

Genomic Insights into Disease Resistance in Sunflower (Helianthus annuus): Identifying Key Regions and Candidate Genes for Verticillium dahliae Resistance


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Plant Materials
2.1.2. Fungal Materials
2.1.3. Genomic Data
2.2. Methods
2.2.1. Pathogen Inoculation
2.2.2. GWA Mapping Using EMMAX
2.2.3. Identification of Candidate Markers and QTLs
2.2.4. From Candidate Regions to Genes and Functions
2.2.5. GO and KEGG Enrichment Analyses
2.2.6. Experimental Validation through Quantitative Real-Time PCR
3. Results
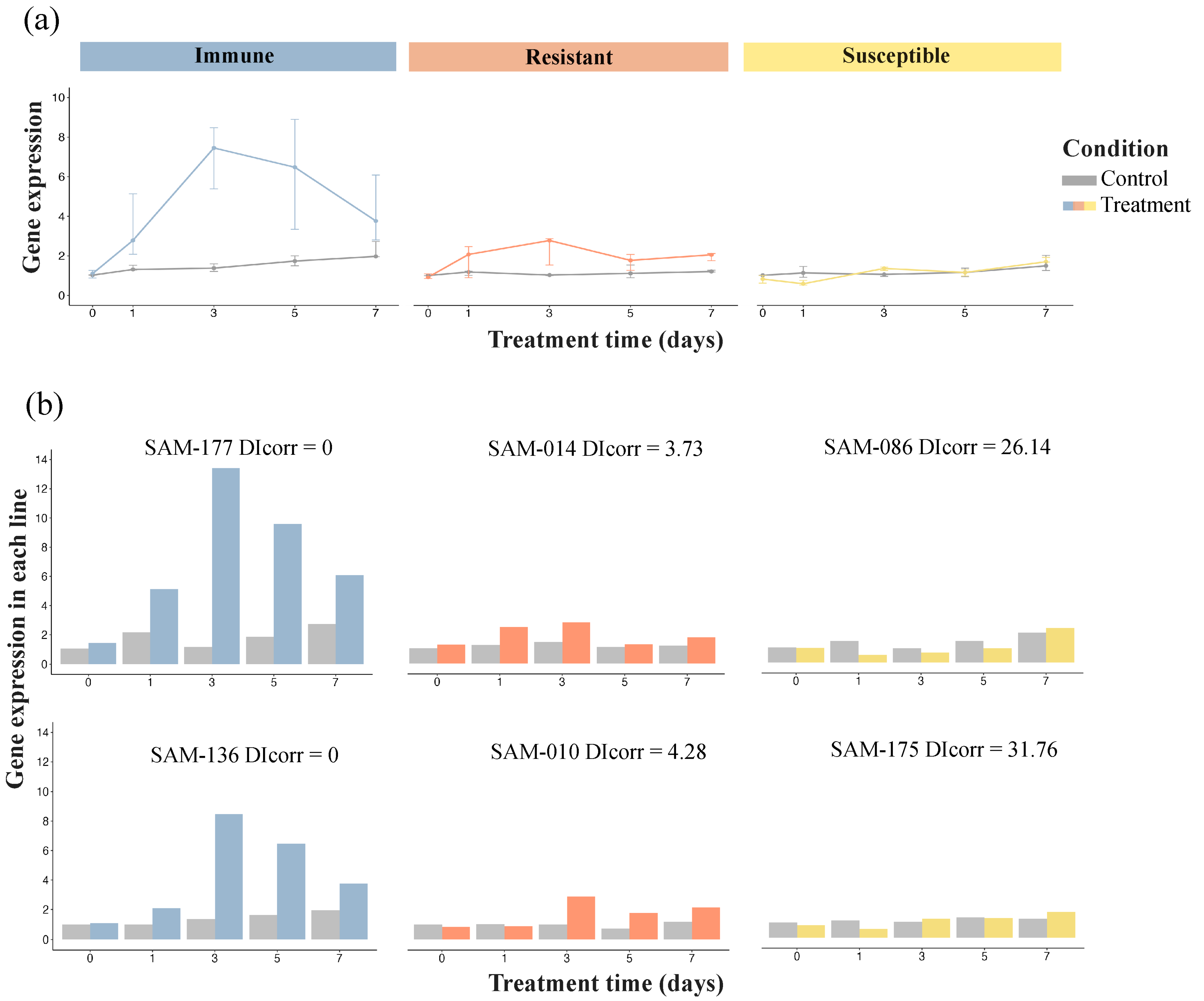
