
Amazon Rainforest Hidden Volatiles—Part I: Unveiling New Compounds from Acmella oleracea (L.) R.K. Jansen Essential Oil

 , , ,
, , , 
Abstract
1. Introduction
2. Results and Discussion
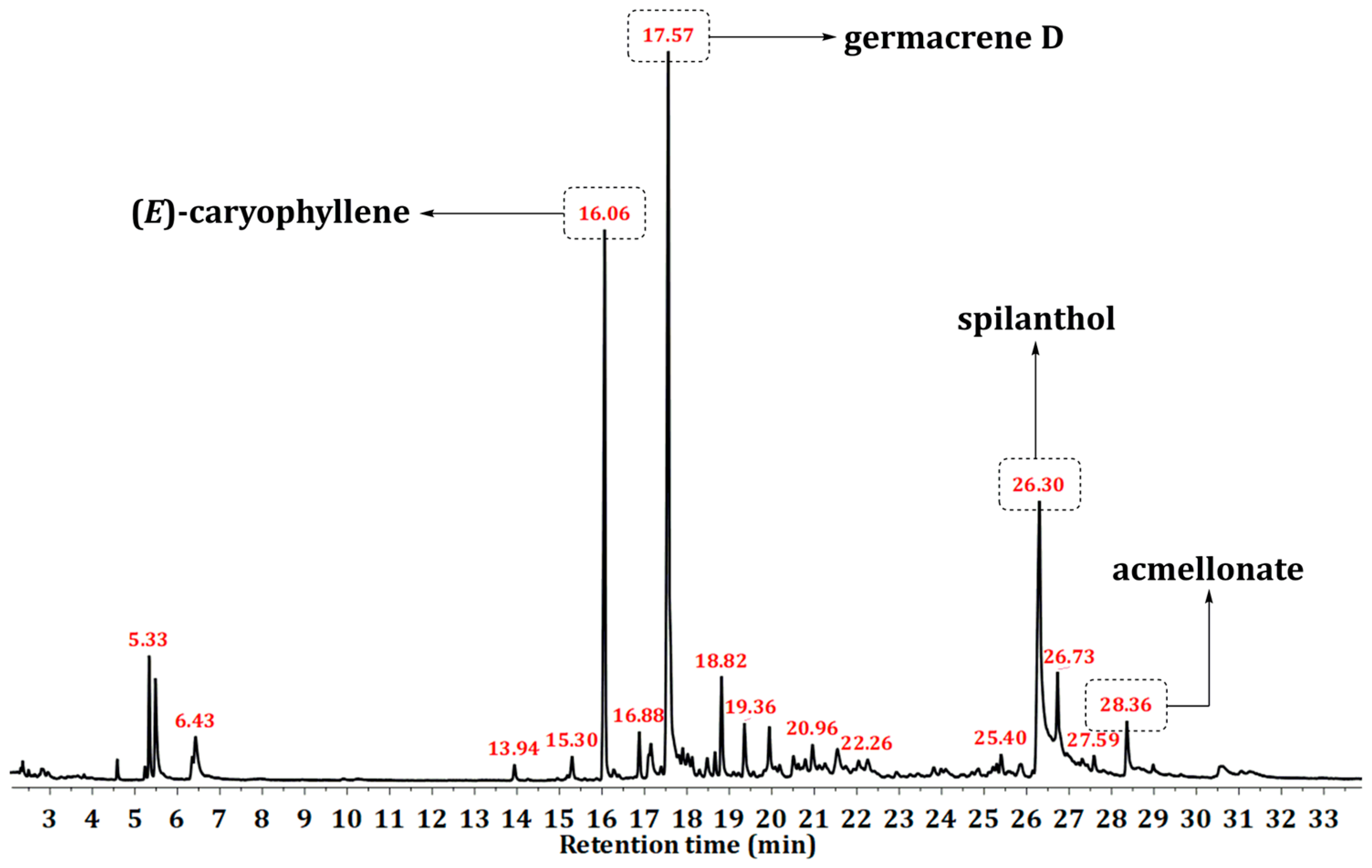
2.1. Composition of A. oleracea Essential Oil and Essential Oil Fractions
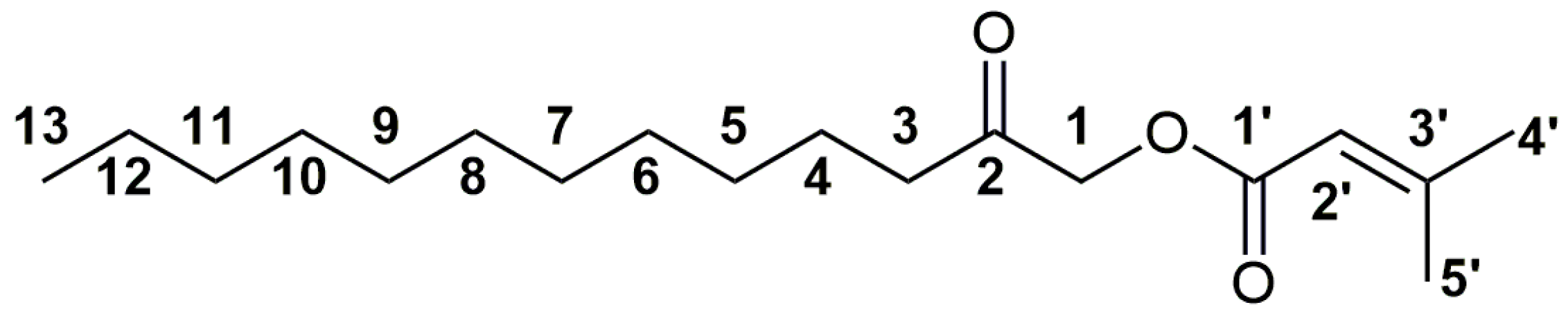
2.2. NMR Spectral Characterization of the New Esters
2.3. Identification of the Isomers/Homologs/Analogs of Acmellonate
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Hydro-Distillation and Chromatographic Fractionation of A. oleracea Essential Oil
3.4. Component Identification
3.5. Gas Chromatography–Mass Spectrometry (GC-MS) Analyses
3.6. NMR Measurements
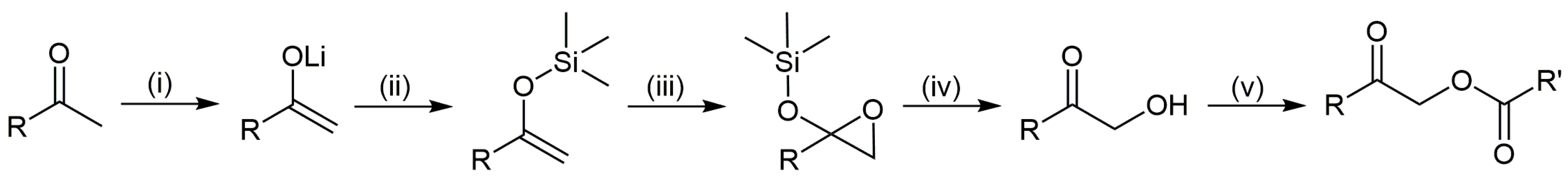
3.7. Synthesis of 1-hydroxyundecan-2-one, 1-hydroxydodecan-2-one, and 1-hydroxytridecan-2-one
3.8. Synthesis of Esters
3.9. Dimethyl Disulfide (DMDS) Derivatization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yáñez-Serrano, A.M.; Bourtsoukidis, E.; Alves, E.G.; Bauwens, M.; Stavrakou, T.; Llusia, J.; Filella, I.; Guenther, A.; Williams, J.; Artaxo, P.; et al. Amazonian biogenic volatile organic compounds under global change. Glob. Chang. Biol. 2020, 26, 4722–4751. [Google Scholar] [CrossRef]
- Uthpala, T.G.G.; Navaratne, S.B. Acmella oleracea plant; identification, applications and use as an emerging food source—Review. Food Rev. Int. 2021, 37, 399–414. [Google Scholar] [CrossRef]
- Spinozzi, E.; Pavela, R.; Bonacucina, G.; Perinelli, D.R.; Cespi, M.; Petrelli, R.; Cappellacci, L.; Fiorini, D.; Scortichini, S.; Garzoli, S.; et al. Spilanthol-rich essential oil obtained by microwave-assisted extraction from Acmella oleracea (L.) R.K. Jansen and its nanoemulsion: Insecticidal, cytotoxic and anti-inflammatory activities. Ind. Crop. Prod. 2021, 172, 114027. [Google Scholar] [CrossRef]
- Benelli, G.; Pavela, R.; Drenaggi, E.; Maggi, F. Insecticidal efficacy of the essential oil of jambú (Acmella oleracea (L.) RK Jansen) cultivated in central Italy against filariasis mosquito vectors, houseflies and moth pests. J. Ethnopharmacol. 2019, 229, 272–279. [Google Scholar] [CrossRef]
- Jirovetz, L.; Buchbauer, G.; Wobus, A.; Shafi, M.P.; Abraham, G.T. Essential oil analysis of Spilanthes acmella Murr. fresh plants from Southern India. J. Essent. Oil Res. 2005, 17, 429–431. [Google Scholar] [CrossRef]
