
Identification of Late Ripening Citrus Mutant, Ara-unshiu (Citrus unshiu), and Its Selectable Marker

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Analysis of Fruit Traits
2.3. Whole Genome Re-Sequencing, Detection of Single Nucleotide Polymorphism (SNP) and Insertion/Deletion (InDel) Variants, and Gene Ontology (GO) Analysis
2.4. Allele-Specific PCR Markers
3. Results and Discussion
3.1. Selection of Mutant Lines by Gamma Irradiation
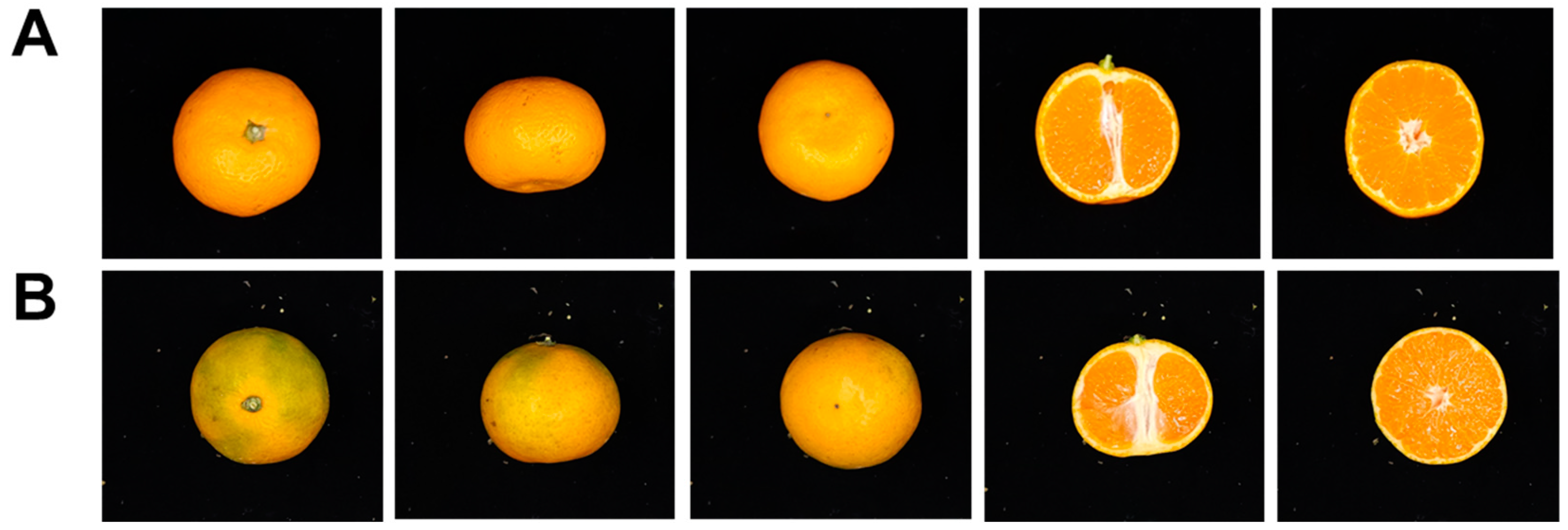
3.2. Comparison of Morphological Traits between Ara-unshiu and Wild Type (WT) Fruit
3.3. Mapping of Re-Sequencing Reads to C. unshiu Marc. Miyagawa-wase CUMW_v1.0
3.4. SNP and InDel Genetic Variation between WT and Ara-unshiu
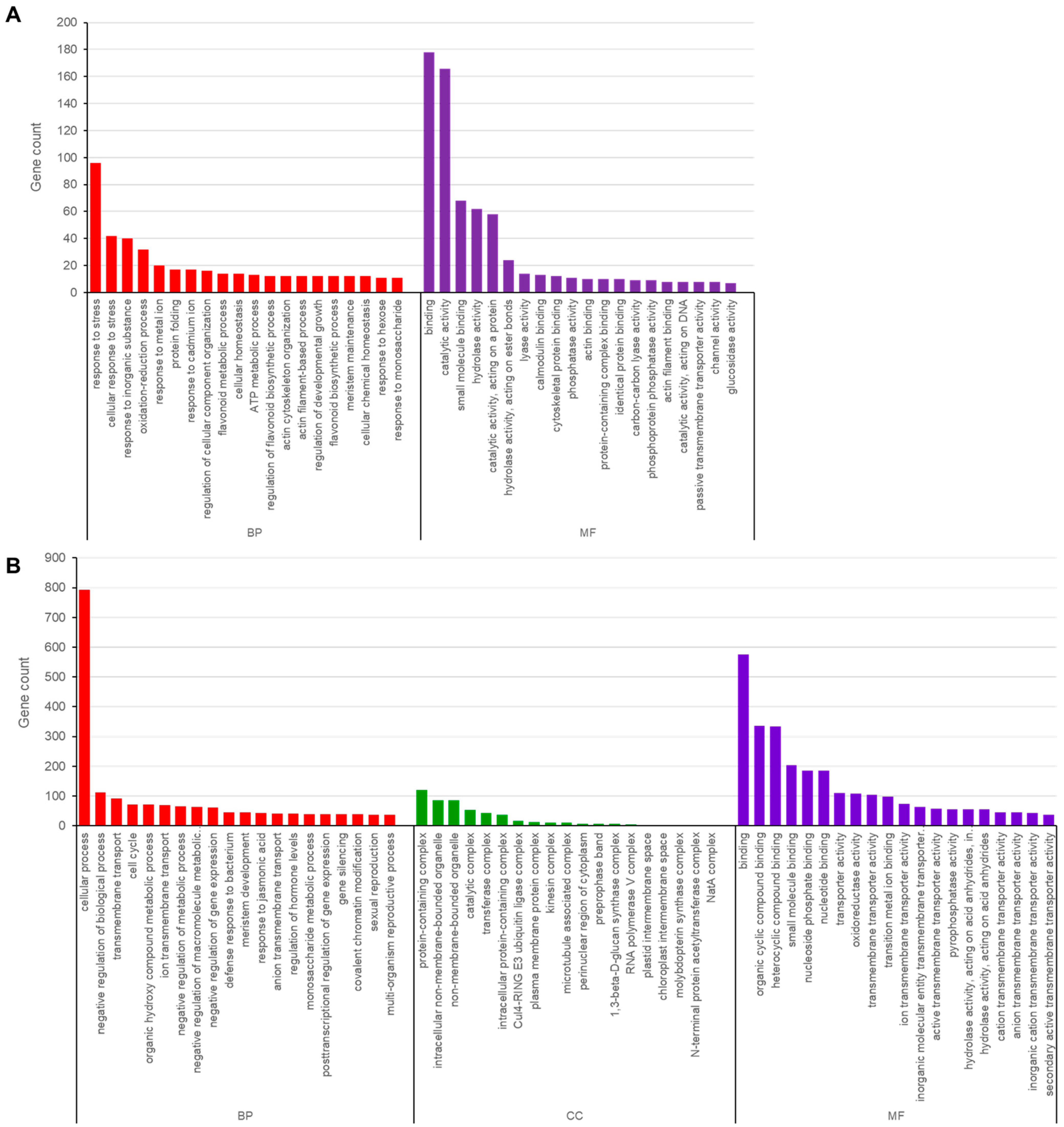
3.5. Functional Annotation of Genes Containing SNPs and InDels in Ara-unshiu
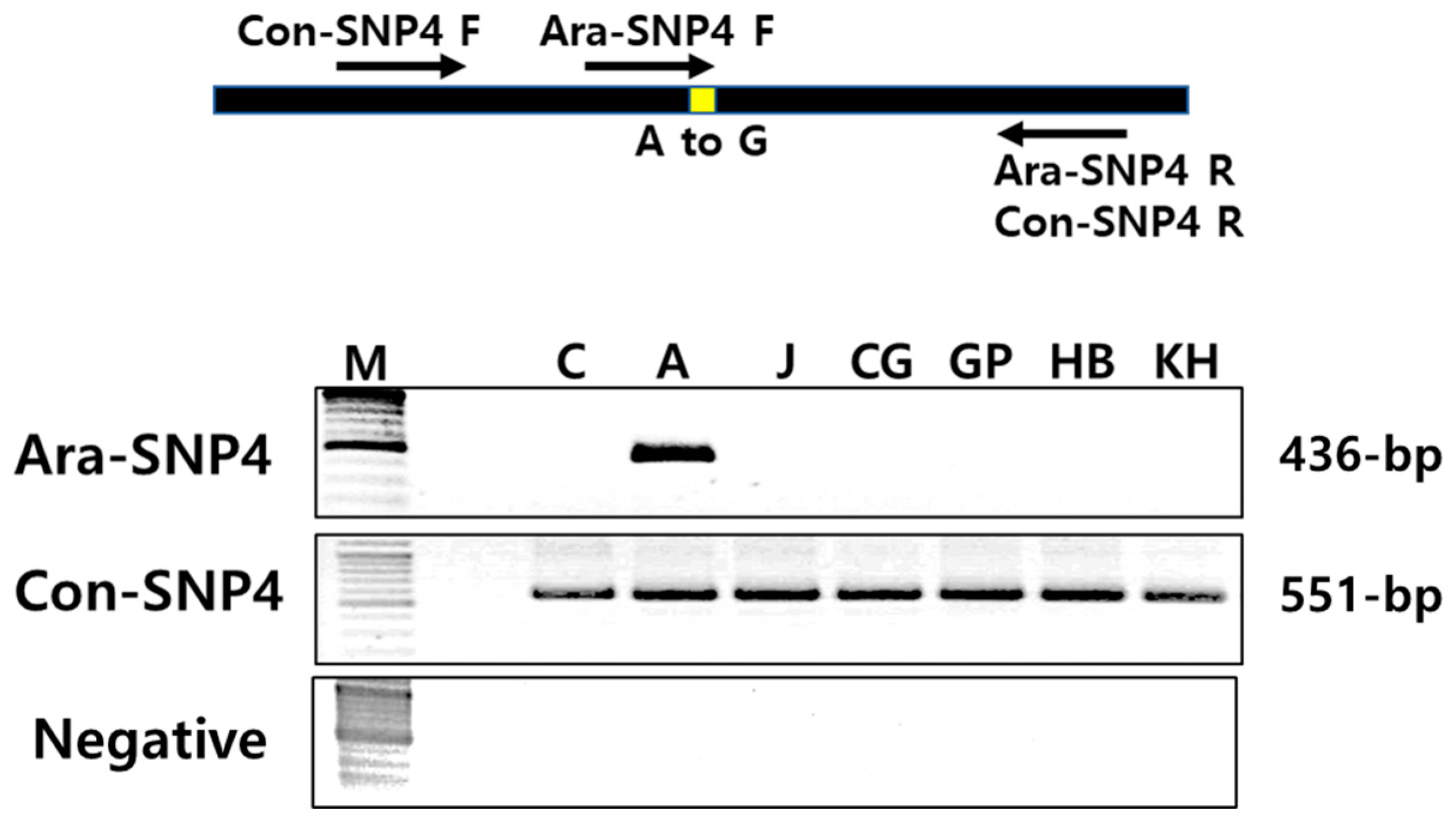
3.6. Identification of Specific Selection Markers for Ara-unshiu
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.H.; Handayani, E.; Wakana, A.; Sato, M.; Miyamoto, M.; Miyazaki, R.; Zhou, X.; Sakai, K.; Mizunoe, Y.; Shigyo, M.; et al. Distribution and evolution of Citrus accessions with S3 and/or S11 alleles for self-incompatibility with an emphasis on sweet orange [Citrus sinensis (L.) Osbeck; SfS3 or SfS3sm]. Genet. Resour. Crop Evol. 2020, 67, 2101–2117. [Google Scholar] [CrossRef]
- Raveh, E.; Goldenberg, L.; Porat, R.; Carmi, N.; Gentile, A.; La Malfa, S. Conventional Breeding of Cultivated Citrus Varieties. In The Citrus Genome; Gentile, A., La Malfa, S., Deng, Z., Eds.; Compendium of Plant Genomes; Springer: Cham, Switzerland, 2020; pp. 33–48. [Google Scholar]
