

Comparative Genome-Wide Analysis of Two Caryopteris x Clandonensis Cultivars: Insights on the Biosynthesis of Volatile Terpenoids



Abstract
:1. Introduction
2. Results and Discussion
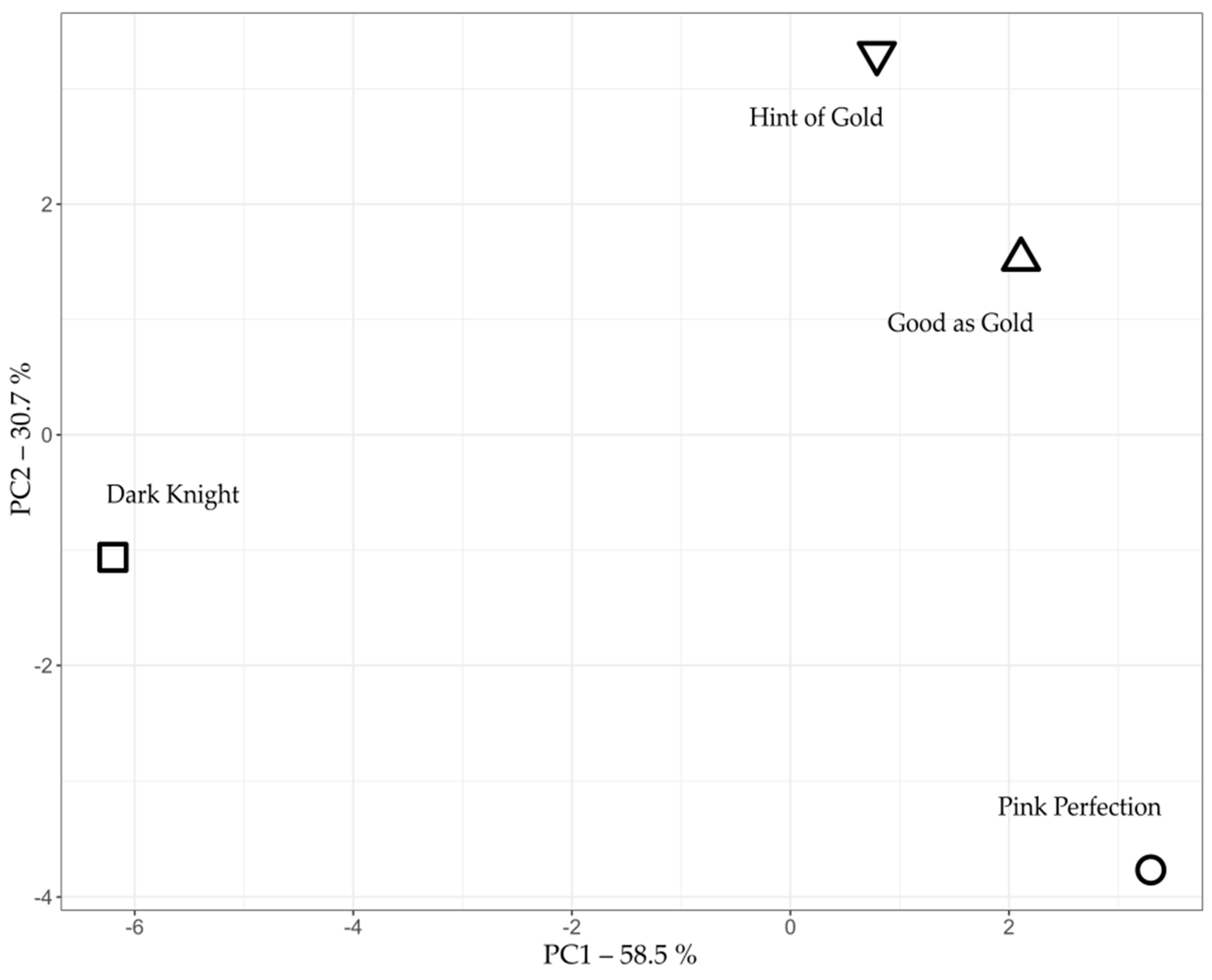
2.1. PCA Analysis of Volatile Compounds
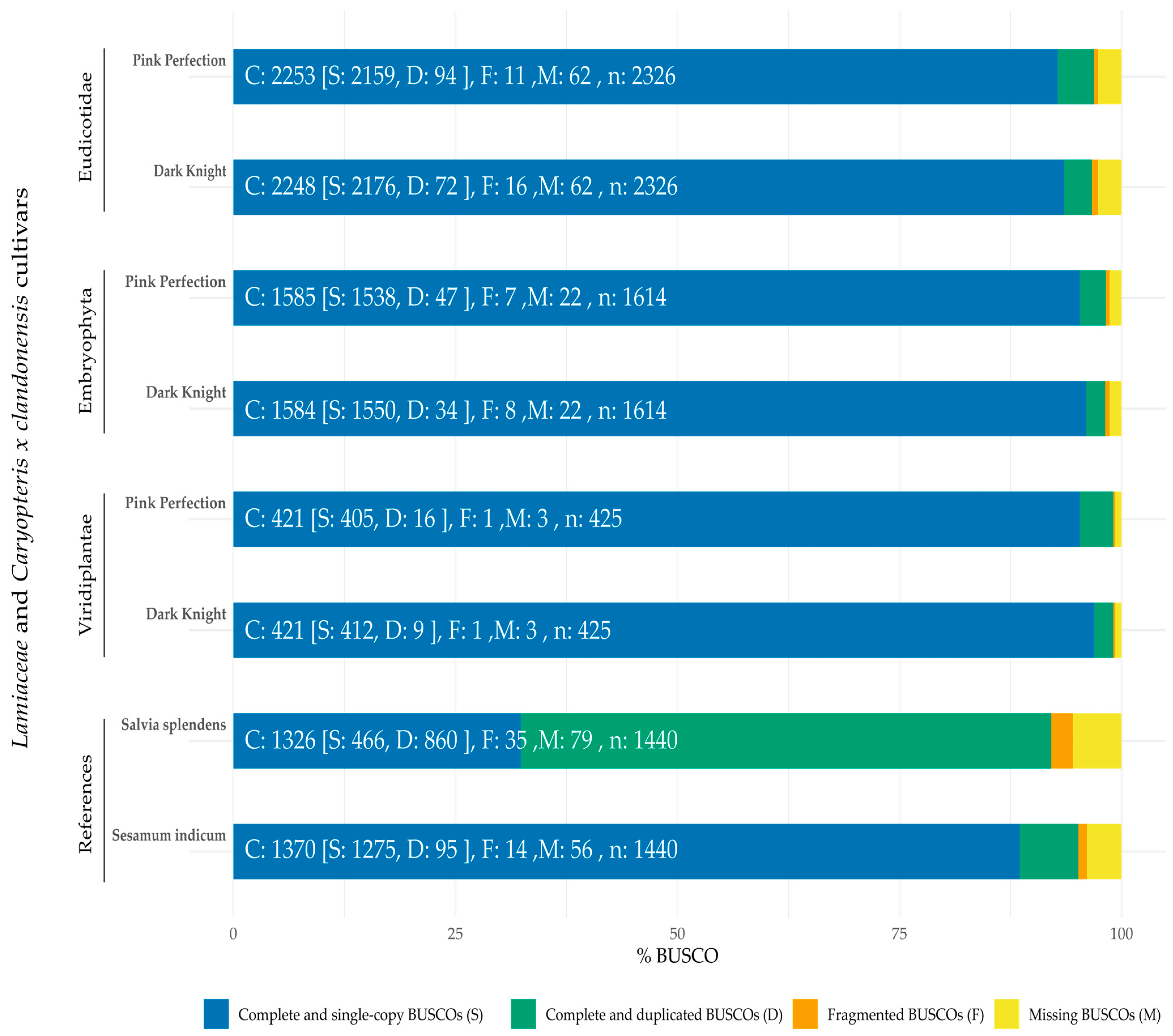
2.2. Genome Sequencing and Quality Assessment
2.3. Evaluation of Structural Differences between Genome Assemblies
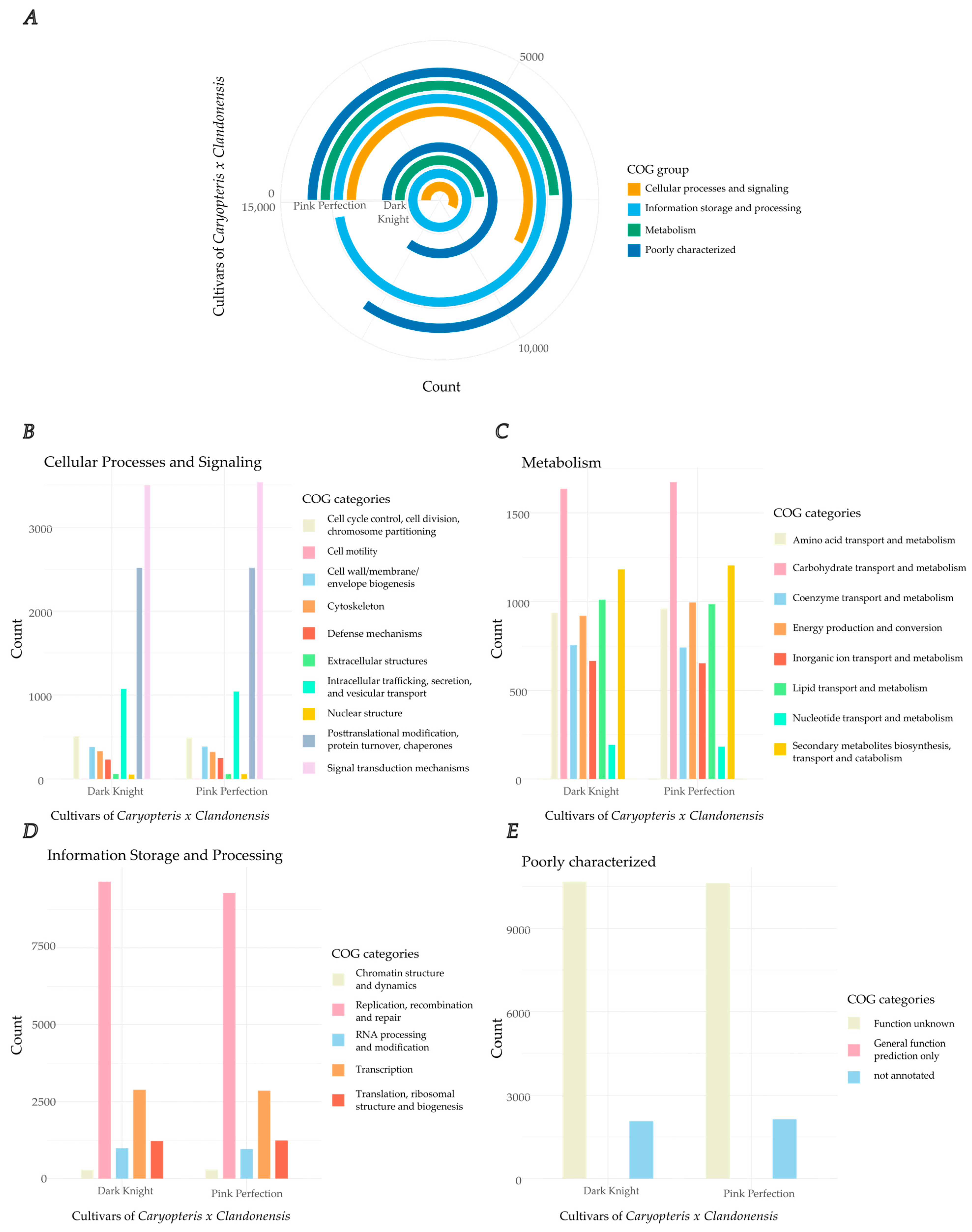
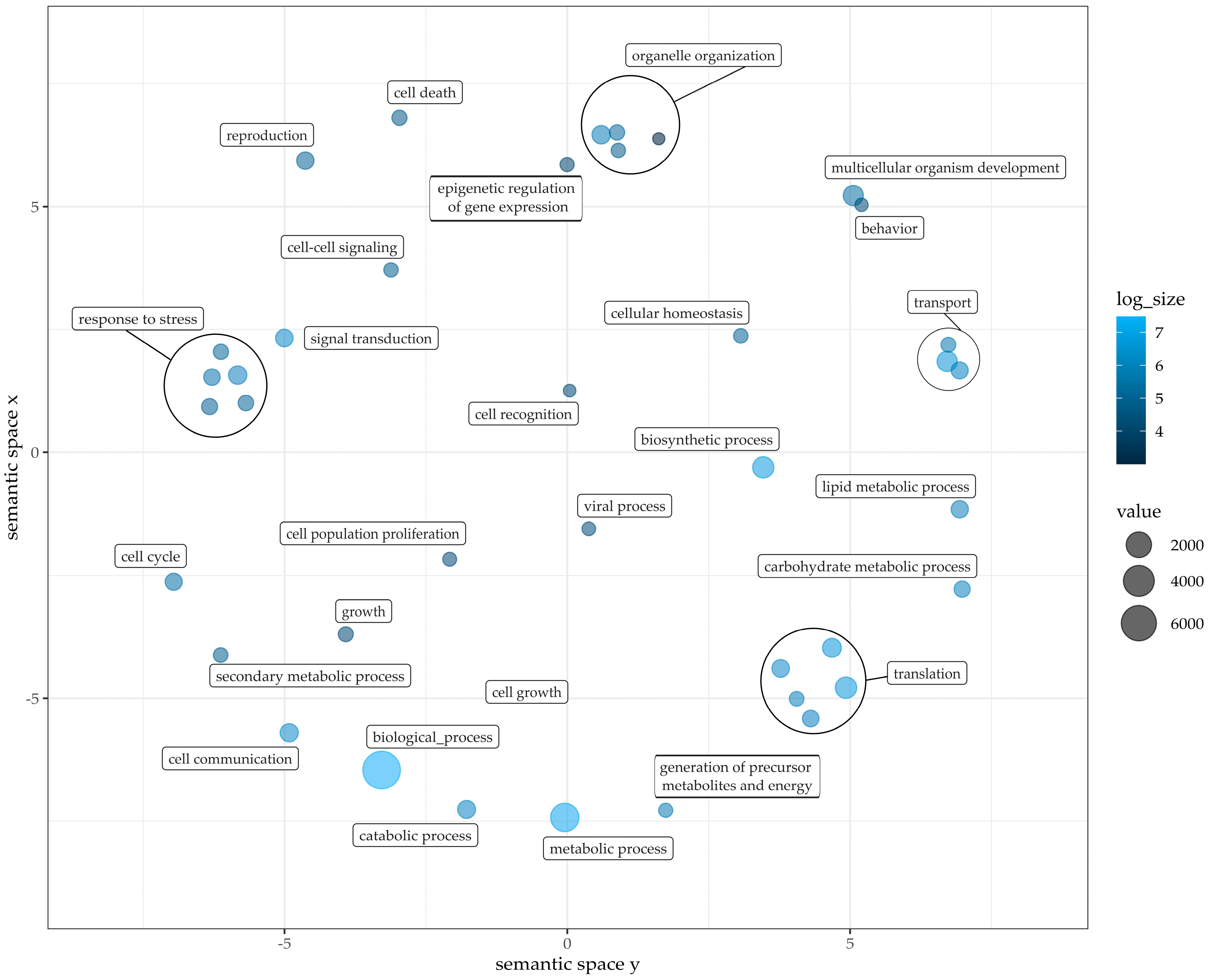
2.4. Gene Models and Functional Annotation
2.5. Identification of Terpenoid Biosynthesis Enzymes
3. Materials and Methods
3.1. Plant Material
3.2. GC-MS Analysis of Volatile Compounds
3.3. High Molecular Weight DNA Extraction and Library Preparation
3.4. Genome Sequencing and Assembly
3.5. RNA Long Read IsoSeq
3.6. Bioinformatic and Statistical Analysis
3.7. Identification of TPS and Cytochrome p450 Enzymes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caputi, L. Use of terpenoids as natural flavouring compounds in food industry. Recent Pat. Food Nutr. Agric. 2011, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Masyita, A.; Sari, R.M.; Astuti, A.D.; Yasir, B.; Rumata, N.R.; Emran, T.B.; Nainu, F.; Simal-Gandara, J. Terpenes and terpenoids as main bioactive compounds of essential oils, their roles in human health and potential application as natural food preservatives. Food Chem. X 2022, 13, 100217. [Google Scholar] [CrossRef] [PubMed]
- da Silva, G.L.; Luft, C.; Lunardelli, A.; Amaral, R.H.; Melo, D.A.D.S.; Donadio, M.V.; Nunes, F.B.; DE Azambuja, M.S.; Santana, J.C.; Moraes, C.M.; et al. Antioxidant, analgesic and anti-inflammatory effects of lavender essential oil. An. Acad. Bras. Cienc. 2015, 87, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Mediratta, P.; Sharma, K.; Singh, S. Evaluation of immunomodulatory potential of Ocimum sanctum seed oil and its possible mechanism of action. J. Ethnopharmacol. 