- Baruah, R.N.; Leclercq, P.A. Characterization of the essential oil from flower heads of Spilanthes acmella. J. Essent. Oil Res. 1993, 5, 693–695. [Google Scholar] [CrossRef]
- Lemos, T.L.G.; Pessoa, O.D.L.; Matos, F.J.A.; Alencar, J.W.; Craveiro, A.A. The essential oil of Spilanthes acmella Murr. J. Essent. Oil Res. 1991, 3, 369–370. [Google Scholar] [CrossRef]
- Dedino, D.B.; de Lima, J.D.; Bortolucci, W.d.C.; Rivadavea, W.R.; Lovato, E.C.W.; Gazim, Z.C.; Gonçalves, J.E.; Monzon, D.L.R.; da Silva, G.J. Red LED light and different cultivation methods changed the essential oil composition of Acmella oleracea. Plant Cell Tissue Organ Cult. 2022, 151, 511–520. [Google Scholar] [CrossRef]
- Radulović, N.S.; Mladenović, M.Z.; Blagojević, P.D.; Stojanović-Radić, Z.Z.; Ilic-Tomic, T.; Senerovic, L.; Nikodinovic-Runic, J. Toxic essential oils. Part III: Identification and biological activity of new allylmethoxyphenyl esters from a Chamomile species (Anthemis segetalis Ten.). Food Chem. Toxicol. 2013, 62, 554–565. [Google Scholar] [CrossRef]
- Ley, J.P.; Blings, M.; Krammer, G.; Reinders, G.; Schmidt, C.O.; Bertram, H.J. Isolation and synthesis of acmellonate, a new unsaturated long chain 2-ketol ester from Spilanthes acmella. Nat. Prod. Res. 2006, 20, 798–804. [Google Scholar] [CrossRef]
- Sojak, L.; Kalovicova, E.; Ostrovsky, I.; Leclercq, P.A. Retention behaviour of conjugated and isolated n-alkadienes: Identification of n-nona- and n-decadienes by capillary gas chromatography using structure-retention correlations and mass spectrometry. J. Chromatogr. A 1984, 1, 241–261. [Google Scholar] [CrossRef]
- Dekić, B.R.; Ristić, M.N.; Mladenović, M.Z.; Dekić, V.S.; Ristić, N.R.; Ranđelović, V.; Radulović, N.S. Diethyl-ether flower washings of Dianthus cruentus GRISEB. (Caryophyllaceae): Derivatization reactions leading to the identification of new wax constituents. Chem. Biodivers. 2019, 16, e1900153. [Google Scholar] [CrossRef]
- Heinzen, V.E.F.; Soares, M.F.; Yunes, R.A. Semi-empirical topological method for the prediction of the chromatographic retention of cis- and trans-alkene isomers and alkanes. J. Chromatogr. A 1999, 849, 495–506. [Google Scholar] [CrossRef]
- Yasuda, I.; Takeya, K.; Itokawa, T. The geometrical structure of spilanthol. Chem. Pharm. Bull. 1980, 28, 2251–2253. [Google Scholar] [CrossRef]