- Ollitrault, P.; Ahmed, D.; Costantino, G.; Evrard, J.C.; Cardi, C.; Mournet, P.; Perdereau, A.; Froelicher, Y. Segregation distortion for male parents in high density genetic maps from reciprocal crosses between two self-incompatible cultivars confirms a gametophytic system for self-incompatibility in citrus. Agreculture 2021, 11, 379. [Google Scholar] [CrossRef]
- Soost, R.K.; Roose, M.L. Fruit breeding. In Tree and Tropical Fruits; Janick, J., Moore, J.N., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 1996; Volume 1, pp. 257–323. [Google Scholar]
- Ge, H.; Li, Y.; Fu, H.; Long, G.; Luo, L.; Li, R.; Deng, Z. Production of sweet orange somaclones tolerant to citrus canker disease by in vitro mutagenesis with EMS. Plant Cell Tissue Org. Cult. 2015, 123, 29–38. [Google Scholar] [CrossRef]
- Saadati, S.; Borzouei, A.; Rahemi, M.R.; Khiabani, B.N. Alteration of physiological and biochemical properties in leaves and fruits of pomegranate in response to gamma irradiation. Sci. Rep. 2022, 12, 4312. [Google Scholar] [CrossRef]
- Sutarto, I.; Agisimanto, D.; Supriyanto, A. Development of Promising Seedless Citrus Mutants through Gamma Irradiation; Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2009. [Google Scholar]
- Mariana, B.D.; Arisah, H.; Yenni, Y.; Selvawajayant, M. Seedless fruit pummelo induced by Gamma Ray irradiation: Fruit morphological characters and stability evaluation. Biodiversitas 2018, 19, 706–711. [Google Scholar] [CrossRef]
- Mahmoud, G.A.; El-Tobgy, K.M.K.; Abo-El-Seoud, M. Application of combined biocides and gamma radiation for keeping good quality stored grapefruits. Arch. Phytopathol. Plant Prot. 2010, 43, 712–721. [Google Scholar] [CrossRef]
- Starrantino, A.; Russo, F.; Donini, B.; Spina, P. Lemon mutants obtained by gamma irradiation of the nucellus cultured in vitro. Proc. Int. Soc. Citric. 1988, 2, 231–235. [Google Scholar]
- Gulsen, O.; Uzun, A.; Pala, H.; Canilhos, E.; Kafa, G. Development of seedless and Mal seco tolerant mutant lemons through budwood irradiation. Sci. Hortic. 2007, 112, 184–190. [Google Scholar] [CrossRef]
