
Genome-Wide Analysis of DNA Demethylases in Land Plants and Their Expression Pattern in Rice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of DNA Demethylases in 12 Land Plants
2.2. Sequence Alignment and Construction of the Phylogenetic Tree
2.3. Protein Structure and Property Analysis
2.4. Gene Structure Analysis
2.5. Gene Duplication Analysis of DNA Demethylase Families
2.6. Promoter Analysis
2.7. Expression Pattern Analysis
3. Results
3.1. Identification and Classification of 48 DNA Demethylases in 12 Plants
3.2. Recent WGD Events Generate Gene Diversification in Few Gene Pairs from Target DNA Demethylases
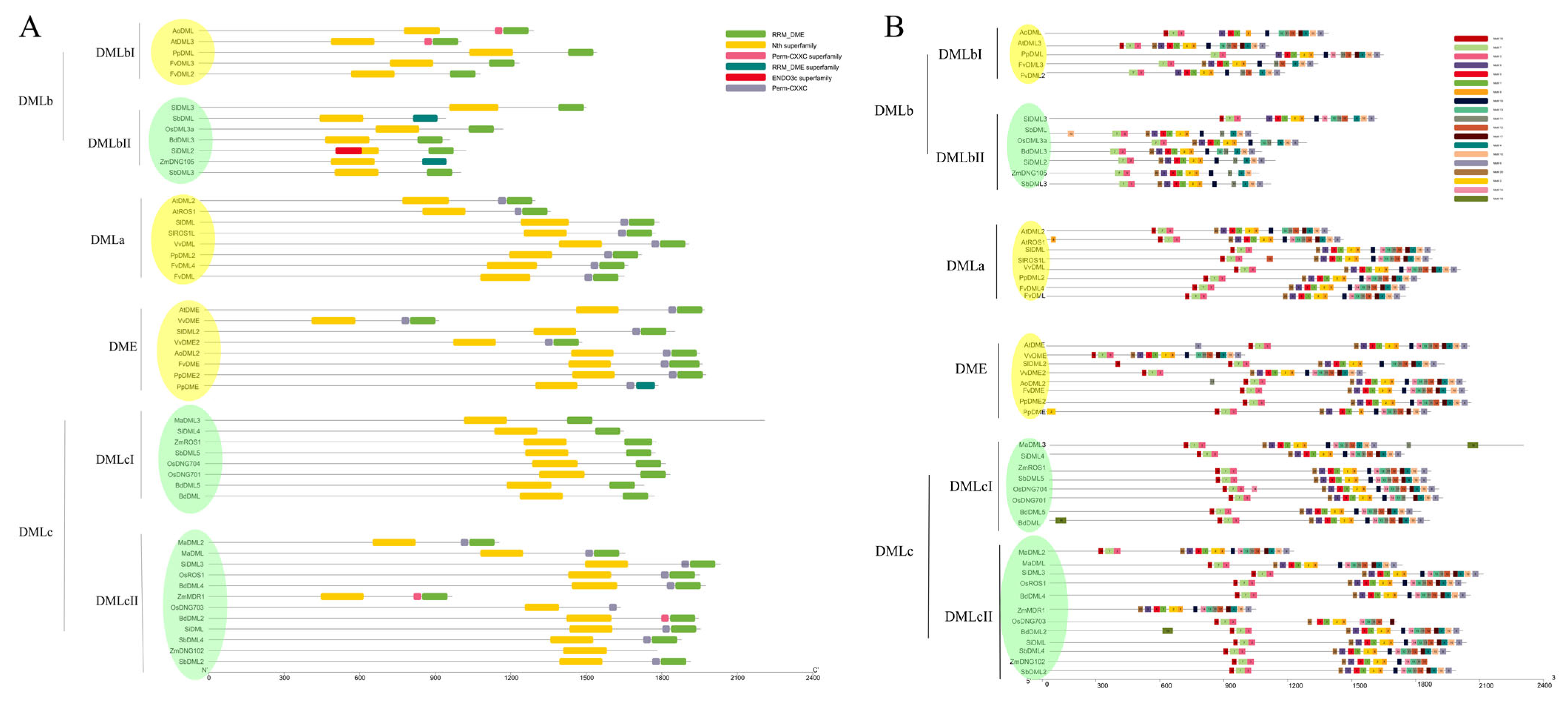
3.3. Protein Structure and Motif Analysis Verify the Classification of DNA Demethylases
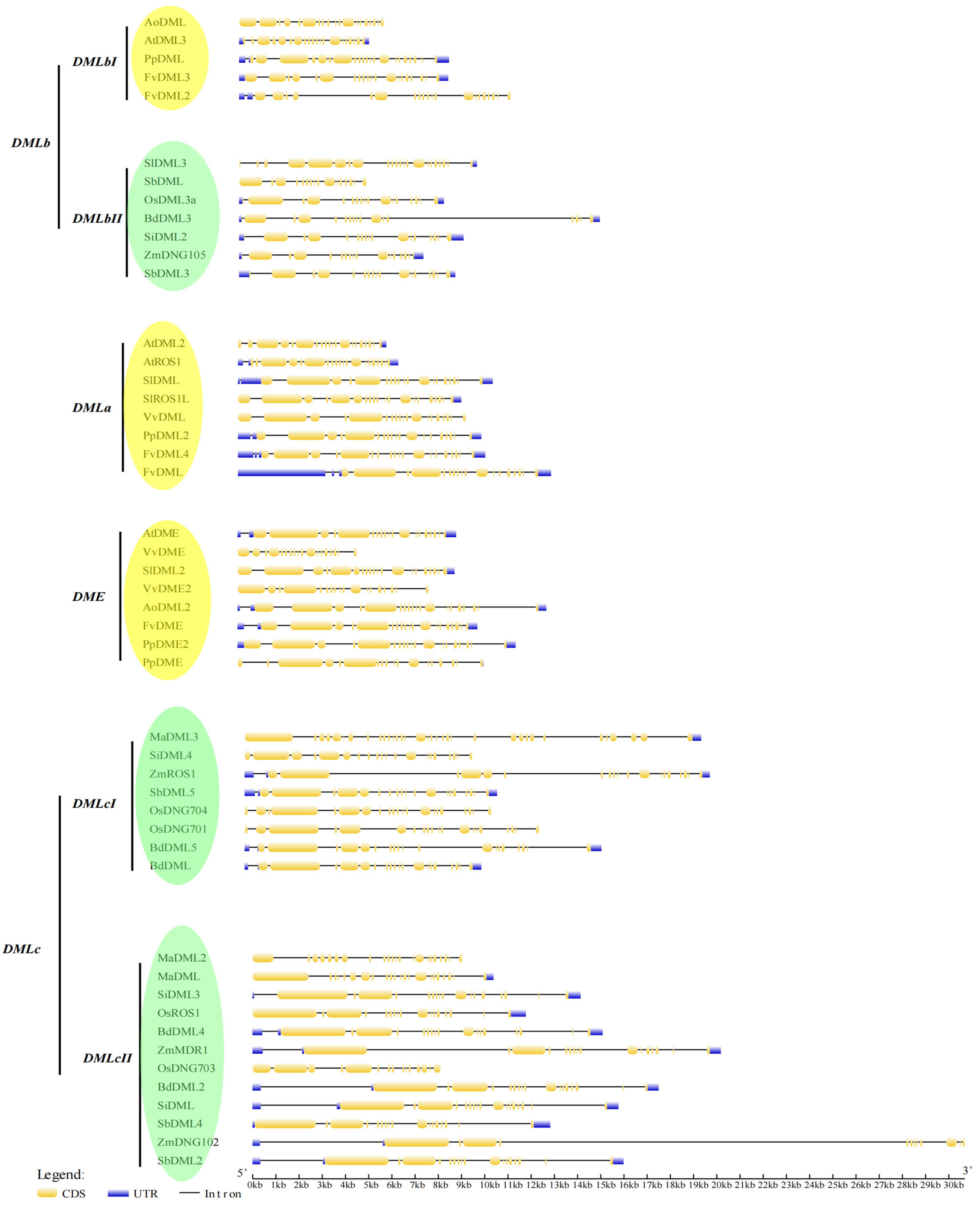
3.4. Gene Structure Analysis Shows the Exon–Intron Patterns of DNA Demethylases
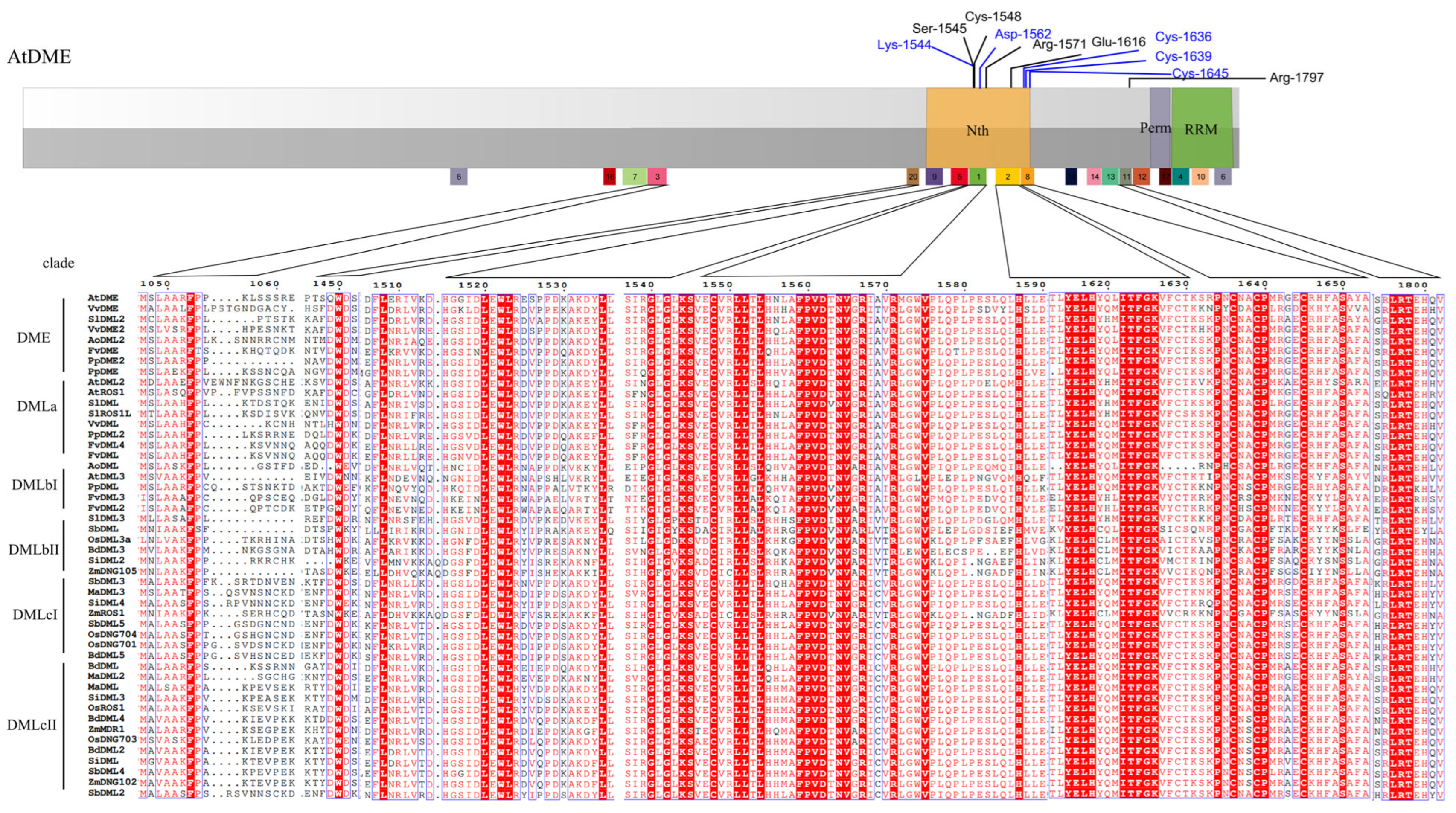
3.5. Sequences Alignment Demonstrates the Conserved Enzymatic Activity Sites
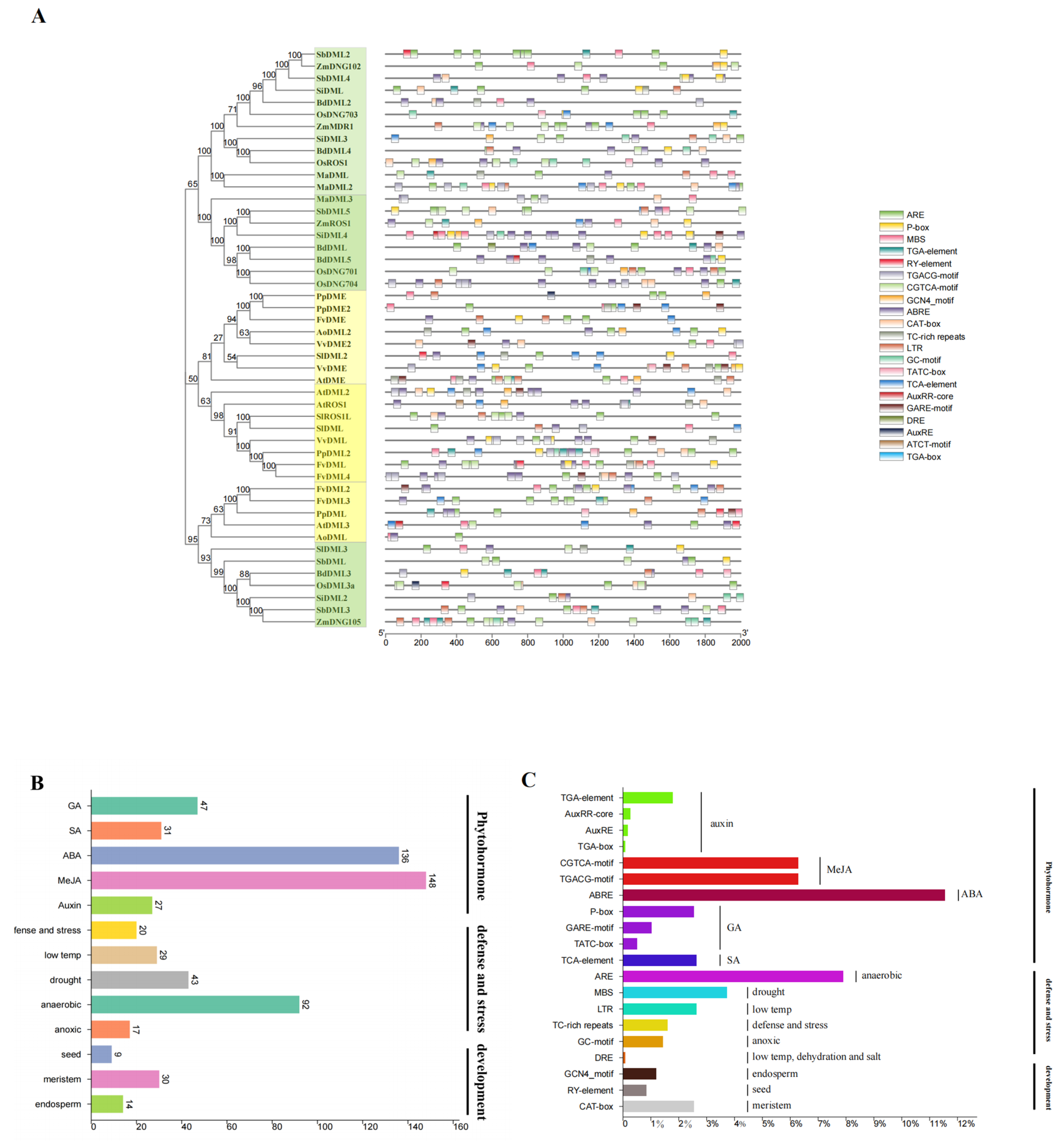
3.6. Cis-Element Analysis Shows Implication for the Future Research of DNA Demethylases in Plants
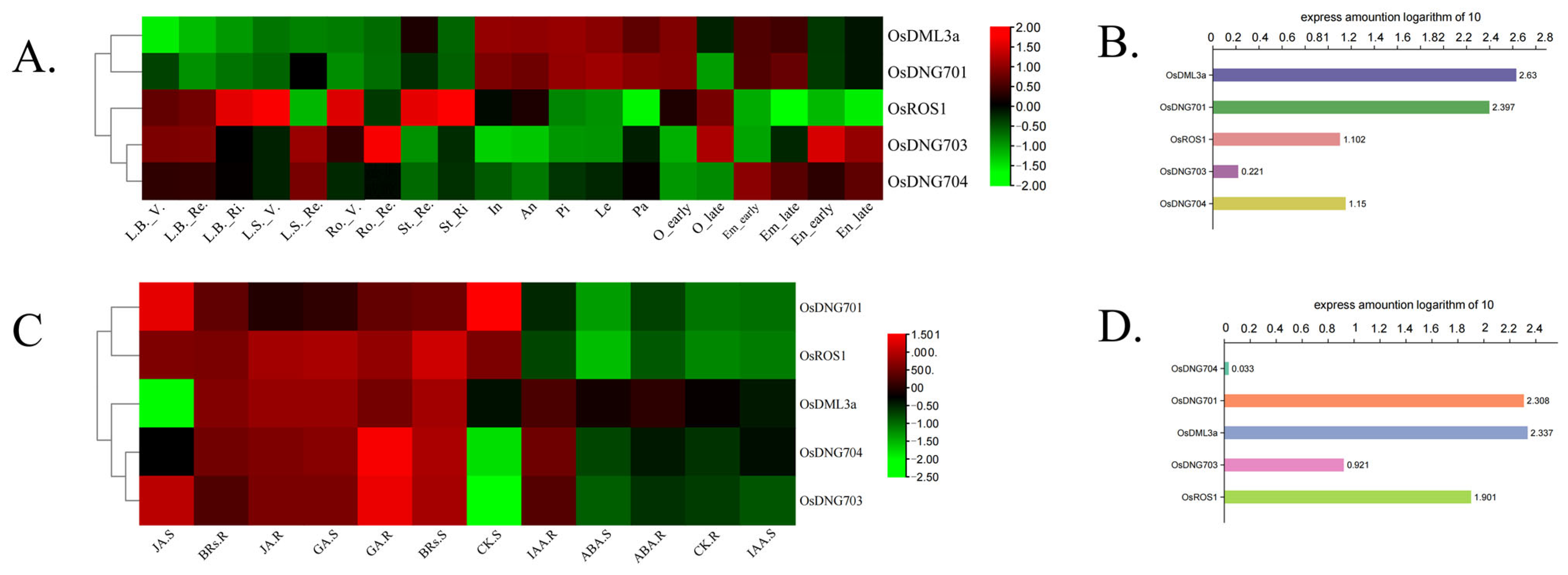
3.7. Expression Patterns of Rice DNA Demethylase Genes in Different Developmental Stages and Tissues and in Response to Phytohormones
4. Discussion
4.1. Construction of Phylogenetic Tree and Further Verification Lead to New Classes of DNA Demethylases
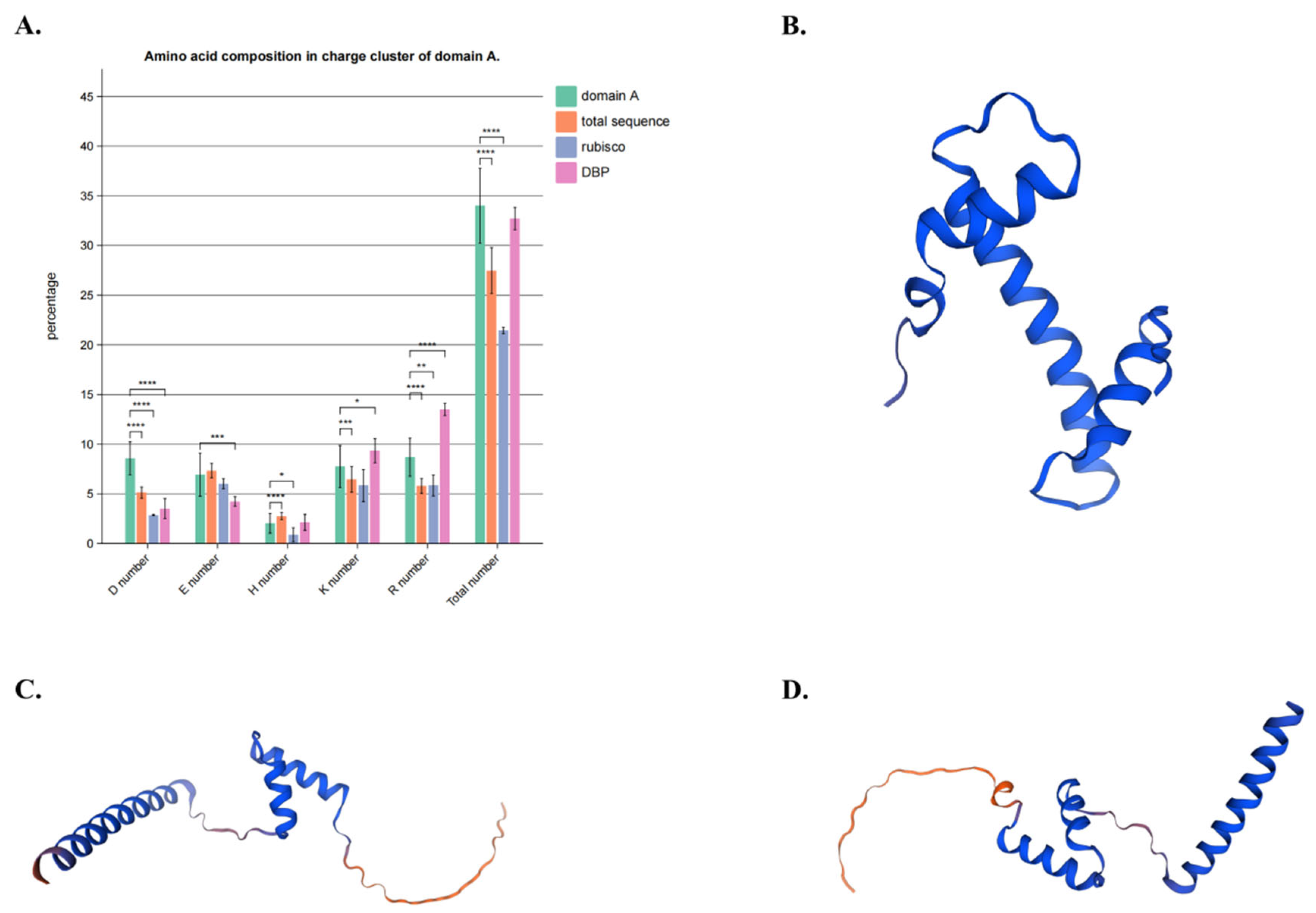
4.2. Multiple Sequence Alignment and Domain and Motif Analysis Discovered a New Functional Region of DNA Demethylases
4.3. Cis-Element Analysis Shows the Future Investigation Field of the Role of DNA Demethylase in Plant Development Regulation and Stress Response
4.4. Expression Patterns Analysis of OsDMLs Shows the Separated Role of Different Rice DNA Demethylases in Response to Specific Phytohormones and in Different Tissues
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, S.; Peng, Y.; Panchenko, A.R. DNA methylation: Precise modulation of chromatin structure and dynamics. Curr. Opin. Struct. Biol. 2022, 75, 102430. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Weiss, T.; Crisp, P.A.; Liang, Z.; Noshay, J.; Menard, C.C.; Wang, C.; Song, M.; Hirsch, C.N.; Springer, N.M.; et al. Genome-wide loss of CHH methylation with limited transcriptome changes in Setaria viridis DOMAINS REARRANGED METHYLTRANSFERASE (DRM) mutants. Plant J. 2022, 111, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef]