- Sut, S.; Ferrarese, I.; Shrestha, S.S.; Kumar, G.; Slaviero, A.; Sello, S.; Altissimo, A.; Pagni, L.; Gattesco, F.; Dall’Acqua, S. Comparison of biostimulant treatments in Acmella oleracea cultivation for alkylamides production. Plants 2020, 9, 818. [Google Scholar] [CrossRef]
- Boonen, J.; Baert, B.; Burvenich, C.; Blondeel, P.; De Saeger, S.; De Spiegeleer, B. LC–MS profiling of N-alkylamides in Spilanthes acmella extract and the transmucosal behavior of its main bio-active spilanthol. J. Pharmaceut. Biomed. 2010, 53, 243–249. [Google Scholar] [CrossRef]
- Savic, S.; Petrovic, S.; Savic, S.; Cekic, N. Identification and photostability of N-alkylamides from Acmella oleracea extract. J. Pharmaceut. Biomed. 2021, 195, 113819. [Google Scholar] [CrossRef]
- Van den Dool, H.; Kratz, P.D. A generalization of the retention index system including linear temperature programmed gas-liquid partition chromatography. J. Chromatogr. 1963, 11, 463–471. [Google Scholar] [CrossRef]
- Radulović, N.S.; Mladenović, M.Z.; Blagojević, P.D. A ‘low-level’ chemotaxonomic analysis of the plant family Apiaceae: The case of Scandix balansae Reut. ex Boiss. (tribe Scandiceae). Chem. Biodivers. 2013, 10, 1202–1219. [Google Scholar] [CrossRef]
- Mladenović, M.Z.; Radulović, N.S. The essential oil of Achillea ageratifolia (Sm.) Boiss. subsp. serbica (Nyman) Heimerl (Asteraceae) revisited: The stereochemical nomenclature issues, structural elucidation and synthesis of (new) sabinyl esters. Flavour Frag. J. 2017, 32, 5–23. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RI a | Constituents b | Samples c | ID d | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EO | F1 | F2 | F3 | F4 | F5 | F6 | F7 | |||
| 786 | Hexan-2-ol | tr | 0.1 | tr | MS, RI, CoI | |||||
| 885 | 2-Butylfuran | tr | tr | tr | 0.1 | MS, RI | ||||
| 928 | α-Pinene | tr | 0.1 | tr | MS, RI, CoI | |||||
| 961 | Sabinene | tr | tr | 0.1 | MS, RI | |||||
| 968 | β-Pinene | 0.2 | 0.8 | 0.2 | MS, RI, CoI | |||||
| 979 | β-Myrcene | 0.5 | 0.9 | 1.7 | MS, RI | |||||
| 995 | p-Mentha-1(7),8-diene | tr | tr | tr | MS, RI | |||||
| 1000 | Decane | tr | tr | MS, RI, CoI | ||||||
| 1015 | p-Cymene | tr | tr | tr | MS, RI, CoI | |||||
| 1019 | Limonene | 0.1 | 0.1 | MS, RI, CoI | ||||||
| 1021 | β-Phellandrene | 0.1 | 0.3 | 0.1 | MS, RI | |||||
| 1025 | (Z)-β-Ocimene | 0.5 | 0.8 | MS, RI | ||||||
| 1028 | Phenylacetaldehyde | tr | 0.1 | tr | MS, RI, CoI | |||||
| 1038 | (E)-β-Ocimene | tr | tr | MS, RI | ||||||