3.1. Evaluation of Disease Resistance of Sunflower Cultivar Lines
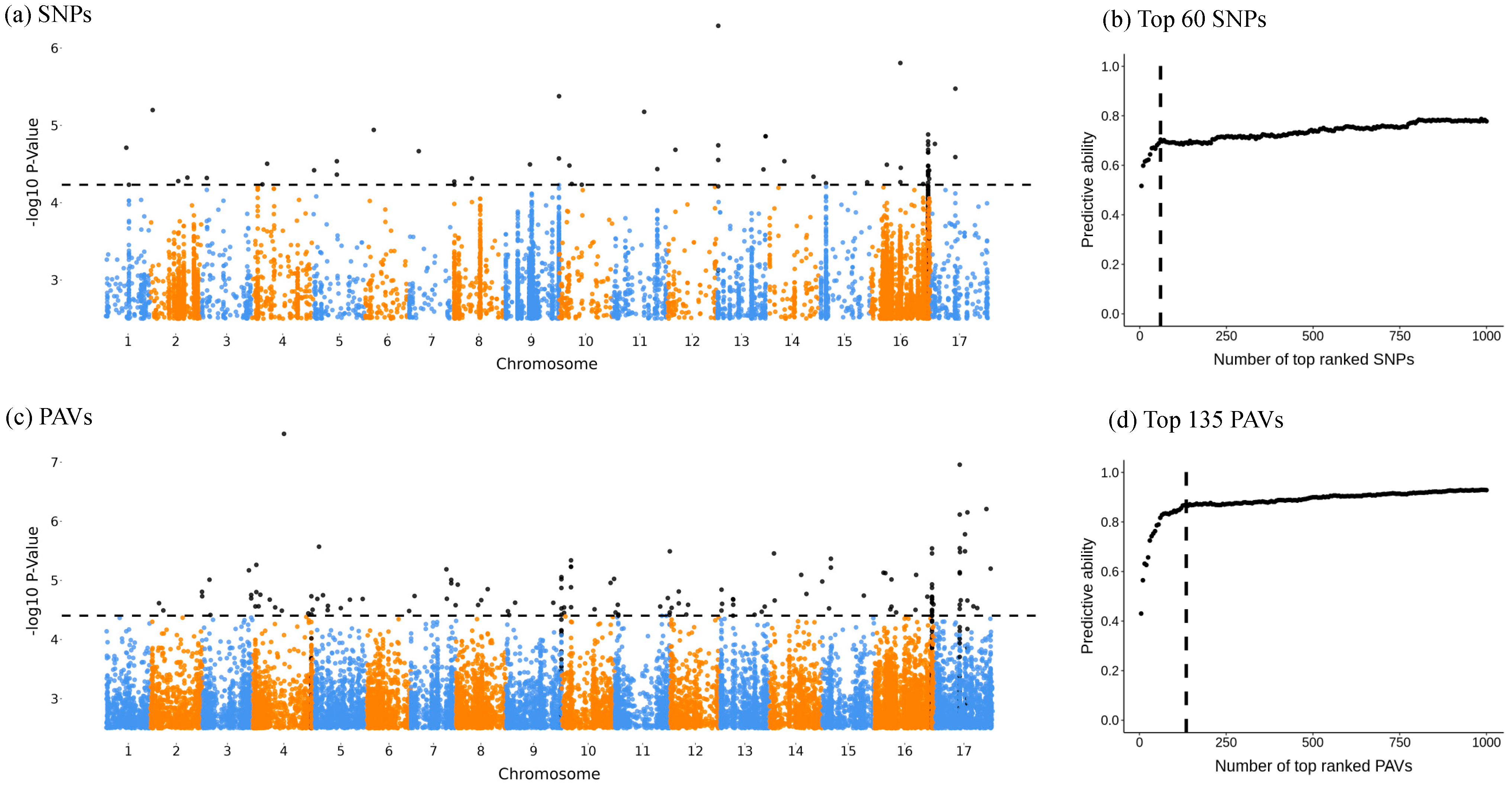
3.2. GWA Mapping and Identification of Top Candidate Markers and QTLs
3.3. From Candidate Regions to Genes and Functions
3.4. Enrichment Analysis
3.5. Experimental Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United States Department of Agriculture (USDA). Oilseeds: World Markets and Trade. United States Department of Agriculture Foreign Agricultural Service. Available online: https://apps.fas.usda.gov/psdonline/circulars/oilseeds.pdf (accessed on 10 July 2024).
