
Syntenic Cell Wall QTLs as Versatile Breeding Tools: Intraspecific Allelic Variability and Predictability of Biomass Quality Loci in Target Plant Species
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. General SQTL Alignment Patterns across Multiple Accessions of Six Angiosperm Species
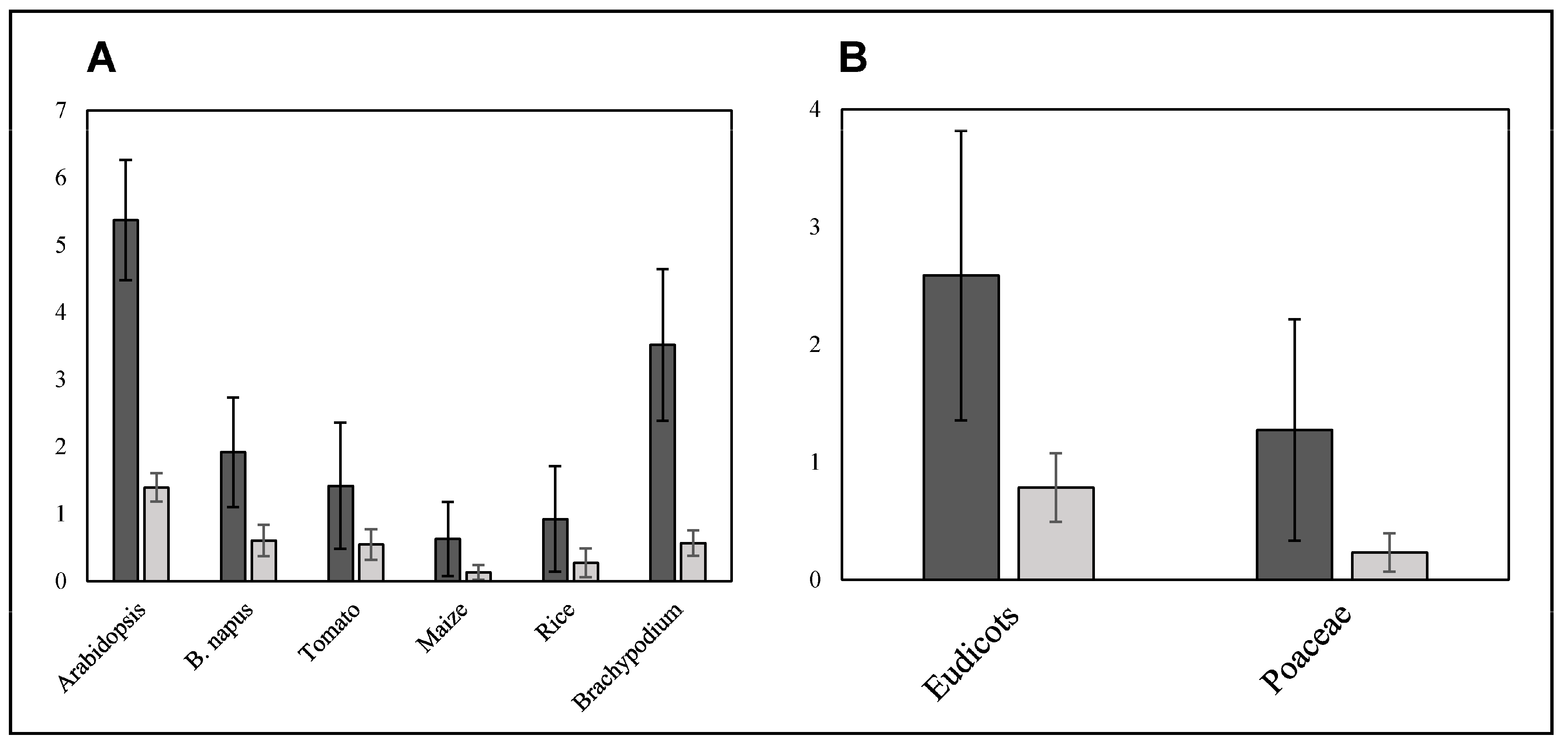
2.2. Large Nucleotide Variation within the Intra-Specific SQTL Aligning Regions
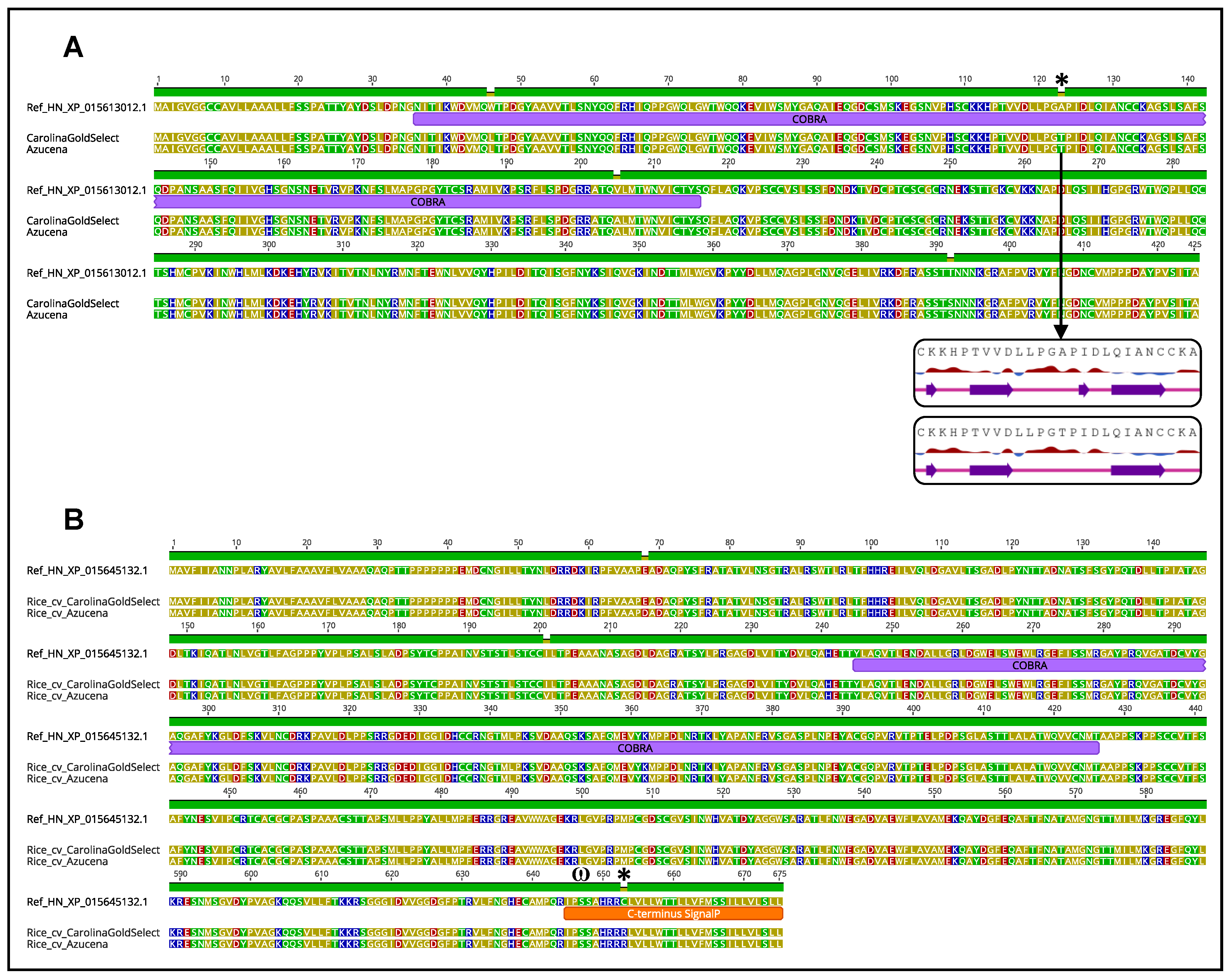
2.3. Intraspecific SQTL Variability Leads to Changes in Cell Wall Protein Sequences with a Potential Functional Impact
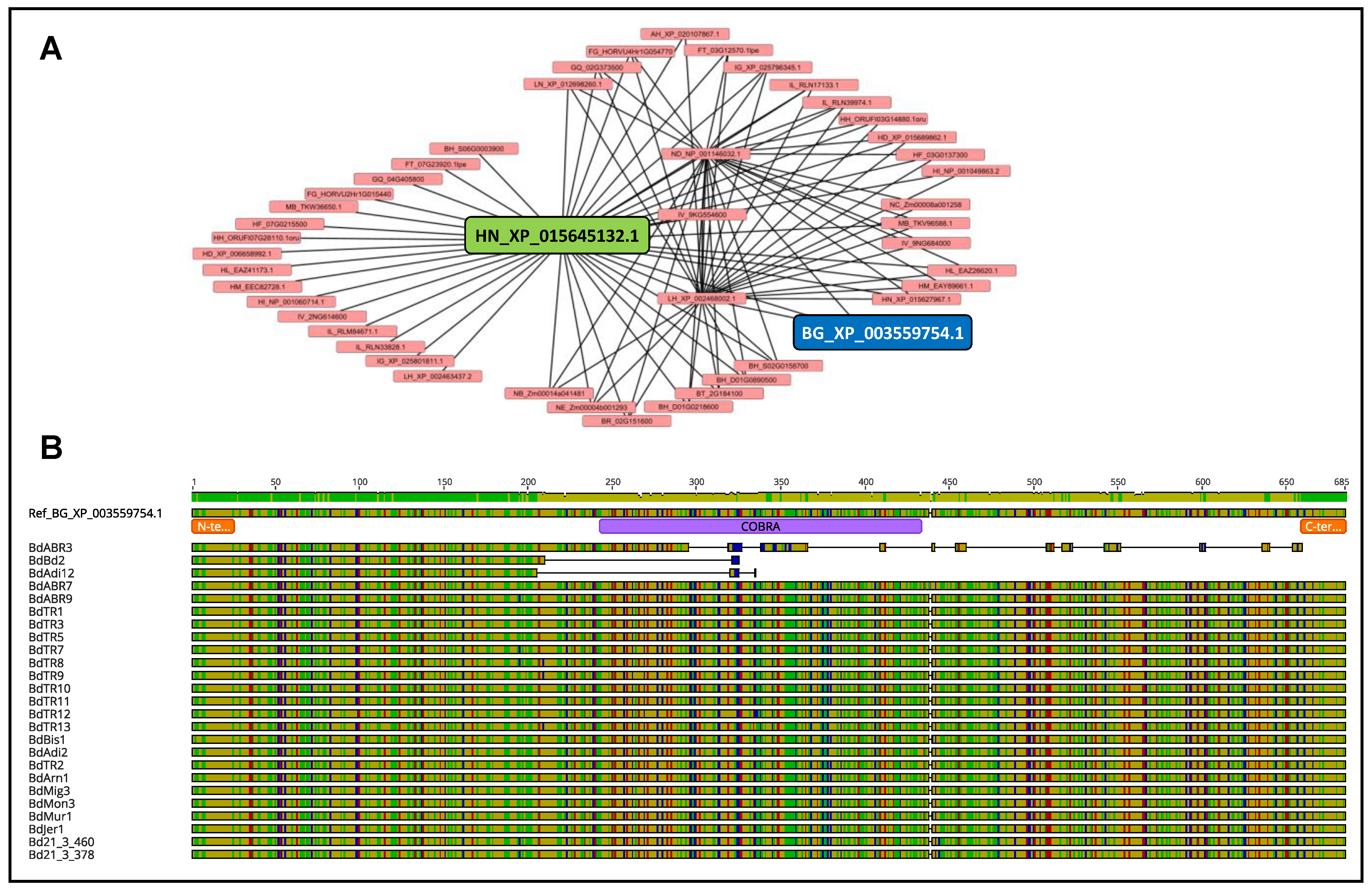
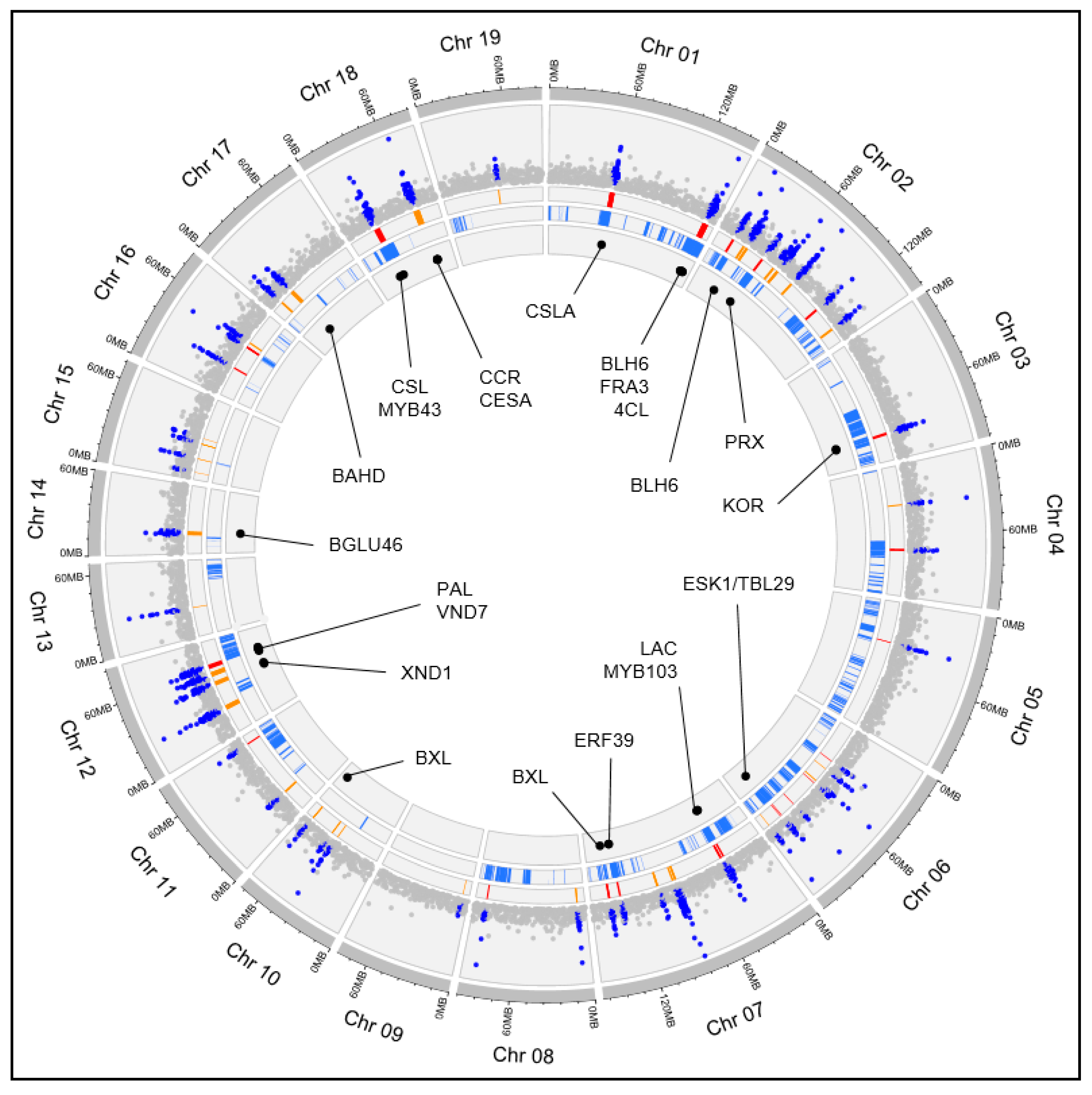
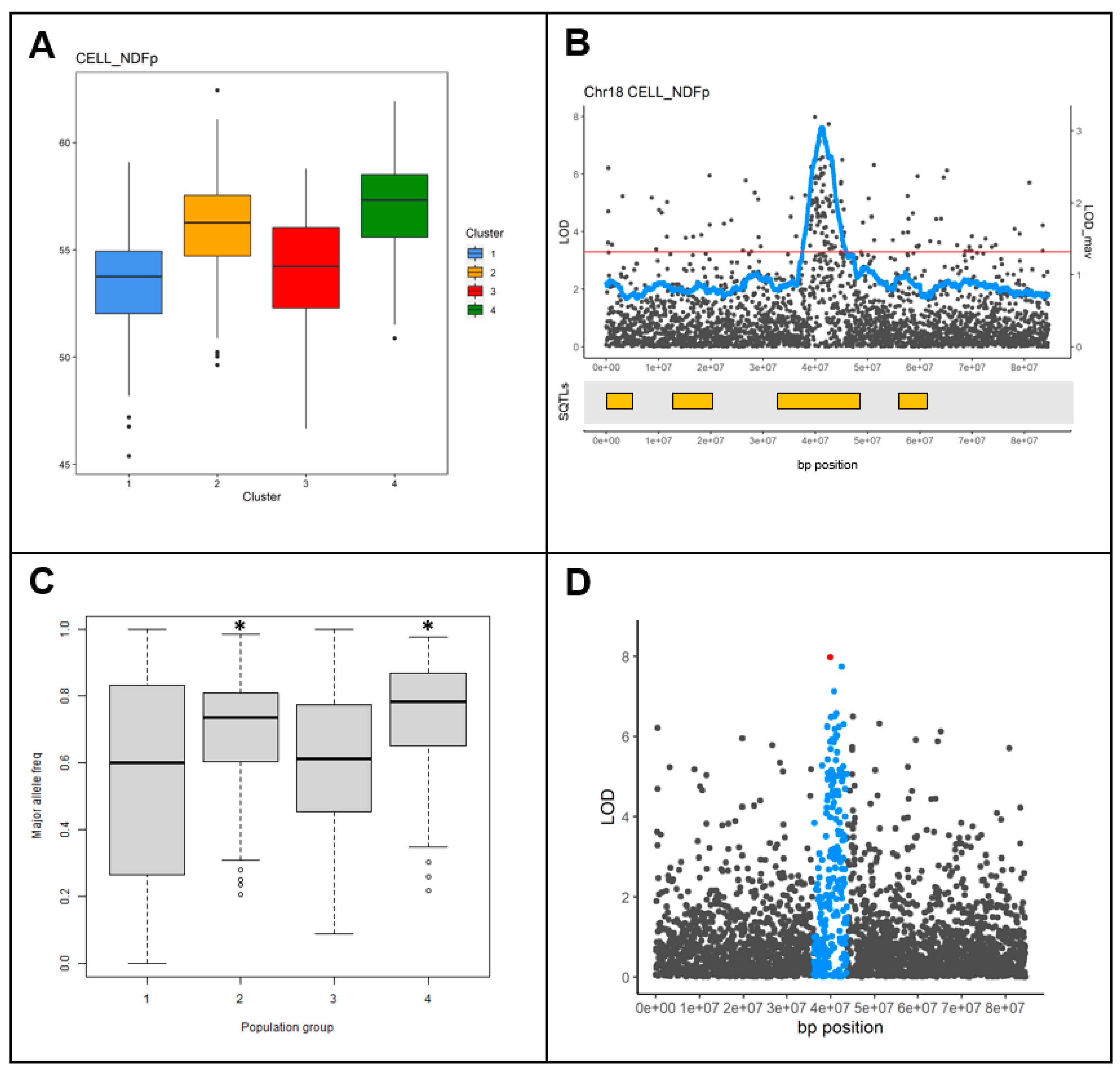
2.4. SQTLs Are Valid Tools to Predict Important Cell Wall Genomic Loci in Miscanthus sinensis and Panicum virgatum
2.5. SQTLs as Tools to Circumvent Limitations of Genetic Mapping Approaches
3. Discussion
3.1. SQTLs as Reservoirs of Allelic Variation with a Potential Use for Biomass Improvement
3.2. SQTLs as Valid Tools toward the Improvement of Breeding Activities in Novel Crops
4. Materials and Methods
4.1. Intraspecific SQTL Alignment
4.2. Analysis of Intraspecific SQTL Alignments
4.3. Analysis of Cell Wall Protein Sequence Changes
4.4. Genome-Wide Association Study on Miscanthus sinensis
4.5. Colocalization of SQTLs and Cell Wall Loci Mapped on M. sinensis
4.6. Retrieval of the P. virgatum Cell Wall QTLs and Analysis of Their Colocalization with SQTLs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pancaldi, F.; Vlegels, D.; Rijken, H.; van Loo, E.N.; Trindade, L.M. Detection and Analysis of Syntenic Quantitative Trait Loci Controlling Cell Wall Quality in Angiosperms. Front. Plant Sci. 2022, 13, 855093. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.N. Positional orthology: Putting genomic evolutionary relationships into context. Briefings Bioinform. 2011, 12, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Schranz, M.E. Network approaches for plant phylogenomic synteny analysis. Curr. Opin. Plant Biol. 2017, 36, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Kerstens, M.H.; Schranz, M.E.; Bouwmeester, K. Phylogenomic analysis of the APETALA2 transcription factor subfamily across angiosperms reveals both deep conservation and lineage-specific patterns. Plant J. 2020, 103, 1516–1524. [Google Scholar] [CrossRef]
- van der Weijde, T.; Alvim Kamei, C.L.; Torres, A.F.; Evermerris, W.; Edolstra, O.; Visser, R.G.F.; Trindade, L.M. The potential of C4 grasses for cellulosic biofuel production. Front. Plant Sci. 2013, 4, 107. [Google Scholar] [CrossRef]