| 1048 | γ-Terpinene | tr | tr | MS, RI | ||||||
| 1089 | Linalool | tr | tr | MS, RI, CoI | ||||||
| 1120 | 2-Phenylethan-1-ol | tr | tr | MS, RI, CoI | ||||||
| 1132 | trans-Pinocarveol | tr | tr | tr | tr | MS, RI | ||||
| 1155 | Pinocarvone | tr | tr | tr | tr | MS, RI, CoI | ||||
| 1158 | Rosefurane epoxide | tr | tr | MS, RI | ||||||
| 1167 | Terpinen-4-ol | 0.5 | 1.0 | MS, RI, CoI | ||||||
| 1177 | Cryptone | tr | 0.1 | tr | MS, RI | |||||
| 1180 | α-Terpineol | 0.1 | 0.2 | MS, RI | ||||||
| 1187 | Myrtenol | tr | tr | tr | MS, RI | |||||
| 1200 | Dodecane | tr | tr | MS, RI, CoI | ||||||
| 1278 | Isobornyl acetate | tr | tr | MS, RI | ||||||
| 1287 | Undecan-2-one | tr | tr | MS, RI, CoI | ||||||
| 1300 | Tridecane | tr | tr | MS, RI, CoI | ||||||
| 1321 | Silphiperfol-5-ene | tr | tr | MS, RI | ||||||
| 1329 | δ-Elemene | 0.3 | 1.4 | 0.9 | MS, RI | |||||
| 1343 | α-Cubebene | tr | tr | 0.1 | MS, RI | |||||
| 1371 | α-Copaene | tr | 0.4 | 0.1 | MS, RI | |||||
| 1374 | (E)-β-Damascenone | tr | 0.1 | MS, RI | ||||||
| 1379 | Modheph-2-ene | tr | tr | MS, RI | ||||||
| 1380 | β-Bourbonene | 0.1 | 1.0 | 0.1 | MS, RI | |||||
| 1389 | Dodecan-2-one | tr | 0.1 | tr | MS, RI, CoI | |||||
| 1395 | β-Elemene | 0.3 | 1.3 | 1.6 | MS, RI | |||||
| 1397 | Cyperene | tr | 0.3 | MS, RI | ||||||
| 1400 | Tetradecane | tr | 0.2 | MS, RI, CoI | ||||||
| 1403 | (Z)-Caryophyllene | tr | 0.1 | tr | MS, RI | |||||
| 1407 | α-Gurjunene | tr | tr | MS, RI | ||||||
| 1411 | β-Ylangene | tr | tr | MS, RI | ||||||
| 1415 | (E)-Caryophyllene | 5.2 | 6.9 | 34.9 | MS, RI, CoI | |||||
| 1426 | Isogermacrene D | 0.1 | 0.5 | MS, RI | ||||||
| 1427 | γ-Elemene | tr | 0.3 | MS, RI | ||||||
| 1430 | β-Copaene | tr | tr | 0.7 | MS, RI | |||||
| 1439 | Neryl acetone | tr | 0.1 | 0.1 | MS, RI, CoI | |||||
| 1447 | (E)-β-Farnesene | tr | tr | MS, RI | ||||||
| 1451 | α-Humulene | 0.5 | 1.1 | 1.3 | MS, RI, CoI | |||||
| 1474 | γ-Muurolene | 0.2 | 0.5 | MS, RI | ||||||
| 1479 | (E)-β-Ionone | tr | 0.1 | 0.5 | 0.3 | MS, RI | ||||
| 1482 | Germacrene D | 17.2 | 42.6 | MS, RI, CoI | ||||||
| 1491 | Tridecan-2-one | 1.4 | 4.4 | 12.2 | 4.6 | 0.1 | MS, RI, CoI | |||
| 1494 | Pentadec-1-ene | 4.8 | 45.9 | MS, RI | ||||||
| 1495 | Bicyclogermacrene | tr | tr | tr | 1.1 | MS, RI | ||||
| 1496 | α-Muurolene | tr | 0.8 | MS, RI | ||||||
| 1498 | (E,E)-α-Farnesene | 0.6 | 1.3 | MS, RI | ||||||