2002, 80, 15–20. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.K.R.; Figueiredo, P.L.B.; Byler, K.G.; Setzer, W.N. Essential oils as antiviral agents, potential of essential oils to treat SARS-CoV-2 infection: An in-silico investigation. Int. J. Mol. Sci. 2020, 21, 3426. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, M.; Karimpour, H.; Monsef-Esfehani, H.R. Antinociceptive effects of Teucrium polium L. total extract and essential oil in mouse writhing test. Pharmacol. Res. 2003, 48, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Đorđević, S.; Petrović, S.; Dobrić, S.; Milenković, M.; Vučićević, D.; Žižić, S.; Kukić, J. Antimicrobial, anti-inflammatory, anti-ulcer and antioxidant activities of Carlina acanthifolia root essential oil. J. Ethnopharmacol. 2007, 109, 458–463. [Google Scholar] [CrossRef]
- Cowen, D.; Wolf, A.; Paige, B.H. Toxoplasmic encephalomyelitis. Arch. Neurol. Psychiatry 1942, 48, 689–739. [Google Scholar] [CrossRef]
- Jantan, I.; Ping, W.O.; Visuvalingam, S.D.; Ahmad, N.W. Larvicidal activity of the essential oils and methanol extracts of Malaysian plants on Aedes aegypti. Pharm. Biol. 2008, 41, 234–236. [Google Scholar] [CrossRef]
- Cox-Georgian, D.; Ramadoss, N.; Dona, C.; Basu, C. Therapeutic and medicinal uses of terpenes. Med. Plants Farm Pharm. 2019, 67, 333–359. [Google Scholar] [CrossRef] [Green Version]
- Sicora, O. The ethanolic stem extract of Caryopteris x Clandonensis Posseses antiproliferative potential by blocking breast cancer cells in mitosis. Farmacia 2019, 67, 1077–1082. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; Mcphail, A.T. Plant antitumor agents. VI. The isolation and structure of Taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Pichersky, E.; Raguso, R.A. Why do plants produce so many terpenoid compounds? New Phytol. 2018, 220, 692–702. [Google Scholar] [CrossRef]