- Shim, S.; Lee, H.G.; Seo, P.J. MET1-Dependent DNA methylation represses light signaling and influences plant regeneration in arabidopsis. Mol. Cells 2021, 44, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Parrilla-Doblas, J.T.; Roldán-Arjona, T.; Ariza, R.R.; Córdoba-Cañero, D. Active DNA demethylation in plants. Int. J. Mol. Sci. 2019, 20, 4683. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T.; David, L.; Zhu, J.K. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Kapoor, A.; Sridhar, V.V.; Agius, F.; Zhu, J.K. The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr. Biol. 2007, 17, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ruiz, T.; Ortega-Galisteo, A.P.; Ponferrada-Marín, M.I.; Martínez-Macías, M.I.; Ariza, R.R.; Roldán-Arjona, T. DEMETER and Repressor of Silencing 1 encode 5-methylcytosine DNA glycosylases. Proc. Natl. Acad. Sci. USA 2006, 103, 6853–6858. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Li, X.; Liu, Q.; Zhao, Y.; Jiang, W.; Wu, A.; Zhou, D.X. DNA demethylases remodel DNA methylation in rice gametes and zygote and are required for reproduction. Mol. Plant 2021, 14, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [PubMed]
- La, H.; Ding, B.; Mishra, G.P.; Zhou, B.; Yang, H.; Bellizzi Mdel, R.; Chen, S.; Meyers, B.C.; Peng, Z.; Zhu, J.K.; et al. A 5-methylcytosine DNA glycosylase/lyase demethylates the retrotransposon Tos17 and promotes its transposition in rice. Proc. Natl. Acad. Sci. USA 2011, 108, 15498–15503. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Yamaguchi, K.; Fukada-Tanaka, S.; Terada, R.; Mitsui, T.; Iida, S. A null mutation of ROS1a for DNA demethylation in rice is not transmittable to progeny. Plant J. 2012, 71, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Ono, A.; Scholten, S.; Kinoshita, T.; Zilberman, D.; Okamoto, T.; Fischer, R.L. DNA demethylation by ROS1a in rice vegetative cells promotes methylation in sperm. Proc. Natl. Acad. Sci. USA 2019, 116, 9652–9657. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wu, L.; Luo, Z.; Zhang, M.; Lai, J.; Li, L.; Springer, N.M.; Li, Q. DNA demethylation affects imprinted gene expression in maize endosperm. Genome Biol. 2022, 23, 77. [Google Scholar] [CrossRef] [PubMed]