- Pancaldi, F.; Trindade, L.M. Marginal Lands to Grow Novel Bio-Based Crops: A Plant Breeding Perspective. Front. Plant Sci. 2020, 11, 227. [Google Scholar] [CrossRef]
- Tadele, Z. Orphan crops: Their importance and the urgency of improvement. Planta 2019, 250, 677–694. [Google Scholar] [CrossRef]
- Salentijn, E.M.; Pereira, A.; Angenent, G.C.; van der Linden, C.G.; Krens, F.; Smulders, M.J.; Vosman, B. Plant translational genomics: From model species to crops. Mol. Breed. 2006, 20, 1–13. [Google Scholar] [CrossRef]
- Armstead, I.; Huang, L.; Ravagnani, A.; Robson, P.; Ougham, H. Bioinformatics in the orphan crops. Briefings Bioinform. 2009, 10, 645–653. [Google Scholar] [CrossRef]
- Kamei, C.L.A.; Severing, E.I.; Dechesne, A.; Furrer, H.; Dolstra, O.; Trindade, L.M. Orphan Crops Browser: A bridge between model and orphan crops. Mol. Breed. 2016, 36, 9. [Google Scholar] [CrossRef] [Green Version]
- Trindade, L.; Dolstra, O.; van Loo, E.R.; Visser, R. Plant Breeding and Its Role in A Biobased Economy. In The Biobased Economy; Routledge: London, UK, 2010; pp. 90–105. [Google Scholar]
- Mehmood, M.A.; Ibrahim, M.; Rashid, U.; Nawaz, M.; Ali, S.; Hussain, A.; Gull, M. Biomass production for bioenergy using marginal lands. Sustain. Prod. Consum. 2016, 9, 3–21. [Google Scholar] [CrossRef]
- Clifton-Brown, J.; Harfouche, A.; Casler, M.D.; Dylan Jones, H.; Macalpine, W.J.; Murphy-Bokern, D.; Smart, L.B.; Adler, A.; Ashman, C.; Awty-Carroll, D. Breeding progress and preparedness for mass-scale deployment of perennial lignocellulosic biomass crops switchgrass, miscanthus, willow and poplar. GCB Bioenergy 2019, 11, 118–151. [Google Scholar] [CrossRef]
- van der Cruijsen, K.; Al Hassan, M.; van Erven, G.; Dolstra, O.; Trindade, L.M. Breeding Targets to Improve Biomass Quality in Miscanthus. Molecules 2021, 26, 254. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, Y. Rice brittleness mutants: A way to open the ‘Black Box’of monocot cell wall biosynthesis free access. J. Integr. Plant Biol. 2011, 53, 136–142. [Google Scholar] [CrossRef]
- Zhong, R.; Cui, D.; Ye, Z.H. Secondary cell wall biosynthesis. New Phytol. 2019, 221, 1703–1723. [Google Scholar] [CrossRef]
- Little, A.; Schwerdt, J.G.; Shirley, N.J.; Khor, S.F.; Neumann, K.; O’Donovan, L.A.; Lahnstein, J.; Collins, H.M.; Henderson, M.; Fincher, G.B.; et al. Revised Phylogeny of the Cellulose Synthase Gene Superfamily: Insights into Cell Wall Evolution. Plant Physiol. 2018, 177, 1124–1141. [Google Scholar] [CrossRef]