- Moussa, J.P. Role of gamma irradiation in regulation of NO3 level in rocket (Eruca vescaria subsp. sativa) plants. Russ. J. Plant Physiol. 2006, 53, 193–197. [Google Scholar] [CrossRef]
- Jankowicz-Cieslak, J.; Tai, T.H.; Kumlehn, J.; Till, B.J. Biotechnologies for Plant Mutation Breeding: Protocols; Springer Nature: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Ashraf, M.; Cheema, A.A.; Rashid, M.; Qamar, Z. Efect of gamma rays on M~ l generation in basmati rice. Pak. J. Bot. 2004, 35, 791–796. [Google Scholar]
- Eun, C.H.; Kim, I.J. The citrus mutant Jedae-unshiu induced by gamma irradiation exhibits a unique fruit shape and increased flavonoid content. Plants 2022, 11, 1337. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, J.; Nguyen, C.; Ampofo, B.; Zhong, S.; Fei, Z. The epigenome and transcriptional dynamics of fruit ripening. Annu. Rev. Plant Biol. 2017, 68, 61–84. [Google Scholar] [CrossRef] [PubMed]
- Brumos, J. Gene regulation in climacteric fruit ripening. Curr. Opin. Plant Biol. 2021, 63, 102042. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, K.; Grierson, D. Molecular and hormonal mechanisms regulating fleshy fruit ripening. Cells 2021, 10, 1136. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Liu, Q.; Xiong, J.J.; Deng, X.X. Difference of a citrus late-ripening mutant (Citrus sinensis) from its parental line in sugar and acid metabolism at the fruit ripening stage. Sci. China Ser. C-Life Sci. 2007, 50, 511–517. [Google Scholar] [CrossRef]