- Gehring, M.; Huh, J.H.; Hsieh, T.F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Agius, F.; Zhu, J.K. Preventing transcriptional gene silencing by active DNA demethylation. FEBS Lett. 2005, 579, 5889–5898. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Frost, J.M.; Park, K.; Ohr, H.; Park, G.T.; Kim, S.; Eom, H.; Lee, I.; Brooks, J.S.; Fischer, R.L.; et al. Control of DEMETER DNA demethylase gene transcription in male and female gamete companion cells in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2017, 114, 2078–2083. [Google Scholar] [CrossRef] [PubMed]
- Schoft, V.K.; Chumak, N.; Choi, Y.; Hannon, M.; Garcia-Aguilar, M.; Machlicova, A.; Slusarz, L.; Mosiolek, M.; Park, J.S.; Park, G.T.; et al. Function of the DEMETER DNA glycosylase in the Arabidopsis thaliana male gametophyte. Proc. Natl. Acad. Sci. USA 2011, 108, 8042–8047. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.H.; Bauer, M.J.; Hsieh, T.F.; Fischer, R.L. Cellular programming of plant gene imprinting. Cell 2008, 132, 735–744. [Google Scholar] [CrossRef]
- Sadhukhan, A.; Prasad, S.S.; Mitra, J.; Siddiqui, N.; Sahoo, L.; Kobayashi, Y.; Koyama, H. How do plants remember drought? Planta 2022, 256, 7. [Google Scholar] [CrossRef] [PubMed]
- Singroha, G.; Kumar, S.; Gupta, O.P.; Singh, G.P.; Sharma, P. Uncovering the epigenetic marks involved in mediating salt stress tolerance in plants. Front. Genet. 2022, 13, 811732. [Google Scholar] [CrossRef] [PubMed]
- Perrella, G.; Bäurle, I.; van Zanten, M. Epigenetic regulation of thermomorphogenesis and heat stress tolerance. New Phytol. 2022, 234, 1144–1160. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; He, Y. Experiencing winter for spring flowering: A molecular epigenetic perspective on vernalization. J. Integr. Plant Biol. 2020, 62, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Hereme, R.; Galleguillos, C.; Morales-Navarro, S.; Molina-Montenegro, M.A. What if the cold days return? Epigenetic mechanisms in plants to cold tolerance. Planta 2021, 254, 46. [Google Scholar] [CrossRef] [PubMed]
- Secco, D.; Whelan, J.; Rouached, H.; Lister, R. Nutrient stress-induced chromatin changes in plants. Curr. Opin. Plant Biol. 2017, 39, 1–7. [Google Scholar] [CrossRef]