- Brown, D.; Wightman, R.; Zhang, Z.; Gomez, L.D.; Atanassov, I.; Bukowski, J.P.; Tryfona, T.; McQueen-Mason, S.J.; Dupree, P.; Turner, S. Arabidopsis genes IRREGULAR XYLEM (IRX15) and IRX15L encode DUF579-containing proteins that are essential for normal xylan deposition in the secondary cell wall. Plant J. 2011, 66, 401–413. [Google Scholar] [CrossRef]
- Zhong, R.; Lee, C.; Zhou, J.; McCarthy, R.L.; Ye, Z.-H. A Battery of Transcription Factors Involved in the Regulation of Secondary Cell Wall Biosynthesis in Arabidopsis. Plant Cell 2008, 20, 2763–2782. [Google Scholar] [CrossRef]
- Rao, X.; Dixon, R.A. Current Models for Transcriptional Regulation of Secondary Cell Wall Biosynthesis in Grasses. Front. Plant Sci. 2018, 9, 399. [Google Scholar] [CrossRef]
- Ali, S.; Serba, D.D.; Walker, D.; Jenkins, J.; Schmutz, J.; Bhamidimarri, S.; Saha, M.C. Genome-wide quantitative trait loci detection for biofuel traits in switchgrass (Panicum virgatum L.). GCB Bioenergy 2020, 12, 923–940. [Google Scholar] [CrossRef]
- Liepman, A.H.; Wilkerson, C.G.; Keegstra, K. Expression of cellulose synthase-like (Csl) genes in insect cells reveals that CslA family members encode mannan synthases. Proc. Natl. Acad. Sci. USA 2005, 102, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Kondo, M.; Aya, K.; Miyao, A.; Sato, Y.; Antonio, B.A.; Namiki, N.; Nagamura, Y.; Matsuoka, M. Identification of Transcription Factors Involved in Rice Secondary Cell Wall Formation. Plant Cell Physiol. 2013, 54, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, B.; van der Hoorn, R.A. Beta galactosidases in Arabidopsis and tomato—A mini review. Biochem. Soc. Trans. 2016, 44, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Burk, D.H.; Morrison, W.H., III; Ye, Z.-H. FRAGILE FIBER3, an Arabidopsis gene encoding a type II inositol polyphosphate 5-phosphatase, is required for secondary wall synthesis and actin organization in fiber cells. Plant Cell 2004, 16, 3242–3259. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Niu, Q.-W.; Nishizawa, N.; Wu, Y.; Kost, B.; Chua, N.-H. KORRIGAN, an Arabidopsis endo-1, 4-β-glucanase, localizes to the cell plate by polarized targeting and is essential for cytokinesis. Plant Cell 2000, 12, 1137–1152. [Google Scholar] [PubMed]
- Glass, M.; Barkwill, S.; Unda, F.; Mansfield, S.D. Endo-β-1,4-glucanases impact plant cell wall development by influencing cellulose crystallization. J. Integr. Plant Biol. 2015, 57, 396–410. [Google Scholar] [CrossRef]
- Öhman, D.; Demedts, B.; Kumar, M.; Gerber, L.; Gorzsás, A.; Goeminne, G.; Hedenström, M.; Ellis, B.; Boerjan, W.; Sundberg, B. MYB 103 is required for FERULATE-5-HYDROXYLASE expression and syringyl lignin biosynthesis in A rabidopsis stems. Plant J. 2013, 73, 63–76. [Google Scholar] [CrossRef]