- Holopainen, J.K.; Himanen, S.J.; Yuan, J.S.; Chen, F.; Stewart, C.N. Ecological Functions of Terpenoids in Changing Climates; Ramawat, K., Mérillon, J.M., Eds.; Natural Products; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Drapeau, J.; Rossano, M.; Touraud, D.; Obermayr, U.; Geier, M.; Rose, A.; Kunz, W. Green synthesis of para-Menthane-3,8-diol from Eucalyptus citriodora: Application for repellent products. Comptes Rendus Chim. 2011, 14, 629–635. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, S.H.; Hong, C.Y.; Park, S.Y.; Choi, I.G. Biotransformation of (-)-α-pinene and geraniol to α-terpineol and p-menthane-3,8-diol by the white rot fungus, Polyporus brumalis. J. Microbiol. 2017, 53, 462–467. [Google Scholar] [CrossRef]
- Drapeau, J.; Verdier, M.; Touraud, D.; Kröckel, U.; Geier, M.; Rose, A.; Kunz, W. Effective insect repellent formulation in both surfactantless and classical microemulsions with a long-lasting protection for human beings. Chem. Biodivers. 2009, 6, 934–947. [Google Scholar] [CrossRef]
- Blythe, E.; Tabanca, N.; Demirci, B.; Bernier, U.; Agramonte, N.; Ali, A.; Khan, I. Composition of the essential oil of Pink ChablisTM bluebeard (Caryopteris × clandonensis ’Durio’) and its biological activity against the yellow fever mosquito Aedes aegypti. Nat. Volatiles Essent. Oils 2015, 2, 11–21. [Google Scholar]
- Bathe, U.; Tissier, A. Cytochrome P450 enzymes: A driving force of plant diterpene diversity. Phytochemistry 2019, 161, 149–162. [Google Scholar] [CrossRef]
- Hernandez-Ortega, A.; Vinaixa, M.; Zebec, Z.; Takano, E.; Scrutton, N.S. A toolbox for diverse Oxyfunctionalisation of monoterpenes. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mabou, F.D.; Belinda, I.; Yossa, N. TERPENES: Structural classification and biological activities. IOSR J. Pharm. Biol. Sci. e-ISSN 2021, 16, 2319–7676. [Google Scholar]