- Inderbitzin, P.; Subbarao, K.V. Verticillium Systematics and Evolution: How Confusion Impedes Verticillium Wilt Management and How to Resolve It. Phytopathology 2014, 104, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Gao, J.; Zhang, G.; Yu, Y.; Zhou, H.; Chen, W.; Zhao, J. The Colonization Process of Sunflower by a Green Fluorescent Protein-Tagged Isolate of Verticillium dahliae and Its Seed Transmission. Plant Dis. 2018, 102, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.; Ingram, T.W.; Oh, Y.; Adhikari, T.B.; Dean, R.A.; Louws, F.J. Opportunities and Challenges in Studies of Host-Pathogen Interactions and Management of Verticillium dahliae in Tomatoes. Plants 2020, 9, 1622. [Google Scholar] [CrossRef] [PubMed]
- Montes-Osuna, N.; Mercado-Blanco, J. Verticillium Wilt of Olive and Its Control: What Did We Learn during the Last Decade? Plants 2020, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Powelson, M.L. Potato Early Dying: Management Challenges in a Changing Production Environment. Plant Dis. 2002, 86, 1184–1193. [Google Scholar] [CrossRef]
- Centre for Agriculture and Bioscience International (CABI). Available online: https://www.cabi.org/isc/datasheet/56275#toDistributionMaps (accessed on 12 July 2024).
- Mathew, F.M.; Mohan, K.; Rafi, N.; Colombo, D.; Block, C.; Gulya, T.; Markell, S.G.; Reyley, M.; Thompson, S.; Harveson, R.M. Verticillium Wilt of Sunflower. Plant Health Instr. 2024, 24. [Google Scholar] [CrossRef]
- García-Ruiz, R.; García-Carneros, A.B.; Molinero-Ruiz, L. A New Race of Verticillium dahliae Causing Leaf Mottle of Sunflower in Europe. Plant Dis. 2014, 98, 1435. [Google Scholar] [CrossRef]
- Gulya, T. New Strain of Verticillium dahliae in North America. Helia 2007, 30, 115–120. [Google Scholar] [CrossRef]
- Sackston, W.E.; McDonald, W.C.; Martens, J. Leaf Mottle or Verticillium Wilt of Sunflower. Plant Dis. Res. Rep. 1957, 41, 337–343. [Google Scholar]
- Markell, S.; Harveson, R.; Block, C.; Gulya, T. Sunflower Disease Diagnostic Series; North Dakota State Cooperative Extension Service Publication; North Dakota State University: Fargo, ND, USA, 2023; p. 1727. [Google Scholar]
- Harveson, R.M.; Markell, S.G.; Block, C.C.; Gulya, T.J. Compendium of Sunflower Diseases: Part I: Biotic Diseases; American Phytopathological Society: St. Paul, MN, USA, 2018. [Google Scholar] [CrossRef]
- Creus, C.; Bazzalo, M.E.; Grondona, M.; Andrade, F.; León, A.J. Disease Expression and Ecophysiological Yield Components in Sunflower Isohybrids with and without Verticillium dahliae Resistance. Crop Sci. 2007, 47, 703–710. [Google Scholar] [CrossRef]
- Hoes, J.A.; Putt, E.D.; Enns, H. Resistance to Verticillium Wilt in Collections of Wild Helianthus in North America. Phytopathology 1973, 63, 1517–1520. [Google Scholar] [CrossRef]
- Wang, D.; Su, Z.; Ning, D.; Zhao, Y.; Meng, H.; Dong, B.; Zhao, J.; Zhou, H. Different Appearance Period of Verticillium Wilt Symptoms Affects Sunflower Growth and Production. J. Plant Pathol. 2021, 103, 513–517. [Google Scholar] [CrossRef]
- Klosterman, S.J.; Atallah, Z.K.; Vallad, G.E.; Subbarao, K.V. Diversity, Pathogenicity, and Management of Verticillium Species. Annu. Rev. Phytopathol. 2009, 47, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Fradin, E.F.; Thomma, B.P. Physiology and Molecular Aspects of Verticillium Wilt Diseases Caused by V. dahliae and V. albo-atrum. Mol. Plant Pathol. 2006, 7, 71–86. [Google Scholar] [CrossRef]