- Saelim, L.; Akiyoshi, N.; Tan, T.T.; Ihara, A.; Yamaguchi, M.; Hirano, K.; Matsuoka, M.; Demura, T.; Ohtani, M. Arabidopsis Group IIId ERF proteins positively regulate primary cell wall-type CESA genes. J. Plant Res. 2018, 132, 117–129. [Google Scholar] [CrossRef]
- Goujon, T.; Minic, Z.; El Amrani, A.; Lerouxel, O.; Aletti, E.; Lapierre, C.; Joseleau, J.P.; Jouanin, L. AtBXL1, a novel higher plant (Arabidopsis thaliana) putative beta-xylosidase gene, is involved in secondary cell wall metabolism and plant development. Plant J. 2003, 33, 677–690. [Google Scholar] [CrossRef]
- Zhong, R.; Ye, Z.-H. Secondary Cell Walls: Biosynthesis, Patterned Deposition and Transcriptional Regulation. Plant Cell Physiol. 2014, 56, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Baldacci-Cresp, F.; Le Roy, J.; Huss, B.; Lion, C.; Créach, A.; Spriet, C.; Duponchel, L.; Biot, C.; Baucher, M.; Hawkins, S.; et al. UDP-GLYCOSYLTRANSFERASE 72E3 Plays a Role in Lignification of Secondary Cell Walls in Arabidopsis. Int. J. Mol. Sci. 2020, 21, 6094. [Google Scholar] [CrossRef]
- Sindhu, A.; Langewisch, T.; Olek, A.; Multani, D.S.; McCann, M.C.; Vermerris, W.; Carpita, N.C.; Johal, G. Maize Brittle stalk2 Encodes a COBRA-Like Protein Expressed in Early Organ Development But Required for Tissue Flexibility at Maturity. Plant Physiol. 2007, 145, 1444–1459. [Google Scholar] [CrossRef]
- Yu, Y.; Hu, R.; Wang, H.; Cao, Y.; He, G.; Fu, C.; Zhou, G. MlWRKY12, a novel Miscanthus transcription factor, participates in pith secondary cell wall formation and promotes flowering. Plant Sci. 2013, 212, 1–9. [Google Scholar] [CrossRef]
- Geng, P.; Zhang, S.; Liu, J.; Zhao, C.; Wu, J.; Cao, Y.; Fu, C.; Han, X.; He, H.; Zhao, Q. MYB20, MYB42, MYB43, and MYB85 Regulate Phenylalanine and Lignin Biosynthesis during Secondary Cell Wall Formation. Plant Physiol. 2019, 182, 1272–1283. [Google Scholar] [CrossRef]
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011, 12, 232. [Google Scholar] [CrossRef]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L.; et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Kondrashov, F.A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc. R. Soc. B Boil. Sci. 2012, 279, 5048–5057. [Google Scholar] [CrossRef]
- Gabur, I.; Chawla, H.S.; Snowdon, R.J.; Parkin, I.A. Connecting genome structural variation with complex traits in crop plants. Theor. Appl. Genet. 2018, 132, 733–750. [Google Scholar] [CrossRef]
- Wagner, A.; Donaldson, L.; Ralph, J. Lignification and lignin manipulations in conifers. In Advances in Botanical Research; Elsevier: Amsterdam, The Netherlands, 2012; Volume 61, pp. 37–76. [Google Scholar]