- Nett, R.S.; Montanares, M.; Marcassa, A.; Lu, X.; Nagel, R.; Charles, T.C.; Hedden, P.; Rojas, M.C.; Peters, R.J. Elucidation of gibberellin biosynthesis in bacteria reveals convergent evolution. Nat. Chem. Biol. 2016, 13, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, L.; Zhuang, W.; Zhang, F.; Shu, X.; Wang, N.; Wang, Z. Recent research progress in Taxol biosynthetic pathway and acylation reactions mediated by Taxus Acyltransferases. Molecules 2021, 26, 2855. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Yu, R. Artemisinin biosynthesis and its regulatory enzymes: Progress and perspective. Pharmacogn. Rev. 2011, 5, 189–194. [Google Scholar] [CrossRef]
- Gershenzon, J.; Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007, 3, 408–414. [Google Scholar] [CrossRef]
- Zhang, X.; Niu, M.; da Silva, J.A.T.; Zhang, Y.; Yuan, Y.; Jia, Y.; Xiao, Y.; Li, Y.; Fang, L.; Zeng, S.; et al. Identification and functional characterization of three new terpene synthase genes involved in chemical defense and abiotic stresses in Santalum album. BMC Plant Biol. 2019, 19, 115. [Google Scholar] [CrossRef]
- Sharma, V.; Sarkar, I.N. Bioinformatics opportunities for identification and study of medicinal plants. Brief. Bioinform. 2013, 14, 238–250. [Google Scholar] [CrossRef]
- Helfrich, E.J.N.; Lin, G.-M.; Voigt, C.A.; Clardy, J. Bacterial terpene biosynthesis: Challenges and opportunities for pathway engineering. Beilstein J. Org. Chem. 2019, 15, 2889–2906. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Mostafa, S.; Lu, Z.; Du, R.; Cui, J.; Wang, Y.; Liao, Q.; Lu, J.; Mao, X.; Chang, B.; et al. The Jasmine (Jasminum sambac) genome provides insight into the biosynthesis of flower fragrances and Jasmonates. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef]
- Degenhardt, J.; Köllner, T.G.; Gershenzon, J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Q.; Li, J.; Luo, J.; Chen, W.; Li, X. Comparative study of volatile compounds in the fruit of two banana cultivars at different ripening stages. Molecules 2018, 23, 2456. [Google Scholar] [CrossRef]