- Guo, S.; Zuo, Y.; Zhang, Y.; Wu, C.; Su, W.; Jin, W.; Yu, H.; An, Y.; Li, Q. Large-Scale Transcriptome Comparison of Sunflower Genes Responsive to Verticillium dahliae. BMC Genom. 2017, 18, 42. [Google Scholar] [CrossRef]
- Mustafa, R.; Hamza, M.; Kamal, H.; Mansoor, S.; Scheffler, J.; Amin, I. Tobacco Rattle Virus-Based Silencing of Enoyl-CoA Reductase Gene and Its Role in Resistance against Cotton Wilt Disease. Mol. Biotechnol. 2017, 59, 241–250. [Google Scholar] [CrossRef]
- de Jonge, R.; van Esse, H.P.; Maruthachalam, K.; Bolton, M.D.; Santhanam, P.; Saber, M.K.; Zhang, Z.; Usami, T.; Lievens, B.; Subbarao, K.V.; et al. Tomato Immune Receptor Ve1 Recognizes Effector of Multiple Fungal Pathogens Uncovered by Genome and RNA Sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 5110–5115. [Google Scholar] [CrossRef]
- Tai, H.H.; De Koeyer, D.; Sønderkær, M.; Hedegaard, S.; Nielsen, K.L. Verticillium dahliae Disease Resistance and the Regulatory Pathway for Maturity and Tuberization in Potato. Plant Genome 2018, 11, 170040. [Google Scholar] [CrossRef]
- Markakis, E.A.; Krasagakis, N.; Manolikaki, I.; Papadaki, A.A.; Kostelenos, G.; Koubouris, G. Evaluation of Olive Varieties Resistance for Sustainable Management of Verticillium Wilt. Sustainability 2022, 14, 9342. [Google Scholar] [CrossRef]
- Song, R.; Li, J.; Xie, C.; Jian, W.; Yang, X. An Overview of the Molecular Genetics of Plant Resistance to the Verticillium Wilt Pathogen Verticillium dahliae. Int. J. Mol. Sci. 2020, 21, 1120. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Tian, L.; Wang, G.; Zhang, X.; Wang, X.; Guo, W. Discovery and Identification of Candidate Genes from the Chitinase Gene Family for Verticillium dahliae Resistance in Cotton. Sci. Rep. 2016, 6, 29022. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, X.; Sun, Y.; Wang, P.; Li, X.; Pei, Y.; Li, F.; Hou, Y. Molecular Evidence for the Involvement of a Polygalacturonase-Inhibiting Protein, GhPGIP1, in Enhanced Resistance to Verticillium and Fusarium Wilts in Cotton. Sci. Rep. 2017, 7, 39840. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nguyen, C.T.; Liang, Y.; Cao, Y.; Stacey, G. Role of LysM Receptors in Chitin-Triggered Plant Innate Immunity. Plant Signal. Behav. 2013, 8, e22598. [Google Scholar] [CrossRef] [PubMed]
- Nazar, R.N.; Xu, X.; Kurosky, A.; Robb, J. Antagonistic Function of the Ve R-Genes in Tomato. Plant Mol. Biol. 2018, 98, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pei, Y.; Sun, Y.; Liu, N.; Wang, P.; Liu, D.; Ge, X.; Li, F.; Hou, Y. A Cotton Cyclin-Dependent Kinase E Confers Resistance to Verticillium dahliae Mediated by Jasmonate-Responsive Pathway. Front. Plant Sci. 2018, 9, 642. [Google Scholar] [CrossRef]
- Gong, Q.; Yang, Z.; Wang, X.; Butt, H.I.; Chen, E.; He, S.; Zhang, C.; Zhang, X.; Li, F. Salicylic Acid-Related Cotton (Gossypium arboreum) Ribosomal Protein GaRPL18 Contributes to Resistance to Verticillium dahliae. BMC Plant Biol. 2017, 17, 59. [Google Scholar] [CrossRef]
- Mandel, J.R.; Dechaine, J.M.; Marek, L.F.; Burke, J.M. Genetic Diversity and Population Structure in Cultivated Sunflower and a Comparison to Its Wild Progenitor, Helianthus annuus L. Theor. Appl. Genet. 2011, 123, 693–704. [Google Scholar] [CrossRef]
- Mandel, J.R.; Nambeesan, S.; Bowers, J.E.; Marek, L.F.; Ebert, D.; Rieseberg, L.H.; Knapp, S.J.; Burke, J.M. Association Mapping and the Genomic Consequences of Selection in Sunflower. PLoS Genet. 2013, 9, e1003378. [Google Scholar] [CrossRef]