- Wu, J.; Xu, Z.; Zhang, Y.; Chai, L.; Yi, H.; Deng, X. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J. Exp. Bot. 2014, 65, 1651–1671. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Wang, X.J.; Wu, J.X.; Chen, S.Y.; Chen, H.; Chai, L.J.; Yi, H.L. Comparative transcriptome analyses between a spontaneous late-ripening sweet orange mutant and its wild type suggest the functions of ABA, sucrose and JA during citrus fruit ripening. PLoS ONE 2014, 9, e116056. [Google Scholar] [CrossRef]
- Alós, E.; Distefano, G.; Rodrigo, M.J.; Gentile, A. Altered sensitivity to ethylene in ‘Tardivo’, a late-ripening mutant of Clementine mandarin. Physiol. Plant 2013, 151, 507–521. [Google Scholar] [CrossRef]
- Pan, H.; Lyu, S.; Chen, Y.; Xu, S.; Ye, J.; Chen, G.; Wu, S.; Li, X.; Chen, J.; Pan, D. MicroRNAs and transcripts associated with an early ripening mutant of Pomelo (Citrus grandis Osbeck). Int. J. Mol. Sci. 2021, 22, 9348. [Google Scholar] [CrossRef]
- Mi, L.; Ma, D.; Lv, S.; Xu, S.; Zhong, B.; Peng, T.; Liu, D.; Liu, Y. Comparative transcriptome and sRNAome analyses reveal the regulatory mechanisms of fruit ripening in a spontaneous early-ripening navel orange mutant and its wild type. Genes 2022, 13, 1706. [Google Scholar] [CrossRef]
- Gao, Y.; Wei, W.; Fan, Z.; Zhao, X.; Zhang, Y.; Jing, Y.; Zhu, B.; Zhu, H.; Shan, W.; Chen, J.; et al. Re-evaluation of the nor mutation and the role of the NAC-NOR transcription factor in tomato fruit ripening. J. Exp. Bot. 2020, 71, 3560–3574. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, C.; Long, D.; Jiang, Y.; He, L.; Wang, Z.; Ma, X.; Bai, F.; Liu, J.; Wu, L.; et al. Genome-wide identification, bioinformatics characterization and functional analysis of pectin methylesterase inhibitors related to low temperature-induced juice sac granulation in navel orange (Citrus sinensis Osbeck). Sci. Hortic. 2022, 298, 110983. [Google Scholar] [CrossRef]
- Juan-Cabot, A.; Galmés, J.; Conesa, M.A. The tomato long shelf-life fruit phenotype: Knowledge, uncertainties and prospects. Sci. Hortic. 2022, 291, 110578. [Google Scholar] [CrossRef]