- Fan, X.; Peng, L.; Zhang, Y. Plant DNA methylation responds to nutrient stress. Genes 2022, 13, 992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gong, Z.; Zhu, J.K. Active DNA demethylation in plants: 20 years of discovery and beyond. J. Integr. Plant Biol. 2022, 64, 2217–2239. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Niu, Q.; Zhang, B.; Chen, K.; Yang, R.; Zhu, J.K.; Zhang, Y.; Lang, Z. Downregulation of RdDM during strawberry fruit ripening. Genome Biol. 2018, 19, 212. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Liu, C.; Qi, Y.; Zhou, X. Geminiviruses employ host DNA glycosylases to subvert DNA methylation-mediated defense. Nat. Commun. 2022, 13, 575. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Huang, H.; Lin, X.; Zhu, C.; Kosami, K.I.; Huang, C.; Zhang, H.; Duan, C.G.; Zhu, J.K.; Miki, D. Roles of DEMETER in regulating DNA methylation in vegetative tissues and pathogen resistance. J. Integr. Plant Biol 2021, 63, 691–706. [Google Scholar] [CrossRef]
- López Sánchez, A.; Stassen, J.H.; Furci, L.; Smith, L.M.; Ton, J. The role of DNA (de)methylation in immune responsiveness of Arabidopsis. Plant J. Cell Mol. Biol. 2016, 88, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lang, C.; Wu, Y.; Meng, D.; Yang, T.; Li, D.; Jin, T.; Zhou, X. ROS1-mediated decrease in DNA methylation and increase in expression of defense genes and stress response genes in Arabidopsis thaliana due to abiotic stresses. BMC Plant Biol. 2022, 22, 104. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Ren, R.; Xiao, H.; Chen, Z.; Yu, J.; Zhang, H.; Shi, Q.; Hou, H.; He, S.; Li, L. Characteristic and evolution of HAT and HDAC genes in Gramineae genomes and their expression analysis under diverse stress in Oryza sativa. Planta 2021, 253, 72. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Mok, Y.G.; Uzawa, R.; Lee, J.; Weiner, G.M.; Eichman, B.F.; Fischer, R.L.; Huh, J.H. Domain structure of the DEMETER 5-methylcytosine DNA glycosylase. Proc. Natl. Acad. Sci. USA 2010, 107, 19225–19230. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Abhiman, S.; Aravind, L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 2011, 101, 25–104. [Google Scholar] [PubMed]
- Du, X.; Yang, Z.; Xie, G.; Wang, C.; Zhang, L.; Yan, K.; Yang, M.; Li, S.; Zhu, J.K.; Du, J. Molecular basis of the plant ROS1-mediated active DNA demethylation. Nat. Plants 2023, 9, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Takehisa, H.; Kamatsuki, K.; Minami, H.; Namiki, N.; Ikawa, H.; Ohyanagi, H.; Sugimoto, K.; Antonio, B.A.; Nagamura, Y. RiceXPro version 3.0: Expanding the informatics resource for rice transcriptome. Nucleic Acids Res. 2013, 41, D1206–D1213. [Google Scholar] [CrossRef]