| 1500 | Pentadecane | tr | 0.9 | MS, RI, CoI | ||||||
| 1511 | γ-Cadinene | 0.2 | 0.2 | 1.1 | MS, RI | |||||
| 1519 | δ-Cadinene | 0.5 | 0.4 | 1.9 | MS, RI | |||||
| 1527 | Isokessane | 0.4 | 15.3 | 1.8 | MS, RI | |||||
| 1530 | trans-Cadina-1,4-diene | tr | tr | MS, RI | ||||||
| 1535 | Kessane | 1.5 | 48.0 | 12.8 | 0.1 | MS, RI | ||||
| 1540 | α-Calacorene | 0.1 | 0.2 | tr | tr | MS, RI | ||||
| 1546 | Elemol | 0.1 | 0.4 | MS, RI | ||||||
| 1553 | Isocaryophyllene oxide | 0.2 | 1.6 | 1.2 | 0.1 | MS, RI | ||||
| 1558 | Germacrene B | 0.9 | 1.4 | MS, RI | ||||||
| 1558 | (E)-Nerolidol | tr | 1.0 | MS, RI | ||||||
| 1562 | β-Calacorene | tr | tr | tr | MS, RI | |||||
| 1566 | 1,5-Epoxysalvial-4(14)-ene | 0.2 | 0.6 | 1.2 | tr | MS, RI | ||||
| 1578 | Spathulenol | 0.3 | 0.4 | MS, RI | ||||||
| 1578 | Germacrene D-4-ol | tr | 0.1 | 0.4 | MS, RI | |||||
| 1587 | Caryophyllene oxide | 1.2 | 14.5 | 15.0 | 2.3 | MS, RI, CoI | ||||
| 1589 | Viridiflorol | tr | 1.0 | tr | MS, RI | |||||
| 1593 | Tetradecan-2-one | tr | 1.0 | MS, RI, CoI | ||||||
| 1594 | Hexadec-1-ene | tr | 0.3 | MS, RI | ||||||
| 1596 | Salvial-4(14)-en-1-one | 0.4 | 2.1 | 2.1 | 0.8 | 0.3 | MS, RI | |||
| 1600 | Humulene epoxide I | tr | 0.4 | 0.3 | MS, RI | |||||
| 1600 | Hexadecane | tr | 0.2 | MS, RI, CoI | ||||||
| 1606 | Tetradecanal | tr | 0.3 | MS, RI | ||||||
| 1610 | β-Oplopenone | 0.8 | 5.2 | 9.4 | 3.2 | 0.4 | MS, RI | |||
| 1611 | Humulene epoxide II | tr | tr | MS, RI | ||||||
| 1617 | 10-epi-γ-Eudesmol | 0.1 | 0.6 | MS, RI | ||||||
| 1624 | Junenol | 0.5 | 2.1 | 1.6 | MS, RI | |||||
| 1630 | 1-epi-Cubenol | tr | 0.4 | MS, RI | ||||||
| 1632 | Guaia-6,10(14)-dien-4β-ol | 1.6 | 1.9 | MS, RI | ||||||
| 1643 | epi-α-Murrolol | 0.1 | 1.0 | MS, RI | ||||||
| 1645 | Cubenol | tr | tr | 0.9 | MS, RI | |||||
| 1646 | α-Muurolol | 0.3 | 0.6 | MS, RI | ||||||
| 1658 | α-Cadinol | 2.2 | 3.3 | MS, RI | ||||||
| 1672 | Tetradecan-1-ol | 0.8 | 1.3 | MS, RI, CoI | ||||||
| 1689 | Germacra-4(15),5,10(14)-trien-1α-ol | 0.6 | 0.7 | MS, RI | ||||||
| 1694 | Heptadec-1-ene | 0.4 | 6.3 | MS, RI | ||||||
| 1695 | Pentadecan-2-one | tr | tr | 1.0 | 0.4 | MS, RI, CoI | ||||
| 1700 | Heptadecane | tr | tr | MS, RI, CoI | ||||||
| 1710 | Pentadecanal | tr | 2.9 | 1.0 | 0.5 | 0.3 | MS, RI, CoI | |||
| 1744 | Mint sulfide | 0.1 | 2.1 | MS, RI | ||||||