- Cramer, A.-C.J.; Mattinson, D.S.; Fellman, J.K.; Baik, B.-K. Analysis of volatile compounds from various types of barley cultivars. J. Agric. Food Chem. 2005, 53, 7526–7531. [Google Scholar] [CrossRef]
- Dong, A.-X.; Xin, H.-B.; Li, Z.-J.; Liu, H.; Sun, Y.-Q.; Nie, S.; Zhao, Z.-N.; Cui, R.-F.; Zhang, R.-G.; Yun, Q.-Z.; et al. High-quality assembly of the reference genome for scarlet sage, Salvia splendens, an economically important ornamental plant. Gigascience 2018, 7, giy068. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Liu, H.; Wang, X.; Li, W.; Chen, M.-S.; Niu, L.-J. Chromatin architectures are associated with response to dark treatment in the oil crop Sesamum indicum, based on a high-quality genome assembly. Plant Cell Physiol. 2020, 61, 978–987. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; A Simão, F.; Zdobnov, E.M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Guan, D.; A McCarthy, S.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Lyons, E.; Pedersen, B.; Schnable, J.C.; Paterson, A.H.; Freeling, M. Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinform. 2011, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hong, W.-Y.; Cho, M.; Sim, M.; Lee, D.; Ko, Y.; Kim, J. Synteny Portal: A web-based application portal for synteny block analysis. Nucleic Acids Res. 2016, 44, W35–W40. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hunt, M.; Tsai, I.J. Inferring synteny between genome assemblies: A systematic evaluation. BMC Bioinform. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Arús, P.; Toshiya, Y.; Elisabeth, D.; Abbott, A.G. Synteny in the Rosaceae. In Plant Breeding Reviews; John and Wiley and Sons: Hoboken, NJ, USA, 2010; pp. 175–211. [Google Scholar]