- Sato, Y.; Antonio, B.A.; Namiki, N.; Takehisa, H.; Minami, H.; Kamatsuki, K.; Sugimoto, K.; Shimizu, Y.; Hirochika, H.; Nagamura, Y. RiceXPro: A platform for monitoring gene expression in japonica rice grown under natural field conditions. Nucleic Acids Res. 2011, 39, D1141–D1148. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Ren, S.; Wang, Y.; Han, Y.; Li, Y. Characterization of DNA methyltransferase and demethylase genes in Fragaria vesca. Mol. Genet. Genom. 2016, 291, 1333–1345. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. (Timetree 5: An expanded resource for species divergence times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Liu, H.L.; Daxinger, L.; Pontes, O.; He, X.; Qian, W.; Lin, H.; Xie, M.; Lorkovic, Z.J.; Zhang, S.; et al. An RNA polymerase II- and AGO4-associated protein acts in RNA-directed DNA methylation. Nature 2010, 465, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Huettel, B.; Kanno, T.; Daxinger, L.; Aufsatz, W.; Matzke, A.J.; Matzke, M. Endogenous targets of RNA-directed DNA methylation and Pol IV in Arabidopsis. EMBO J. 2006, 25, 2828–2836. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, O.; Reinders, J.; Caikovski, M.; Smathajitt, C.; Paszkowski, J. Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 2007, 130, 851–862. [Google Scholar] [CrossRef]
- Khouider, S.; Borges, F.; LeBlanc, C.; Ungru, A.; Schnittger, A.; Martienssen, R.; Colot, V.; Bouyer, D. Male fertility in Arabidopsis requires active DNA demethylation of genes that control pollen tube function. Nat. Commun. 2021, 12, 410. [Google Scholar] [CrossRef]
- Tang, D.; Gallusci, P.; Lang, Z. Fruit development and epigenetic modifications. New Phytol. 2020, 228, 839–844. [Google Scholar] [CrossRef]
- Arora, K.; Panda, K.K.; Mittal, S.; Mallikarjuna, M.G.; Thirunavukkarasu, N. In silico characterization and functional validation of cell wall modification genes imparting waterlogging tolerance in maize. Bioinform. Biol. Insights 2017, 11, 1177932217747277. [Google Scholar] [CrossRef]
- Pan, J.; Sharif, R.; Xu, X.; Chen, X. Mechanisms of Waterlogging Tolerance in Plants: Research Progress and Prospects. Front. Plant Sci. 2021, 11, 627331. [Google Scholar] [CrossRef] [PubMed]