- Preisner, M.; Kulma, A.; Zebrowski, J.; Dymińska, L.; Hanuza, J.; Arendt, M.; Starzycki, M.; Szopa, J. Manipulating cinnamyl alcohol dehydrogenase (CAD) expression in flax affects fibre composition and properties. BMC Plant Biol. 2014, 14, 50. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Shi, Y.; Li, W.; Chen, S.; Wang, Y.; He, X.; Yin, X. RcPAL, a key gene in lignin biosynthesis in Ricinus communis L. BMC Plant Biol. 2019, 19, 181. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and Biological Functions in Plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Zhang, N.; Severing, E.I.; Nijveen, H.; Cheng, F.; Visser, R.G.; Wang, X.; de Ridder, D.; Bonnema, G. Beyond genomic variation-comparison and functional annotation of three Brassica rapagenomes: A turnip, a rapid cycling and a Chinese cabbage. BMC Genom. 2014, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qian, Q.; Zhou, Y.; Yan, M.; Sun, L.; Zhang, M.; Fu, Z.; Wang, Y.; Han, B.; Pang, X. BRITTLE CULM1, Which Encodes a COBRA-Like Protein, Affects the Mechanical Properties of Rice Plants. Plant Cell 2003, 15, 2020–2031. [Google Scholar] [CrossRef]
- Dai, X.; You, C.; Wang, L.; Chen, G.; Zhang, Q.; Wu, C. Molecular characterization, expression pattern, and function analysis of the OsBC1L family in rice. Plant Mol. Biol. 2009, 71, 469–481. [Google Scholar] [CrossRef]
- Jahn, C.E.; Mckay, J.K.; Mauleon, R.; Stephens, J.; McNally, K.L.; Bush, D.R.; Leung, H.; Leach, J.E. Genetic Variation in Biomass Traits among 20 Diverse Rice Varieties. Plant Physiol. 2010, 155, 157–168. [Google Scholar] [CrossRef]
- Liu, Y.; You, S.; Taylor-Teeples, M.; Li, W.L.; Schuetz, M.; Brady, S.M.; Douglas, C.J. BEL1-LIKE HOMEODOMAIN6 and KNOTTED ARABIDOPSIS THALIANA7 interact and regulate secondary cell wall formation via repression of REVOLUTA. Plant Cell 2014, 26, 4843–4861. [Google Scholar] [CrossRef]
- Wang, H.; Avci, U.; Nakashima, J.; Hahn, M.G.; Chen, F.; Dixon, R.A. Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proc. Natl. Acad. Sci. USA 2010, 107, 22338–22343. [Google Scholar] [CrossRef]
- Zhou, J.; Zhong, R.; Ye, Z.-H. Arabidopsis NAC domain proteins, VND1 to VND5, are transcriptional regulators of secondary wall biosynthesis in vessels. PLoS ONE 2014, 9, e105726. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, X.; Yang, F.; Fan, D.; Jiang, Y.; Luo, K. PtrWRKY19, a novel WRKY transcription factor, contributes to the regulation of pith secondary wall formation in Populus trichocarpa. Sci. Rep. 2016, 6, 18643. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, M.; Zhang, R.; Yu, H.; Wang, H.; Li, J.; Wang, Y.; Hu, Z.; Wang, Y.; Luo, Z. Down-regulation of OsMYB103L distinctively alters beta-1, 4-glucan polymerization and cellulose microfibers assembly for enhanced biomass enzymatic saccharification in rice. Biotechnol. Biofuels 2021, 14, 1–15. [Google Scholar] [CrossRef]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2017, 16, 1265–1274. [Google Scholar] [CrossRef]
- Jiao, W.-B.; Schneeberger, K. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Delcher, A.L.; Phillippy, A.; Carlton, J.; Salzberg, S.L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002, 30, 2478–2483. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gíslason, M.H.; Nielsen, H.; Armenteros, J.J.A.; Johansen, A.R. Prediction of GPI-anchored proteins with pointer neural networks. Curr. Res. Biotechnol. 2021, 3, 6–13. [Google Scholar] [CrossRef]
- Thumuluri, V.; Almagro Armenteros, J.J.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 2022, 50, W228–W234. [Google Scholar] [CrossRef] [PubMed]
- Høie, M.H.; Kiehl, E.N.; Petersen, B.; Nielsen, M.; Winther, O.; Nielsen, H.; Hallgren, J.; Marcatili, P. NetSurfP-3.0: Accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic Acids Res. 2022, 50, W510–W515. [Google Scholar] [CrossRef]
- Tai, T.H.; Tanksley, S.D. A rapid and inexpensive method for isolation of total DNA from dehydrated plant tissue. Plant Mol. Biol. Rep. 1990, 8, 297–303. [Google Scholar] [CrossRef]
- Mitros, T.; Session, A.M.; James, B.T.; Wu, G.A.; Belaffif, M.B.; Clark, L.V.; Shu, S.; Dong, H.; Barling, A.; Holmes, J.R. Genome biology of the paleotetraploid perennial biomass crop Miscanthus. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef]
- van Rossum, B.-J.; Kruijer, W.; van Eeuwijk, F.; Boer, M.; Malosetti, M.; Bustos-Korts, D.; Millet, E.; Paulo, J.; Verouden, M.; Wehrens, R. Package ‘statgenGWAS’. R Package, version 1.0.7, 2020. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Pancaldi, F.; van Loo, E.N.; Schranz, M.E.; Trindade, L.M. Genomic Architecture and Evolution of the Cellulose synthase Gene Superfamily as Revealed by Phylogenomic Analysis. Front. Plant Sci. 2022, 13, 870818. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
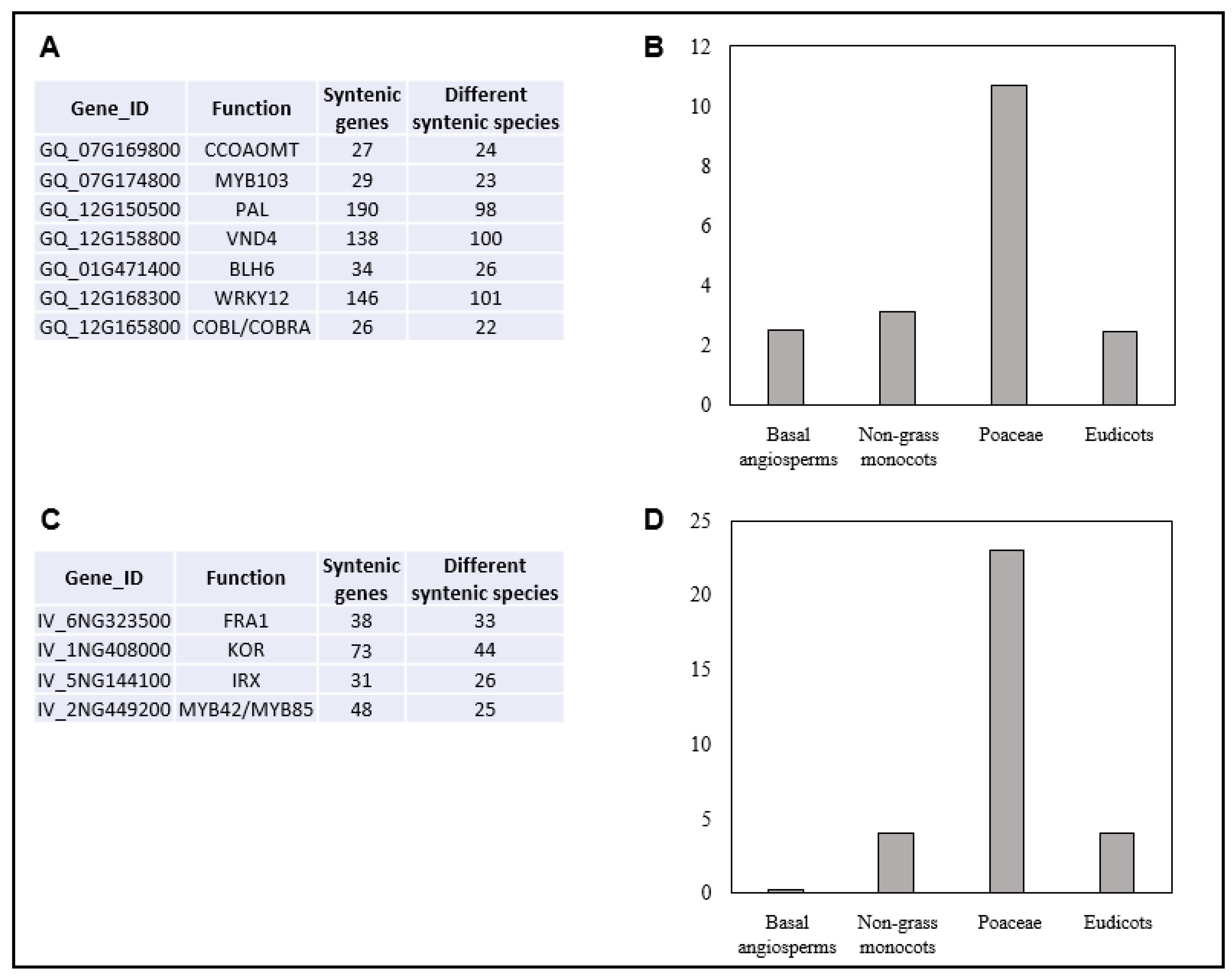
| Gene_ID | Chrom | Conserved in SQTL | Cell Wall Function | Notes | References |
|---|---|---|---|---|---|
| GQ_01G229400 | Chr01 | MSQTL_122 | CSL | Homolog of AtCSLA9, involved in the synthesis of mannans. | [22] |
| GQ_01G468200 | Chr01 | MSQTL_122 | CSL | Homolog of AtCSLA9, involved in the synthesis of mannans. | [22] |