- Devos, K.M.; Moore, G.; Gale, M.D. Conservation of marker synteny during evolution. Euphytica 1995, 85, 67–372. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-genome annotation with BRAKER. Methods Mol. Biol. 2019, 1962, 65–95. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS: Table 1. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Ge, X.; Yang, Z.; Qin, W.; Sun, G.; Wang, Z.; Li, Z.; Liu, J.; Wu, J.; Wang, Y.; et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nat. Commun. 2019, 10, 2989. [Google Scholar] [CrossRef]
- Liu, S.; An, Y.; Tong, W.; Qin, X.; Samarina, L.; Guo, R.; Xia, X.; Wei, C. Characterization of genome-wide genetic variations between two varieties of tea plant (Camellia sinensis) and development of InDel markers for genetic research. BMC Genom. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Raina, A.; Sahu, P.K.; Laskar, R.A.; Rajora, N.; Sao, R.; Khan, S.; Ganai, R.A. Mechanisms of genome maintenance in plants: Playing it safe with breaks and bumps. Front. Genet. 2021, 12, 675686. [Google Scholar] [CrossRef]
- Lim, C.; Pratama, M.Y.; Rivera, C.; Silvestro, M.; Tsao, P.S.; Maegdefessel, L.; Gallagher, K.A.; Maldonado, T.; Ramkhelawon, B. Linking single nucleotide polymorphisms to signaling blueprints in abdominal aortic aneurysms. Sci. Rep. 2022, 12, 20990. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Butler, J.B.; Freeman, J.; Potts, B.M.; Vaillancourt, R.; Grattapaglia, D.; Silva-Junior, O.B.; Simmons, B.; Healey, A.L.; Schmutz, J.; Barry, K.; et al. Annotation of the Corymbia terpene synthase gene family shows broad conservation but dynamic evolution of physical clusters relative to Eucalyptus. Heredity 2018, 121, 87–104. [Google Scholar] [CrossRef]
- Warren, R.L.; Keeling, C.I.; Yuen, M.M.S.; Raymond, A.; Taylor, G.A.; Vandervalk, B.P.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.D.; et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015, 83, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.-H.; Liu, H.; Zhang, R.-G.; Xu, J.; Zhou, S.-S.; Jiao, S.-Q.; Yan, X.-M.; Tian, X.-C.; Shi, T.