder, P.P.J.; Heijmans, J.; Hoogenboom, A.; Agalou, A.; Michel, C.; Morel, J.-B.; Dreni, L.; et al. OsJAR1 is required for JA-regulated floret opening and anther dehiscence in rice. Plant Mol. Biol. 2014, 86, 19–33. [Google Scholar] [CrossRef]
- Delfin, J.C.; Kanno, Y.; Seo, M.; Kitaoka, N.; Matsuura, H.; Tohge, T.; Shimizu, T. AtGH3.10 is another jasmonic acid-amido synthetase in Arabidopsis thaliana. Plant J. 2022, 110, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y. Study on the double petal formation mechanism of Grandiflora clematis. Master’s Thesis, Southwest Forestry University, Kunming, China, 4 June 2013. [Google Scholar]
- González, R.M.; Iusem, N.D. Twenty years of research on Asr (ABA-stress-ripening) genes and proteins. Planta 2014, 239, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Kim, S.H.; Berberich, T.; Kusano, T. Identification and properties of a small protein that interacts with a tobacco bZIPtype transcription factor TBZF. Plant Biotechnol. 2012, 29, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Han, J.; Deng, X.; Tan, S.; Li, L.; Li, L.; Zhou, J.; Peng, H.; Yang, G.; He, G.; et al. Expansion and stress responses of AP2/EREBP superfamily in Brachypodium distachyon. Sci. Rep. 2016, 6, 21623. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Luo, F.; Hochholdinger, F. LOB domain proteins: Beyond lateral organ boundaries. Trends Plant Sci. 2016, 21, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Zobell, O.; Faigl, W.; Saedler, H.; Münster, T. MIKC* MADS-box proteins: Conserved regulators of the gametophytic generation of land plants. Mol. Biol. Evol. 2010, 27, 1201–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Qi, S.; Touqeer, A.; Li, H.; Zhang, X.; Liu, X.; Wu, S. SlGT11 controls floral organ patterning and floral determinacy in tomato. BMC Plant Biol 2020, 20, 562. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 4, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads (M) | Total Clean Reads (M) | Total Clean Bases (Gb) | Clean Bases of Q20 (%) | Mapped Reads |
|---|---|---|---|---|---|
| PeSi1 | 47.33 | 43.59 | 6.54 | 96.34 | 40.63 (93.22%) |
| PeSi2 | 49.08 | 43.74 | 6.56 | 97.08 | 40.85 (93.39%) |
| PeSi3 | 45.57 | 42.04 | 6.31 | 96.89 | 39.67 (94.37%) |
| StSi1 | 47.33 | 43.19 | 6.48 | 96.72 | 40.34 (93.39%) |
| StSi2 | 47.33 | 43.30 | 6.50 | 96.86 | 40.55 (93.65%) |
| StSi3 | 49.08 | 43.98 | 6.60 | 96.16 | 40.30 (91.63%) |
| PeDo1 | 49.08 | 43.53 | 6.53 | 96.75 | 40.62 (93.31%) |
| PeDo2 | 49.08 | 43.38 | 6.51 | 96.88 | 40.41 (93.15%) |
| PeDo3 | 50.83 | 44.70 | 6.70 | 96.63 | 41.56 (92.98%) |
| StDo1 | 49.08 | 44.07 | 6.61 | 97.06 | 40.66 (92.27%) |
| StDo2 | 47.33 | 43.10 | 6.47 | 97.03 | 39.99 (92.79%) |
| StDo3 | 47.33 | 42.50 | 6.38 | 97.03 | 39.23 (92.31%) |
| Function Category | Gene Name (Gene ID) |
|---|---|
| Signal transduction | serine/threonine-protein phosphatase (CDL15_Pgr004407) |
| mitogen-activated protein kinase (CDL15_Pgr001413, CDL15_Pgr003484, CDL15_Pgr019927, CDL15_Pgr006403) | |
| interleukin-1 receptor-associated kinase 4 (CDL15_Pgr008721, CDL15_Pgr012996, CDL15_Pgr014246) | |
| 1-phosphatidylinositol-4-phosphate 5-kinase (CDL15_Pgr003539, CDL15_Pgr019098, CDL15_Pgr026956, CDL15_Pgr013697, CDL15_Pgr018872) | |
| DNA repair | centromere protein S (CDL15_Pgr011715, CDL15_Pgr015888, CDL15_Pgr023953) |
| ATP-dependent DNA helicase 2 (CDL15_Pgr007504, CDL15_Pgr025038, CDL15_Pgr028666) | |
| RuvB-like protein 1 (CDL15_Pgr006382) | |
| DNA replication licensing factor (CDL15_Pgr024576, CDL15_Pgr007997) | |
| DNA repair protein (CDL15_Pgr011609) | |
| Translation release | peptide chain release factor 1 (CDL15_Pgr018587, CDL15_Pgr021630) |
| peptide chain release factor 2 (CDL15_Pgr003092, CDL15_Pgr018186) | |
| peptidyl-tRNA hydrolase (CDL15_Pgr003817) | |
| Transmembrane transport | protein transport protein (BGI_novel_G000167) |
| mitochondrial import inner membrane translocase subunit (CDL15_Pgr012086, CDL15_Pgr026447) | |
| Redox balance | peroxidase (CDL15_Pgr017144) |
| laccase (CDL15_Pgr000587, CDL15_Pgr006286, CDL15_Pgr006287, CDL15_Pgr007746, CDL15_Pgr007747, CDL15_Pgr007840) | |
| Lignin metabolism | cinnamyl-alcohol dehydrogenase (CDL15_Pgr003366) |
| caffeic acid 3-O-methyltransferase (CDL15_Pgr023102) | |
| laccase (CDL15_Pgr000587, CDL15_Pgr006286, CDL15_Pgr006287, CDL15_Pgr007746, CDL15_Pgr007747, CDL15_Pgr007840) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, Y.; Yang, H.; Ding, W.; Huang, T.; Yuan, Z.; Zhu, Z. Combined Transcriptome and Proteome Analysis Provides Insights into Petaloidy in Pomegranate. Plants 2023, 12, 2402. https://doi.org/10.3390/plants12132402
Huo Y, Yang H, Ding W, Huang T, Yuan Z, Zhu Z. Combined Transcriptome and Proteome Analysis Provides Insights into Petaloidy in Pomegranate. Plants. 2023; 12(13):2402. https://doi.org/10.3390/plants12132402
Chicago/Turabian StyleHuo, Yan, Han Yang, Wenjie Ding, Tao Huang, Zhaohe Yuan, and Zunling Zhu. 2023. "Combined Transcriptome and Proteome Analysis Provides Insights into Petaloidy in Pomegranate" Plants 12, no. 13: 2402. https://doi.org/10.3390/plants12132402