- Dinolfo, M.I.; Martínez, M.; Castañares, E.; Vanzetti, L.S.; Rossi, F.; Stenglein, S.A.; Arata, A.F. Interaction of methyl-jasmonate and Fusarium poae in bread wheat. Fungal Biol. 2022, 126, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nie, Y.; Zhang, Z.; Ye, Z.; Zou, X.; Zhang, L.; Wang, Z. Comparative proteomic analysis of methyl jasmonate-induced defense responses in different rice cultivars. Proteomics 2014, 14, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.J.; Fischer, R.L. Genome demethylation and imprinting in the endosperm. Curr. Opin. Plant Biol. 2011, 14, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Lepère, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; et al. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Cleaves, K.; Hewezi, T. Expression patterns of DNA methylation and demethylation genes during plant development and in response to phytohormones. Int. J. Mol. Sci. 2021, 22, 9681. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Ju, Z.; Gao, C.; Mei, X.; Fu, D.; Zhu, H.; Luo, Y.; Zhu, B. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in Solanum lycopersicum. Gene 2014, 550, 230–237. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monocots | Eudicots | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Musaceae | Gramineae | Brassicaceae | Anacardiaceae | Rosaceae | Poaceae | Vitaceae | |||||||
| Ma | Bd | Os | Sb | Si | Zm | At | Ao | Fv | Pp | Sl | Vv | ||
| Demethylase | |||||||||||||
| DMLb class | 0 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 0 | 12 |
| DMLa class | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 | 1 | 2 | 1 | 8 |
| DME class | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 2 | 1 | 2 | 8 |
| DMLc class | 3 | 4 | 4 | 3 | 3 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 20 |
| Total | 3 | 5 | 5 | 5 | 4 | 4 | 4 | 2 | 4 | 4 | 4 | 3 | 48 |
| Species | Gene Pair | Ka/Ks | Date (Mya) |
|---|---|---|---|
| Ao | AoDML vs. AoDML2 | 0.192689736 | 166.914916 |
| Ma | MaDML2 vs. MaDML3 | 0.232780074 | 90.99861036 |
| Pp | PpDML vs. PpDML2 | 0.295172933 | 108.1189753 |
| Sl | SlROS1L vs. SlDML | 0.393649662 | 34.13422417 |
| Sl | SlROS1L vs. SlDML2 | 0.188789595 | 149.3784079 |
| Vv | VvDME vs. VvDML | 0.207862231 | 99.7488283 |