-L.; Luo, H.; et al. Chromosome-scale assembly and evolution of the tetraploid Salvia splendens (Lamiaceae) genome. Hortic. Res. 2021, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Vining, K.J.; Qi, X.; Yu, X.; Zheng, Y.; Liu, Z.; Fang, H.; Li, L.; Bai, Y.; Liang, C.; et al. Genome-wide analysis of terpene synthase gene family in Mentha longifolia and catalytic activity analysis of a single terpene synthase. Genes 2021, 12, 518. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P.; Godden, G.T.; Lanier, E.; Bhat, W.W.; Kinser, T.J.; Vaillancourt, B.; Wang, H.; Wood, J.C.; Jiang, J.; Soltis, P.S.; et al. Generation of a chromosome-scale genome assembly of the insect-repellent terpenoid-producing Lamiaceae species, Callicarpa americana. Gigascience 2020, 9, giaa093. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Shalev, T.J.; Yuen, M.M.S.; Gesell, A.; Yuen, A.; Russell, J.H.; Bohlmann, J. An annotated transcriptome of highly inbred Thuja plicata (Cupressaceae) and its utility for gene discovery of terpenoid biosynthesis and conifer defense. Tree Genet. Genomes 2018, 14, 35. [Google Scholar] [CrossRef]
- Ringel, M.; Reinbold, M.; Hirte, M.; Haack, M.; Huber, C.; Eisenreich, W.; Masri, M.A.; Schenk, G.; Guddat, L.W.; Loll, B.; et al. Towards a sustainable generation of pseudopterosin-type bioactives. Green Chem. 2020, 22, 6033–6046. [Google Scholar] [CrossRef]
- Inglis, P.W.; Pappas, M.; Resende, L.V.; Grattapaglia, D. Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PLoS ONE 2018, 13, e0206085. [Google Scholar] [CrossRef]
- Healey, A.; Furtado, A.; Cooper, T.; Henry, R.J. Protocol: A simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 2014, 10, 21. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Extraction of total cellular DNA from plants, algae and fungi. Plant Mol. Biol. Man. 1994, 2, 183–190. [Google Scholar] [CrossRef]
- GitHub—PacificBiosciences/pbipa: Improved Phased Assembler. Available online: https://github.com/PacificBiosciences/pbipa (accessed on 11 December 2022).