| GQ_01G471400 | Chr01 | MSQTL_5 | BLH6 | Transcription factor promoting secondary cell wall synthesis in grasses | [20,23] |
| GQ_01G471500 | Chr01 | MSQTL_5 | BGAL | Beta-galactosidase involved in the degradation of several polysaccharides and cell wall remodeling | [24] |
| GQ_01G474600 | Chr01 | MSQTL_122 | FRA3 | Homolog of AtFRA3, which coordinates actin organization during cellulose deposition | [25] |
| GQ_01G478800 | Chr01 | MSQTL_212 | 4CL | Important gene of the lignin synthesis pathway | [16] |
| GQ_03G236400 | Chr03 | MSQTL_216 | Endoglucanase/KOR | Important genes for cellulose and plant cell wall metabolism | [26,27] |
| GQ_03G240500 | Chr03 | MSQTL_248 | Endoglucanase/KOR | Important genes for cellulose and plant cell wall metabolism | [26,27] |
| GQ_07G169800 | Chr07 | MSQTL_180 | CCOAOMT | Central gene for lignin synthesis, affecting both lignin amount and monolignols ratio | [16] |
| GQ_07G169900 | Chr07 | MSQTL_180 | LAC | Gene involved in the in muro lignin deposition | [16] |
| GQ_07G174800 | Chr07 | MSQTL_180 | MYB103 | Regulates F5H expression and S-lignin deposition in arabidopsis | [28] |
| GQ_07G435600 | Chr07 | MSQTL_245 | ERF39 | Binds to promoters of CESA1/3/6, synthesizing primary-like cell walls | [29] |
| GQ_07G435700 | Chr07 | MSQTL_245 | ERF39 | Binds to promoters of CESA1/3/6, synthesizing primary-like cell walls | [29] |
| GQ_07G477100 | Chr07 | MSQTL_445 | BXL | Inhibits xylan synthesis | [30] |
| GQ_12G086100 | Chr12 | MSQTL_118 | XND1/WND1A | Major transcription factor in regulating secondary cell wall synthesis | [31] |
| GQ_12G091100 | Chr12 | MSQTL_118 | Endoglucanase/KOR | Important genes for cellulose and plant cell wall metabolism | [26,27] |
| GQ_12G150500 | Chr12 | MSQTL_38 | PAL | First step of the lignin pathway | [16] |
| GQ_12G153400 | Chr12 | MSQTL_38 | UGT72E3 | Gene influencing the kinetics of lignin deposition | [32] |
| GQ_12G158800 | Chr12 | MSQTL_2 | VND4 | Master regulator of secondary cell walls | [20,31] |
| GQ_12G165800 | Chr12 | MSQTL_91 | COBL/COBRA | Homolog of brittle stalk2 locus of maize | [33] |
| GQ_12G168300 | Chr12 | MSQTL_2 | WRKY12 | Involved in regulating secondary cell wall and flowering in Miscanthus lutarioriparius | [20,34] |
| GQ_18G102600 | Chr18 | MSQTL_178 | CSL | Gene involved in hemicellulose biosynthesis | [17] |
| GQ_18G114300 | Chr18 | MSQTL_185 | MYB20/MYB43 | MYB20, MYB43, and MYB85 regulate secondary cell wall formation | [20,31,35] |
| Gene_ID | Chrom | QTL Trait(s) [21] | Conserved in SQTL | Cell Wall Function |
|---|---|---|---|---|