| Gene Name | Light | Auxin | GA | SA | ABA | MeJA | Defense and Stress | Low Temp | Drought | Seed | Meristem | Endosperm | Anaerobic | Anoxic |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AoDML | 13 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| AtDML3 | 11 | 1 | 0 | 2 | 3 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PpDML | 13 | 1 | 2 | 0 | 2 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 2 | 0 |
| FvDML3 | 5 | 1 | 0 | 2 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 6 | 0 |
| FvDML2 | 16 | 0 | 2 | 2 | 6 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 4 | 0 |
| SlDML3 | 9 | 1 | 1 | 0 | 1 | 2 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| SbDML | 7 | 0 | 1 | 0 | 1 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| OsDML3a | 7 | 1 | 0 | 0 | 0 | 8 | 0 | 0 | 1 | 1 | 2 | 0 | 3 | 0 |
| BdDML3 | 10 | 2 | 2 | 0 | 3 | 2 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 |
| SiDML2 | 15 | 0 | 0 | 0 | 2 | 2 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 2 |
| ZmDNG105 | 11 | 3 | 0 | 0 | 2 | 8 | 0 | 2 | 2 | 0 | 1 | 0 | 2 | 3 |
| SbDML3 | 10 | 1 | 0 | 0 | 4 | 4 | 0 | 2 | 2 | 0 | 2 | 0 | 2 | 0 |
| AtDML2 | 24 | 0 | 0 | 2 | 10 | 2 | 1 | 0 | 0 | 0 | 2 | 1 | 0 | 0 |
| AtROS1 | 15 | 1 | 0 | 1 | 3 | 4 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 1 |
| SlDML | 8 | 1 | 0 | 1 | 2 | 2 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| SlROS1L | 11 | 0 | 0 | 0 | 3 | 4 | 0 | 1 | 0 | 0 | 1 | 0 | 4 | 0 |
| VvDML | 14 | 0 | 3 | 0 | 4 | 8 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| PpDML2 | 9 | 3 | 2 | 1 | 3 | 2 | 0 | 0 | 2 | 0 | 2 | 0 | 3 | 1 |
| FvDML4 | 17 | 0 | 1 | 0 | 8 | 8 | 1 | 1 | 0 | 2 | 1 | 0 | 2 | 0 |
| FvDML | 13 | 0 | 2 | 0 | 7 | 4 | 1 | 1 | 2 | 3 | 1 | 0 | 3 | 0 |
| AtDME | 10 | 1 | 1 | 0 | 1 | 4 | 3 | 0 | 2 | 0 | 0 | 1 | 2 | 0 |
| VvDME | 5 | 0 | 5 | 2 | 1 | 2 | 1 | 1 | 0 | 0 | 1 | 1 | 2 | 0 |
| SlDML2 | 12 | 0 | 1 | 3 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 3 | 0 |
| VvDME2 | 4 | 0 | 1 | 0 | 1 | 4 | 0 | 0 | 1 | 0 | 2 | 0 | 1 | 0 |
| AoDML2 | 16 | 0 | 1 | 2 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 3 | 0 |
| FvDME | 11 | 0 | 1 | 1 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 2 | 0 |
| PpDME2 | 14 | 0 | 2 | 2 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 2 | 0 |
| PpDME | 7 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 2 | 0 |
| MaDML3 | 9 | 0 | 0 | 0 | 1 | 6 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| SiDML4 | 14 | 1 | 4 | 0 | 6 | 2 | 1 | 0 | 5 | 0 | 0 | 1 | 0 | 1 |