- Masalia, R.R.; Temme, A.A.; Torralba, N.L.; Burke, J.M. Multiple Genomic Regions Influence Root Morphology and Seedling Growth in Cultivated Sunflower (Helianthus annuus L.) under Well-Watered and Water-Limited Conditions. PLoS ONE 2018, 13, e0204279. [Google Scholar] [CrossRef]
- Gao, L.; Lee, J.S.; Hübner, S.; Hulke, B.S.; Qu, Y.; Rieseberg, L.H. Genetic and Phenotypic Analyses Indicate That Resistance to Flooding Stress Is Uncoupled from Performance in Cultivated Sunflower. New Phytol. 2019, 223, 1657–1670. [Google Scholar] [CrossRef]
- Temme, A.A.; Kerr, K.L.; Masalia, R.R.; Burke, J.M.; Donovan, L.A. Key Traits and Genes Associate with Salinity Tolerance Independent from Vigor in Cultivated Sunflower. Plant Physiol. 2020, 184, 865–880. [Google Scholar] [CrossRef]
- Temme, A.A.; Kerr, K.L.; Nolting, K.M.; Dittmar, E.L.; Masalia, R.R.; Bucksch, A.K.; Burke, J.M.; Donovan, L.A. The Genomic Basis of Nitrogen Utilization Efficiency and Trait Plasticity to Improve Nutrient Stress Tolerance in Cultivated Sunflower. J. Exp. Bot. 2024, 75, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Hübner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Rieseberg, L.H. Sunflower Pan-Genome Analysis Shows That Hybridization Altered Gene Content and Disease Resistance. Nat. Plants 2019, 5, 54–62. [Google Scholar] [CrossRef]
- Guidini, R.; Jahani, M.; Huang, K.; Rieseberg, L.; Mathew, F.M. Genome-Wide Association Mapping in Sunflower (Helianthus annuus) Reveals Common Loci and Putative Candidate Genes for Resistance to Diaporthe gulyae and D. helianthi Causing Phomopsis Stem Canker. Plant Dis. 2023, 107, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Todesco, M.; Owens, G.L.; Bercovich, N.; Légaré, J.S.; Soudi, S.; Burge, D.O.; Huang, K.; Ostevik, K.L.; Drummond, E.B.M.; Imerovski, I.; et al. Massive Haplotypes Underlie Ecotypic Differentiation in Sunflowers. Nature 2020, 584, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Lee, J.S.; Jahani, M.; Huang, K.; Mandel, J.R.; Marek, L.F.; Burke, J.M.; Langlade, N.B.; Owens, G.L.; Rieseberg, L.H. Expression Complementation of Gene Presence/Absence Polymorphisms in Hybrids Contributes Importantly to Heterosis in Sunflower. J. Adv. Res. 2022, 42, 83–98. [Google Scholar] [CrossRef]
- Peng, S.; Xue-Lian, L.; Gao, F.; Guo-Ying, L.I.; Hui, L.I. Study on a New Rapid Inoculation Method for Verticillium Wilt and Fusarium Wilt of Cotton. Cotton Sci. 2008, 20, 174–178. [Google Scholar] [CrossRef]
- Jie, Y.; Ying, W.; Zhao, J.; Lan, J.; Zhou, H. Study on Inoculation Methods of Sunflower Verticillium Wilt. China Plant Protect. 2015, 12, 16–20. (In Chinese) [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance Component Model to Account for Sample Structure in Genome-Wide Association Studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.S. How Does Multiple Testing Correction Work? Nat. Biotechnol. 2009, 27, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Endelman, J.B.; Jannink, J.L. Shrinkage Estimation of the Realized Relationship Matrix. G3 2012, 2, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Huang, K.; Jahani, M.; Gouzy, J.; Legendre, A.; Carrere, S.; Lázaro-Guevara, J.M.; González Segovia, E.G.; Todesco, M.; Mayjonade, B.; Rodde, N.; et al. The Genomics of Linkage Drag in Inbred Lines of Sunflower. Proc. Natl. Acad. Sci. USA 2023, 120, e2205783119. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein Domains Identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology; R Package Version 2.56.0; Bioconductor: Dortmund, Germany, 2024. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 1 June 2024).