| IV_1KG461400 | Chr_01K | Glucose; total cell wall sugar | MSQTL_195; | 4CL |
| IV_6NG354300 | Chr_06N | Total cell wall sugar | MSQTL_80; | BGAL |
| IV_3NG135100 | Chr_03N | Glucose | MSQTL_25; | BGLU46 |
| IV_9KG034800 | Chr_09K | Xylose | MSQTL_102; | BGLU46 |
| IV_1NG408000 | Chr_01N | Total cell wall sugar | MSQTL_185; | Endoglucanase/KOR |
| IV_6NG323500 | Chr_06N | Glucose; total cell wall sugar | MSQTL_45; | FRA1 |
| IV_1NG306100 | Chr_01N | Total cell wall sugar | MSQTL_18; | GALT/HPGT |
| IV_2NG448300 | Chr_02N | Klason lignin | MSQTL_54; | GAUT1 |
| IV_2KG434900 | Chr_02K | Glucose | MSQTL_230; | GH17 |
| IV_5NG130900 | Chr_05N | Klason lignin | MSQTL_123; | GLCAT |
| IV_5NG144100 | Chr_05N | Glucose; xylose | MSQTL_130; | IRX10/10L |
| IV_2KG435600 | Chr_02K | Glucose | MSQTL_230; | LAC |
| IV_5KG613400 | Chr_05K | Glucose | MSQTL_350; | LAC |
| IV_1NG407200 | Chr_01N | Total cell wall sugar | MSQTL_185; | MED/REF |
| IV_1NG407100 | Chr_01N | Total cell wall sugar | MSQTL_185; | MYB20/MYB43 |
| IV_6NG354000 | Chr_06N | Total cell wall sugar | MSQTL_80; | MYB4/MYB6/MYB7/MYB21/MYB32 |
| IV_2NG449200 | Chr_02N | Klason lignin | MSQTL_248; | MYB42/MYB85 |
| IV_6NG352900 | Chr_06N | Total cell wall sugar | MSQTL_80; | MYB42/MYB85 |
| IV_6NG354100 | Chr_06N | Total cell wall sugar | MSQTL_80; | PRX |
| IV_9KG292900 | Chr_09N | Klason lignin | MSQTL_122; | RHM |
| IV_1KG460000 | Chr_01K | Glucose; total cell wall sugar | MSQTL_164; | XET/XTH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pancaldi, F.; van Loo, E.N.; Senio, S.; Al Hassan, M.; van der Cruijsen, K.; Paulo, M.-J.; Dolstra, O.; Schranz, M.E.; Trindade, L.M. Syntenic Cell Wall QTLs as Versatile Breeding Tools: Intraspecific Allelic Variability and Predictability of Biomass Quality Loci in Target Plant Species. Plants 2023, 12, 779. https://doi.org/10.3390/plants12040779
Pancaldi F, van Loo EN, Senio S, Al Hassan M, van der Cruijsen K, Paulo M-J, Dolstra O, Schranz ME, Trindade LM. Syntenic Cell Wall QTLs as Versatile Breeding Tools: Intraspecific Allelic Variability and Predictability of Biomass Quality Loci in Target Plant Species. Plants. 2023; 12(4):779. https://doi.org/10.3390/plants12040779
Chicago/Turabian StylePancaldi, Francesco, Eibertus N. van Loo, Sylwia Senio, Mohamad Al Hassan, Kasper van der Cruijsen, Maria-João Paulo, Oene Dolstra, M. Eric Schranz, and Luisa M. Trindade. 2023. "Syntenic Cell Wall QTLs as Versatile Breeding Tools: Intraspecific Allelic Variability and Predictability of Biomass Quality Loci in Target Plant Species" Plants 12, no. 4: 779. https://doi.org/10.3390/plants12040779