| ZmROS1 | 14 | 1 | 1 | 1 | 2 | 2 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 |
| SbDML5 | 9 | 0 | 1 | 1 | 1 | 8 | 0 | 1 | 1 | 0 | 1 | 0 | 4 | 0 |
| OsDNG704 | 16 | 1 | 0 | 0 | 6 | 8 | 0 | 1 | 0 | 0 | 2 | 0 | 1 | 0 |
| OsDNG701 | 9 | 0 | 0 | 1 | 3 | 6 | 0 | 2 | 1 | 0 | 0 | 1 | 2 | 1 |
| BdDML5 | 10 | 1 | 1 | 0 | 6 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| BdDML | 9 | 1 | 0 | 1 | 4 | 2 | 1 | 1 | 0 | 0 | 1 | 0 | 2 | 0 |
| MaDML2 | 6 | 0 | 2 | 2 | 1 | 10 | 0 | 1 | 3 | 0 | 1 | 0 | 3 | 1 |
| MaDML | 7 | 1 | 0 | 0 | 1 | 2 | 1 | 1 | 2 | 0 | 0 | 0 | 1 | 0 |
| SiDML3 | 8 | 0 | 0 | 1 | 1 | 2 | 0 | 1 | 0 | 0 | 1 | 1 | 2 | 2 |
| OsROS1/DNG702 | 17 | 0 | 1 | 0 | 11 | 6 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 3 |
| BdDML4 | 10 | 0 | 1 | 0 | 3 | 2 | 0 | 2 | 0 | 0 | 1 | 0 | 1 | 0 |
| ZmMDR1 | 9 | 0 | 1 | 2 | 3 | 6 | 0 | 1 | 1 | 0 | 0 | 1 | 3 | 0 |
| OsDNG703 | 7 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1 |
| BdDML2 | 19 | 0 | 0 | 0 | 3 | 4 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| SiDML | 10 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 3 | 0 |
| SbDML4 | 14 | 0 | 2 | 0 | 11 | 2 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| ZmDNG102 | 7 | 0 | 1 | 0 | 0 | 4 | 0 | 0 | 2 | 0 | 0 | 1 | 3 | 0 |
| SbDML2 | 7 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 7 | 0 |
| Total No. | Protein Length (aa) | Molecular Weight (kDa) | pI | No. of Genes with pI > 7 | |
|---|---|---|---|---|---|
| DME gene family | 8 | 1793 ± 371 | 196 ± 40 | 7.5 ± 0.6 | 7 |
| DMLa gene family | 8 | 1680 ± 212 | 188 ± 22 | 6.8 ± 0.9 | 2 |
| DMLb gene family | 12 | 1179 ± 210 | 133 ± 24 | 7.9 ± 1.0 | 10 |
| DMLc gene family | 20 | 1775 ± 283 | 197 ± 31 | 6.5 ± 0.5 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, S.; Xiao, J.; Zhao, Y.; Hou, J.; Li, L. Genome-Wide Analysis of DNA Demethylases in Land Plants and Their Expression Pattern in Rice. Plants 2024, 13, 2068. https://doi.org/10.3390/plants13152068
Mao S, Xiao J, Zhao Y, Hou J, Li L. Genome-Wide Analysis of DNA Demethylases in Land Plants and Their Expression Pattern in Rice. Plants. 2024; 13(15):2068. https://doi.org/10.3390/plants13152068
Chicago/Turabian StyleMao, Shengxin, Jian Xiao, Yating Zhao, Jiaqi Hou, and Lijia Li. 2024. "Genome-Wide Analysis of DNA Demethylases in Land Plants and Their Expression Pattern in Rice" Plants 13, no. 15: 2068. https://doi.org/10.3390/plants13152068