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Millard, S.P. EnvStats: An R Package for Environmental Statistics; Springer: New York, NY, USA, 2013; Available online: https://link.springer.com/book/10.1007/978-1-4614-8456-1 (accessed on 13 June 2024).
- Qin, T.; Zhao, P.; Sun, J.; Zhao, Y.; Zhang, Y.; Yang, Q.; Wang, W.; Chen, Z.; Mai, T.; Zou, Y.; et al. Research Progress of PPR Proteins in RNA Editing, Stress Response, Plant Growth and Development. Front. Genet. 2021, 12, 765580. [Google Scholar] [CrossRef] [PubMed]
- Laluk, K.; AbuQamar, S.; Mengiste, T. The Arabidopsis Mitochondria-Localized Pentatricopeptide Repeat Protein PGN Functions in Defense Against Necrotrophic Fungi and Abiotic Stress Tolerance. Plant Physiol. 2011, 156, 2053–2068. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Rai, A.K.; Kanwar, S.S.; Sharma, T.R. Comparative Analysis of Zinc Finger Proteins Involved in Plant Disease Resistance. PLoS ONE 2012, 7, e42578. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Sato, K.; Takeuchi, M. Molecular Mechanisms of Sar/Arf GTPases in Vesicular Trafficking in Yeast and Plants. Front. Plant Sci. 2014, 5, 411. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.M.; Kuhn, H.; Micali, C.; Liller, C.; Kwaaitaal, M.; Panstruga, R. Interaction of a Blumeria graminis f. sp. hordei Effector Candidate with a Barley ARF-GAP Suggests That Host Vesicle Trafficking Is a Fungal Pathogenicity Target. Mol. Plant Pathol. 2014, 15, 535–549. [Google Scholar] [CrossRef]
- Gu, Y.; Zavaliev, R.; Dong, X. Membrane Trafficking in Plant Immunity. Mol. Plant 2017, 10, 1026–1034. [Google Scholar] [CrossRef]
- Gillingham, A.K.; Pfeifer, A.C.; Munro, S. CASP, the Alternatively Spliced Product of the Gene Encoding the CCAAT-Displacement Protein Transcription Factor, Is a Golgi Membrane Protein Related to Giantin. Mol. Biol. Cell 2002, 13, 3761–3774. [Google Scholar] [CrossRef]
- Chaliha, C.; Rugen, M.D.; Field, R.A.; Kalita, E. Glycans as Modulators of Plant Defense Against Filamentous Pathogens. Front. Plant Sci. 2018, 9, 928. [Google Scholar] [CrossRef]
- Kaya, Y. Sunflower. In Breeding Oilseed Crops for Sustainable Production; Academic Press: Cambridge, MA, USA, 2016; pp. 55–88. [Google Scholar] [CrossRef]
- Neupane, S.; Andersen, E.J.; Neupane, A.; Nepal, M.P. Genome-Wide Identification of NBS-Encoding Resistance Genes in Sunflower (Helianthus annuus L.). Genes 2018, 9, 384. [Google Scholar] [CrossRef]
- DeYoung, B.J.; Innes, R.W. Plant NBS-LRR Proteins in Pathogen Sensing and Host Defense. Nat. Immunol. 2006, 7, 1243–1249. [Google Scholar] [CrossRef]
- Li, B.; Lu, D.; Shan, L. Ubiquitination of Pattern Recognition Receptors. Mol. Plant Pathol. 2014, 15, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant Receptor-Like Serine Threonine Kinases: Roles in Signaling and Plant Defense. Mol. Plant-Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Li, Y.; Wu, L.; Zhou, H.; Zhang, G.; Ma, Z. Ectopic Expression of a Novel Ser/Thr Protein Kinase from Cotton (Gossypium barbadense) Enhances Resistance to Verticillium dahliae Infection and Oxidative Stress in Arabidopsis. Plant Cell Rep. 2013, 32, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Chezem, W.R.; Clay, N.K. Ternary WD40 Repeat-Containing Protein Complexes: Evolution, Composition, and Roles in Plant Immunity. Front. Plant Sci. 2016, 6, 1108. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; He, C.; Zhang, D.; Liu, X.; Xu, Z.; Tian, Y.; Liu, X.H.; Zang, S.; Pauly, M.; Zhou, Y.; et al. Two Trichome Birefringence-Like Proteins Mediate Xylan Acetylation, Which Is Essential for Leaf Blight Resistance in Rice. Plant Physiol. 2017, 173, 470–481. [Google Scholar] [CrossRef]
- Lv, T.; Li, X.; Fan, T.; Luo, H.; Xie, C.; Zhou, Y.; Tian, C.E. The Calmodulin-Binding Protein IQM1 Interacts with CATALASE2 to Affect Pathogen Defense. Plant Physiol. 2019, 181, 1314–1327. [Google Scholar] [CrossRef]
- Ribone, A.I.; Fass, M.; Gonzalez, S.; Lia, V.; Paniego, N.; Rivarola, M. Co-Expression Networks in Sunflower: Harnessing the Power of Multi-Study Transcriptomic Public Data to Identify and Categorize Candidate Genes for Fungal Resistance. Plants 2023, 12, 2767. [Google Scholar] [CrossRef]
- Yuen, E.L.H.; Shepherd, S.; Bozkurt, T.O. Traffic Control: Subversion of Plant Membrane Trafficking by Pathogens. Annu. Rev. Phytopathol. 2023, 61, 325–350. [Google Scholar] [CrossRef]
- Yun, H.S.; Sul, W.J.; Chung, H.S.; Lee, J.-H.; Kwon, C. Secretory Membrane Traffic in Plant–Microbe Interactions. New Phytol. 2023, 237, 53–59. [Google Scholar] [CrossRef]
- Bhandari, D.D.; Brandizzi, F. Logistics of Defense: The Contribution of Endomembranes to Plant Innate Immunity. J. Cell Biol. 2024, 223, e202307066. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Resistance Level | Corrected Disease Index (DIcorr) | Number of SAM Accessions |
|---|---|---|
| Immunity (I) | DI = 0 | 18 |
| High Resistance (HR) | 0 < DI ≤ 10 | 102 |
| Medium Resistance (MR) | 10 < DI ≤ 25 | 103 |
| Medium Susceptibility (MS) | 25 < DI ≤ 40 | 8 |
| High Susceptibility (HS) | DI > 40 | 0 |
| Resistance Classification | Resistance Gene Function | Annotated Ha412 Gene | Quantitative Trait Loci (QTLs) | ||||
|---|---|---|---|---|---|---|---|
| Name | Chromosome | Start | End | Range | Length (kbp) | ||
| Receptor-like kinases | Leucine-rich repeat (LRR) receptor-like protein kinases domain | Ha412HOChr09g0430181 | 9 | 199,673,064 | 199,676,740 | Chr9:199587300..199671800 | 84.501 |
| Ha412HOChr11g0485191 | 11 | 12,489,907 | 12,491,965 | Chr11:12496400..12496400 | 0.001 | ||
| THH1/TOM1/TOM3_domain | Ha412HOChr05g0201031 | 5 | 2,135,315 | 2,140,825 | Chr05:2142873..2142873 | 0.001 | |
| Enzymes | Plant glutathione S-transferases (GSTs) | Ha412HOChr04g0161611 | 4 | 54,865,558 | 54,867,010 | Chr04:54869161..54869161 | 0.001 |
| Pectinesterase | Ha412HOChr09g0430121 | 9 | 199,621,706 | 199,624,213 | Chr9:199587300..199671800 | 84.501 | |
| Ubiquitin conjugating enzyme E2 | Ha412HOChr09g0430131 | 9 | 199,654,317 | 199,657,697 | Chr9:199587300..199671800 | 84.501 | |
| GDP-fucose protein O-fucosyl transferase | Ha412HOChr09g0430141 | 9 | 199,661,153 | 199,666,513 | Chr9:199587300..199671800 | 84.501 | |