| 1762 | 2-Oxoundec-7-en-1-yl isobutyrate * | tr | 0.4 | NEW | ||||||
| 1769 | 2-Oxoundecyl isobutyrate | tr | 0.3 | NEW | ||||||
| 1779 | 14-Oxy-α-muurolene | tr | 0.6 | 1.3 | 0.5 | 0.4 | MS, RI | |||
| 1800 | Octadecane | tr | tr | MS, RI, CoI | ||||||
| 1815 | (7Z,9E)-2-Oxoundeca-7,9-dien-1-yl isobutyrate # | 0.8 | 0.9 | NEW | ||||||
| 1842 | Spilanthol isomer e | tr | 1.7 | MS | ||||||
| 1856 | 2-Oxoundec-7-en-1-yl isovalerate * | 0.2 | 1.3 | 0.9 | NEW | |||||
| 1863 | 2-Oxoundecyl isovalerate | tr | tr | NEW | ||||||
| 1869 | 2-Oxododecyl isobutyrate | tr | tr | NEW | ||||||
| 1876 | Hexadecan-1-ol | 0.3 | 0.6 | MS, RI, CoI | ||||||
| 1891 | (2E,6Z,8E)-N-Isobutyldeca-2,6,8-trienamide (syn. spilanthol) | 28.9 | 56.0 | MS, RI | ||||||
| 1894 | Nonadec-1-ene | tr | 0.1 | MS, RI | ||||||
| 1898 | Heptadecan-2-one | tr | tr | tr | MS, RI, CoI | |||||
| 1900 | Nonadecane | tr | 0.2 | MS, RI, CoI | ||||||
| 1900 | 2-Oxoundec-7-en-1-yl angelate * | 0.4 | 0.7 | 0.5 | NEW | |||||
| 1908 | 2-Oxoundecyl angelate | tr | tr | 0.1 | NEW | |||||
| 1909 | Spilanthol isomer f | 1.2 | 2.9 | MS | ||||||
| 1910 | (7Z,9E)-2-Oxoundeca-7,9-dien-1-yl isovalerate # | 2.4 | 6.6 | 18.0 | NEW | |||||
| 1912 | Spilanthol isomer g | 1.1 | 1.8 | MS | ||||||
| 1916 | (5E,9E)-Farnesyl acetone | tr | 0.3 | tr | MS, RI | |||||
| 1917 | Spilanthol isomer h | 1.4 | 2.1 | MS | ||||||
| 1923 | (7E,9E)-2-Oxoundeca-7,9-dien-1-yl isovalerate # | tr | 0.8 | 2.0 | NEW | |||||
| 1925 | (7E,9Z)-2-Oxoundeca-7,9-dien-1-yl isovalerate # | tr | 0.4 | 0.9 | NEW | |||||
| 1929 | (7Z,9Z)-2-Oxoundeca-7,9-dien-1-yl isovalerate # | tr | 0.4 | 1.3 | NEW | |||||
| 1940 | 2-Oxoundec-7-en-1-yl senecioate * | 0.4 | 0.6 | 3.8 | 3.2 | NEW | ||||
| 1948 | 2-Oxoundecyl senecioate | 0.2 | 0.7 | 1.9 | 1.3 | NEW | ||||
| 1952 | 2-Oxotridec-7-en-1-yl isobutyrate * | tr | tr | NEW | ||||||
| 1956 | (7Z,9E)-2-Oxoundeca-7,9-dien-1-yl angelate # | 0.8 | 1.9 | 8.1 | 3.7 | NEW | ||||
| 1964 | 2-Oxododecyl isovalerate | tr | tr | tr | tr | NEW | ||||
| 1966 | N-(2-Methylbutyl)deca-2,6,8-trienamide isomer i | tr | 0.1 | MS | ||||||
| 1969 | (7E,9E)-2-Oxoundeca-7,9-dien-1-yl angelate # | tr | 0.6 | 1.5 | 0.9 | NEW | ||||
| 1969 | 2-Oxotridecyl isobutyrate | tr | 0.3 | 1.2 | tr | NEW | ||||
| 1971 | (7E,9Z)-2-Oxoundeca-7,9-dien-1-yl angelate # | tr | tr | tr | NEW | |||||
| 1975 | (7Z,9Z)-2-Oxoundeca-7,9-dien-1-yl angelate # | tr | 0.3 | 0.8 | 0.4 | NEW | ||||