- Healey, A.; Furtado, A.; Cooper, T.; Henry, R.J. Protocol: A simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 2014, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Eun, C.H.; Kim, I.J. Genome-wide DNA polymorphisms of Citrus unshiu Marc. cv. Miyagawa-wase cultivated in different regions based on whole-genome re-sequencing. Plant Biotechnol. Rep. 2021, 15, 551–559. [Google Scholar] [CrossRef]
- Bui, M.; Liu, Z. Simple allele-discriminating PCR for cost-effective and rapid genotyping and mapping. Plant Methods 2009, 5, 1. [Google Scholar] [CrossRef]
- Kim, I.J.; Kim, O.R.; Kim, H.W.; Lee, S.H.; Kim, K.M.; Lee, H.Y. Status of citrus mutation breeding with gamma ray irradiation. J. Subtrop. Agri. Biotechnol. 2008, 24, 37–42. [Google Scholar]
- Kim, I.J.; Song, S.Y.; Lee, H.Y. Putative induced by gamma ray irradiation. J. Asian Agric. Biotechnol. 2009, 25, 1–4. [Google Scholar]
- Shimizu, T.; Tanizawa, Y.; Mochizuki, T.; Nagasaki, H.; Yoshioka, T.; Toyoda, A.; Fujiyama, A.; Kaminuma, E.; Nakamura, Y. Draft sequencing of the heterozygous diploid genome of satsuma (Citrus unshiu Marc.) using a hybrid assembly approach. Front. Genet. 2017, 8, 180. [Google Scholar] [CrossRef]
- Torkamaneh, D.; Boyle, B.; Belzile, F. Efficient genome-wide genotyping strategies and data integration in crop plants. Theor. Appl. Genet. 2018, 131, 499–511. [Google Scholar] [CrossRef]
- Liu, J.; Huang, S.M.; Sun, M.Y.; Liu, S.Y.; Liu, Y.M.; Wang, W.X.; Zhang, X.R.; Wang, H.Z.; Hua, W. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods 2012, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, J.; Fan, F.J.; Zhu, J.Y.; Chen, T.; Wang, C.L.; Zheng, T.Q.; Zhang, J.; Zhong, W.G.; Xu, J.L. Development of AS-PCR marker based on a key mutation confirmed by resequencing of Wx-mp in milky princess and its application in japonica soft rice (Oryza sativa L.) breeding. Plant Breed. 2013, 132, 595–603. [Google Scholar] [CrossRef]