| Transcription regulator | Pentatricopeptide Repeat (PPR) | Ha412HOChr05g0201051 | 5 | 2,144,526 | 2,145,764 | Chr05:2142873..2142873 | 0.001 |
| Ha412HOChr13g0624831 | 13 | 159,187,929 | 159,189,605 | Chr13:159192700..159192700 | 0.001 | ||
| Signalling | WD40 repeat domain | Ha412HOChr04g0149911 | 4 | 12,506,690 | 12,515,912 | Chr4:12515100..12515100 | 0.001 |
| Ankyrin repeat domain | Ha412HOChr04g0193401 | 4 | 209,950,163 | 209,950,678 | Chr4:209869900..209957500 | 87.601 | |
| Signal response regulator receiver domain | Ha412HOChr05g0212311 | 5 | 47,237,986 | 47,241,942 | Chr5:47234800..47234800 | 0.001 | |
| IQ Calmodulin binding motif (IQM) | Ha412HOChr07g0294431 | 7 | 34,499,627 | 34,502,094 | Chr07:34502762..34502762 | 0.001 | |
| Trichome Birefringence-Like (TBL) proteins | Ha412HOChr08g0326731 | 8 | 385,886 | 387,955 | Chr08:391677..391677 | 0.001 | |
| Protein kinase domain | Ha412HOChr11g0531821 | 11 | 195,939,617 | 195,939,979 | Chr11:195944400..195944400 | 0.001 | |
| Ha412HOChr11g0531831 | 11 | 195,940,158 | 195,946,483 | Chr11:195944400..195944400 | 0.001 | ||
| Serine/threonine-protein kinase (STK) | Ha412HOChr11g0531841 | 11 | 195,949,268 | 195,951,584 | Chr11:195944400..195944400 | 0.001 | |
| Defense regulation | Small GTPase superfamily ARF/SAR related protein | Ha412HOChr08g0327051 | 8 | 812,081 | 815,534 | Chr08:815128..815128 | 0.001 |
| Ha412HOChr16g0793171 | 16 | 190,225,952 | 190,245,150 | Chr16:190241501..190241501 | 0.001 | ||
| CASP C-terminal domain | Ha412HOChr17g0837651 | 17 | 106,817,961 | 106,823,156 | Chr17:106827000..106827100 | 0.101 | |
| Zinc finger domain | Ha412HOChr01g0016781 | 1 | 73,395,376 | 73,396,787 | Chr01:73392344..73392344 | 0.001 | |
| Ha412HOChr11g0485201 | 11 | 12,494,087 | 12,496,285 | Chr11:12496400..12496400 | 0.001 | ||
| Ha412HOChr16g0802371 | 16 | 212,155,400 | 212,161,057 | Chr16:212158239..212187958 | 29.720 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Yang, J.; Zhang, J.; Rieseberg, L.H.; Zhao, J. Genomic Insights into Disease Resistance in Sunflower (Helianthus annuus): Identifying Key Regions and Candidate Genes for Verticillium dahliae Resistance. Plants 2024, 13, 2582. https://doi.org/10.3390/plants13182582
Yu Y, Yang J, Zhang J, Rieseberg LH, Zhao J. Genomic Insights into Disease Resistance in Sunflower (Helianthus annuus): Identifying Key Regions and Candidate Genes for Verticillium dahliae Resistance. Plants. 2024; 13(18):2582. https://doi.org/10.3390/plants13182582
Chicago/Turabian StyleYu, Yue, Jianfeng Yang, Jian Zhang, Loren H. Rieseberg, and Jun Zhao. 2024. "Genomic Insights into Disease Resistance in Sunflower (Helianthus annuus): Identifying Key Regions and Candidate Genes for Verticillium dahliae Resistance" Plants 13, no. 18: 2582. https://doi.org/10.3390/plants13182582
APA StyleYu, Y., Yang, J., Zhang, J., Rieseberg, L. H., & Zhao, J. (2024). Genomic Insights into Disease Resistance in Sunflower (Helianthus annuus): Identifying Key Regions and Candidate Genes for Verticillium dahliae Resistance. Plants, 13(18), 2582. https://doi.org/10.3390/plants13182582