| 1979 | Hexadecanoic acid | tr | 6.1 | MS, RI, CoI | ||||||
| 1995 | (7Z,9E)-2-Oxoundeca-7,9-dien-1-yl senecioate (syn. acmellonate) | 4.7 | 0.1 | 5.8 | 30.8 | MS, RI | ||||
| 2000 | Eicosane | tr | 0.7 | MS, RI, CoI | ||||||
| 2009 | (7E,9E)-2-Oxoundeca-7,9-dien-1-yl senecioate # | 0.3 | 1.5 | 4.1 | NEW | |||||
| 2009 | 2-Oxododecyl angelate | 0.8 | 0.1 | tr | NEW | |||||
| 2011 | (7E,9Z)-2-Oxoundeca-7,9-dien-1-yl senecioate # | 0.2 | tr | 1.8 | NEW | |||||
| 2014 | N-(2-Methylbutyl)deca-2,6,8-trienamide isomer j | tr | 3.5 | MS | ||||||
| 2015 | (7Z,9Z)-2-Oxoundeca-7,9-dien-1-yl senecioate # | 0.3 | 0.8 | 2.5 | NEW | |||||
| 2023 | N-(2-Methylbutyl)deca-2,6,8-trienamide isomer k | tr | 0.2 | MS | ||||||
| 2026 | N-(2-Methylbutyl)deca-2,6,8-trienamide isomer l | tr | 0.2 | MS | ||||||
| 2030 | N-(2-Methylbutyl)deca-2,6,8-trienamide isomer m | tr | 0.2 | MS | ||||||
| 2035 | N-(2-Methylbutyl)deca-2,6,8-trienamide isomer n | tr | 0.2 | MS | ||||||
| 2031 | 2-Oxotridec-6-en-1-yl isovalerate * | tr | 0.6 | 1.5 | tr | NEW | ||||
| 2047 | 2-Oxotridec-7-en-1-yl isovalerate * | tr | tr | tr | NEW | |||||
| 2049 | 2-Oxododecyl senecioate | 0.3 | 0.7 | 1.8 | 0.4 | NEW | ||||
| 2065 | 2-Oxotridecyl isovalerate | 0.1 | 2.1 | 2.2 | 0.3 | NEW | ||||
| 2078 | 2-Oxotridec-6-en-1-yl angelate * | 0.1 | 0.9 | 0.3 | tr | NEW | ||||
| 2092 | 2-Oxotridec-7-en-1-yl angelate * | tr | 0.3 | 0.1 | NEW | |||||
| 2100 | Heneicosane | tr | 1.1 | MS, RI, CoI | ||||||
| 2110 | 2-Oxotridecyl angelate | tr | 0.9 | tr | NEW | |||||
| 2113 | (E)-Phytol | 2.3 | 1.3 | 2.6 | MS, RI | |||||
| 2119 | 2-Oxotridec-6-en-1-yl senecioate * | 0.8 | 2.8 | 6.3 | 2.8 | tr | NEW | |||
| 2133 | 2-Oxotridec-7-en-1-yl senecioate * | 0.1 | 0.8 | 1.2 | 0.6 | NEW | ||||
| 2146 | (9Z,12Z)-Octadeca-9,12-dienoic acid | tr | 1.1 | MS, RI, CoI | ||||||
| 2150 | 2-Oxotridecyl senecioate | 0.8 | 10.1 | 13.2 | 2.8 | NEW | ||||
| 2151 | (9Z,12Z,15Z)-Octadeca-9,12,15-trienoic acid | tr | 1.4 | MS, RI | ||||||
| 2200 | Docosane | tr | 1.5 | tr | tr | tr | MS, RI, CoI | |||
| 2300 | Tricosane | 0.1 | 2.8 | tr | tr | tr | MS, RI, CoI | |||
| 2400 | Tetracosane | tr | 2.1 | 0.3 | 0.3 | MS, RI, CoI | ||||
| 2430 | Docosanal | tr | tr | MS, RI | ||||||
| 2462 | 2-Methyltetracosane | tr | 0.1 | MS, RI | ||||||
| 2474 | 3-Methyltetracosane | tr | 0.1 | MS, RI | ||||||
| 2500 | Pentacosane | 0.5 | 6.4 | tr | tr | MS, RI, CoI | ||||