- GitHub—dfguan/purge_dups: Haplotypic Duplication Identification Tool. Available online: https://github.com/dfguan/purge_dups (accessed on 11 December 2022).
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; A Simão, F.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Bao, J.; Reecy, J. CateGOrizer: A web-based program to batch analyze gene ontology classification categories. Online J. Bioinform. 2008, 9, 108–112. Available online: http://www.animalgenome.org/bioinfo/tools/catego/ (accessed on 11 December 2022).
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dark Knight | Good as Gold | Hint of Gold | Pink Perfection |
|---|---|---|---|
| α-pinene ** | D-limonene * | D-limonene * | D-limonene * |
| trans-pinocarveol ** | Cubebol | Cubebol | cis-p-mentha-1(7),8-dien-2-ol * |
| Pinocarvone ** | Carvone * | trans-carveol * | trans-p-mentha-2,8-dien-1-ol * |
| Caryophyllene oxide | trans-carveol * | Carvone * | Caryophyllene oxide |
| β-pinene ** | cis-p-mentha-1(7),8-dien-2-ol * | Caryophyllene oxide | trans-carveol * |
| (E,E)-α-farnesene | Caryophyllene oxide | trans-p-mentha-1(7),8-dien-2-ol * | cis-p-mentha-2,8-dien-1-ol * |
| α-campholenal | α-copaene | cis-p-mentha-1(7),8-dien-2-ol * | Carvone |
| α-copaene | β-pinene ** | cis-p-mentha-2,8-dien-1-ol * | α-pinene ** |
| Caryophyllene | cis-p-mentha-2,8-dien-1-ol * | α-copaene | β-pinene ** |
| D-limonene * | trans-p-mentha-2,8-dien-1-ol * | trans-p-mentha-2,8-dien-1-ol * | Caryophyllene |
| Analysis Metric | Dark Knight | Pink Perfection |
|---|---|---|
| Total Bases (Gb) | 444.13 | 229.43 |
| HiFi Reads | 1,823,939 | 843,632 |
| HiFi Yield (Gb) | 27.28 | 12.92 |
| HiFi Read Length (mean, bp) | 14,954 | 15,312 |
| HiFi Read Quality (median) | Q35 | Q34 |
| HiFi Number of Passes (mean) | 12 | 13 |
| Assembly | Dark Knight | Pink Perfection |
|---|---|---|
| # contigs | 1183 | 782 |
| Largest contig | 29,672,976 | 31,977,049 |
| Total length | 366,625,098 | 344,117,456 |
| Estimated reference length | 300,000,000 | 300,000,000 |
| GC (%) | 31.50 | 31.77 |
| N50 | 8,177,750 | 7,086,741 |
| L50 | 13 | 14 |
| # N’s per 100 kbp | 0.41 | 0.44 |
| TPS Subfamily | Dark Knight | Pink Perfection |
|---|---|---|
| a (green) | 16 | 14 |
| b (black) | 7 | 7 |
| c (purple) | 10 | 10 |
| d (blue) | - | - |
| e (turquoise) | 2 | 2 |
| f (petrol) | 5 | 5 |
| g (red) | 3 | 3 |
| h (pink) | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritz, M.; Ahmad, N.; Brueck, T.; Mehlmer, N. Comparative Genome-Wide Analysis of Two Caryopteris x Clandonensis Cultivars: Insights on the Biosynthesis of Volatile Terpenoids. Plants 2023, 12, 632. https://doi.org/10.3390/plants12030632
Ritz M, Ahmad N, Brueck T, Mehlmer N. Comparative Genome-Wide Analysis of Two Caryopteris x Clandonensis Cultivars: Insights on the Biosynthesis of Volatile Terpenoids. Plants. 2023; 12(3):632. https://doi.org/10.3390/plants12030632
Chicago/Turabian StyleRitz, Manfred, Nadim Ahmad, Thomas Brueck, and Norbert Mehlmer. 2023. "Comparative Genome-Wide Analysis of Two Caryopteris x Clandonensis Cultivars: Insights on the Biosynthesis of Volatile Terpenoids" Plants 12, no. 3: 632. https://doi.org/10.3390/plants12030632
APA StyleRitz, M., Ahmad, N., Brueck, T., & Mehlmer, N. (2023). Comparative Genome-Wide Analysis of Two Caryopteris x Clandonensis Cultivars: Insights on the Biosynthesis of Volatile Terpenoids. Plants, 12(3), 632. https://doi.org/10.3390/plants12030632