- Chen, H.; Shan, J.; Yang, K.; Wang, Y.Y.; Lu, C.M. Abundant variation of Waxy gene in Yunnan rice landraces and molecular characterization of a novel Wxzm allele. Crop Sci. 2014, 54, 2152–2159. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, Y.; Feng, L.; Hao, W.; Li, C.; Yang, Y.; Fan, X.; Li, Q.; Zhang, C.; Liu, Q. Genetic dissection and functional differentiation of ALKa and ALKb, two natural alleles of the ALK/SSIIa gene, responding to low gelatinization temperature in rice. Rice 2020, 13, 39. [Google Scholar] [CrossRef]
- Polo-Oltra, Á.; Romero, C.; López, I.; Badenes, M.L.; Zuriaga, E. Cost-effective and time-efficient molecular assisted selection for PPV resistance in Apricot based on ParPMC2 allele-specific PCR. Agronomy 2020, 10, 1292. [Google Scholar] [CrossRef]
- Wang, F.Q.; Xu, Y.; Li, W.Q.; Chen, Z.H.; Wang, J.; Fan, F.J.; Tao, Y.J.; Jiang, Y.J.; Zhu, Q.H.; Yang, J. Creating a novel herbicide-tolerance OsALS allele using CRISPR/Cas9-mediated gene editing. Crop J. 2021, 9, 305–312. [Google Scholar] [CrossRef]
- Jiang, Y.; Ren, Y.; Xu, X.; Wang, H.; Wei, C. Application of allele specific PCR in identifying offspring genotypes of bi-allelic SbeIIb mutant lines in rice. Plants 2022, 11, 524. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Year | Horizontal Length | Vertical Length | Single Fruit Weight | Peel Thickness | Hardness | |
|---|---|---|---|---|---|---|
| (mm) | (mm) | (g) | (mm) | (G) | ||
| WT | 2021 | 46.27 ± 3.89 | 58.45 ± 4.03 | 87.60 ± 13.73 | 2.12 ± 0.22 | 937 ± 58 |
| 2022 | 47.83 ± 3.68 | 62.32 ± 6.64 | 98.94 ± 29.29 | 2.65 ± 0.43 | 862 ± 119 | |
| Ara-unshiu | 2021 | 50.78 ± 2.39 | 58.82 ± 3.18 | 95.41 ± 15.19 | 2.27 ± 0.55 | 1352 ± 86 |