| 2600 | Hexacosane | tr | 1.2 | 0.4 | 0.5 | 0.1 | MS, RI, CoI | |||
| 2663 | 2-Methylhexacosane | tr | 0.2 | MS, RI | ||||||
| 2674 | 3-Methylhexacosane | tr | 0.1 | MS, RI | ||||||
| 2700 | Heptacosane | 0.1 | 2.8 | 0.5 | 0.5 | 0.1 | MS, RI, CoI | |||
| 2763 | 2-Methylheptacosane | tr | 0.1 | MS, RI | ||||||
| 2774 | 3-Methylheptacosane | tr | 0.1 | MS, RI | ||||||
| 2800 | Octacosane | tr | 0.8 | 0.2 | 0.3 | 0.1 | MS, RI, CoI | |||
| 2824 | Supraene | tr | 1.5 | MS, RI, CoI | ||||||
| 2864 | 2-Methyloctacosane | tr | 0.1 | MS, RI | ||||||
| 2900 | Nonacosane | 0.1 | 3.3 | 0.5 | 0.5 | tr | MS, RI, CoI | |||
| 2964 | 2-Methylnonacosane | tr | tr | MS, RI | ||||||
| 2974 | 3-Methylnonacosane | tr | 0.1 | MS, RI | ||||||
| 3000 | Triacontane | tr | 0.4 | 0.1 | 0.2 | tr | MS, RI, CoI | |||
| 3100 | Hentriacontane | 0.1 | 1.5 | tr | 0.3 | tr | MS, RI, CoI | |||
| 3200 | Dotriacontane | tr | tr | MS, RI, CoI | ||||||
| 3300 | Tritriacontane | tr | tr | MS, RI, CoI | ||||||
| Total identified (%) | 97.0 | 95.2 | 90.4 | 91.2 | 89.5 | 92.7 | 90.9 | 89.2 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radulović, N.S.; Mladenović, M.Z.; Lima, C.S.; Müller, E.C.A.; da Costa, E.V.M.; Martins, R.V.; Boylan, F. Amazon Rainforest Hidden Volatiles—Part I: Unveiling New Compounds from Acmella oleracea (L.) R.K. Jansen Essential Oil. Plants 2024, 13, 1690. https://doi.org/10.3390/plants13121690
Radulović NS, Mladenović MZ, Lima CS, Müller ECA, da Costa EVM, Martins RV, Boylan F. Amazon Rainforest Hidden Volatiles—Part I: Unveiling New Compounds from Acmella oleracea (L.) R.K. Jansen Essential Oil. Plants. 2024; 13(12):1690. https://doi.org/10.3390/plants13121690
Chicago/Turabian StyleRadulović, Niko S., Marko Z. Mladenović, Clarissa Silva Lima, Elza Caroline Alves Müller, Elizabeth Vianna Moraes da Costa, Rozilene Valadares Martins, and Fabio Boylan. 2024. "Amazon Rainforest Hidden Volatiles—Part I: Unveiling New Compounds from Acmella oleracea (L.) R.K. Jansen Essential Oil" Plants 13, no. 12: 1690. https://doi.org/10.3390/plants13121690
APA StyleRadulović, N. S., Mladenović, M. Z., Lima, C. S., Müller, E. C. A., da Costa, E. V. M., Martins, R. V., & Boylan, F. (2024). Amazon Rainforest Hidden Volatiles—Part I: Unveiling New Compounds from Acmella oleracea (L.) R.K. Jansen Essential Oil. Plants, 13(12), 1690. https://doi.org/10.3390/plants13121690