| 2022 | 53.06 ± 8.76 | 60.28 ± 9.96 | 108.9 ± 30.60 | 2.28 ± 0.76 | 1145 ± 444 |
| Year | Hunter Color Values | Total Soluble Solid | Acidity | |||
|---|---|---|---|---|---|---|
| L (Light) | a (Red) | b (Yellow) | (Brix) | (wt%) | ||
| WT | 2021 | 59.36 ± 1.50 | 25.22 ± 0.72 | 35.47 ± 0.59 | 9.38 ± 0.27 | 0.72 ± 0.08 |
| 2022 | 59.47 ± 1.62 | 25.92 ± 2.01 | 35.28 ± 1.05 | 9.41 ± 0.32 | 0.46 ± 0.03 | |
| Ara-unshiu | 2021 | 63.04 ± 3.33 | 13.76 ± 2.52 | 37.21 ± 2.58 | 8.96 ± 0.27 | 0.78 ± 0.07 |
| 2022 | 62.94 ± 9.52 | 6.84 ± 8.89 | 37.17 ± 5.85 | 9.27 ± 1.48 | 0.73 ± 014 | |
| Sample | Clean Reads 1 | Mapped Reads 2 | Mapped Regions 3 (%) | Coverage 4 |
|---|---|---|---|---|
| WT | 80,693,250 | 71,296,288 (88.35%) | 310,983,030 (86.75%) | ≒27.49× |
| Ara-unshiu | 80,976,944 | 77,318,919 (88.90%) | 312,053,993 (86.77%) | ≒29.50× |
| Number 1 | Homozygous | Heterozygous | ||
|---|---|---|---|---|
| WT vs. Ara-unshiu | SNP | 3469 | 46 | 3423 |
| InDel | 7162 | 45 | 7117 |
| Number | Within Genes 1 | GO Genes 2 | ||
|---|---|---|---|---|
| WT vs. Ara-unshiu | SNP | 2057 | 323 | 364 |
| InDel | 3612 | 1180 | 1108 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, J.-M.; Eun, C.-H.; Kim, I.-J. Identification of Late Ripening Citrus Mutant, Ara-unshiu (Citrus unshiu), and Its Selectable Marker. Plants 2023, 12, 3355. https://doi.org/10.3390/plants12193355
Heo J-M, Eun C-H, Kim I-J. Identification of Late Ripening Citrus Mutant, Ara-unshiu (Citrus unshiu), and Its Selectable Marker. Plants. 2023; 12(19):3355. https://doi.org/10.3390/plants12193355
Chicago/Turabian StyleHeo, Ji-Man, Chang-Ho Eun, and In-Jung Kim. 2023. "Identification of Late Ripening Citrus Mutant, Ara-unshiu (Citrus unshiu), and Its Selectable Marker" Plants 12, no. 19: 3355. https://doi.org/10.3390/plants12193355