
Integrated Analysis of microRNAs and Transcription Factor Targets in Floral Transition of Pleioblastus pygmaeus

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Preparation and sRNA Library Generation
2.3. Data Control and miRNA Annotation
2.4. DEM Identification
2.5. RT-qPCR Validation
2.6. TG Prediction and DEM-TG Interaction Validation
2.7. GO and KEGG Enrichment Analysis
3. Results
3.1. Identification and Classification of sRNAs in P. pygmaeus
3.2. Identification and Classification of miRNAs in P. pygmaeus
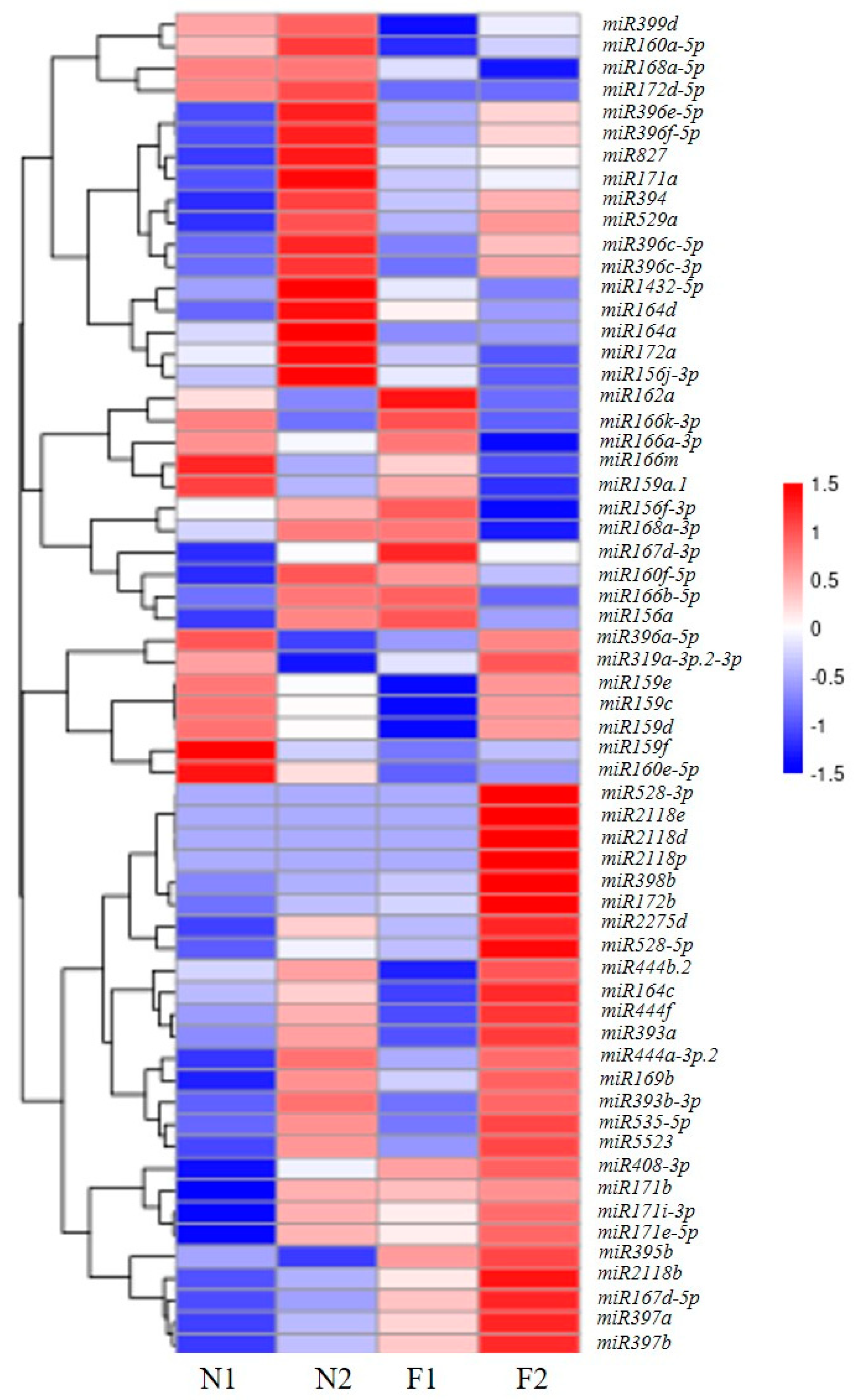
3.3. Identification of Differentially Expressed miRNAs in P. pygmaeus
3.4. Go and KEGG Analysis of Candidate TGs of DEMs in P. pygmaeus
3.5. Identification of DEMs and TGs in Flowering and Flower Organ Development in P. pygmaeus
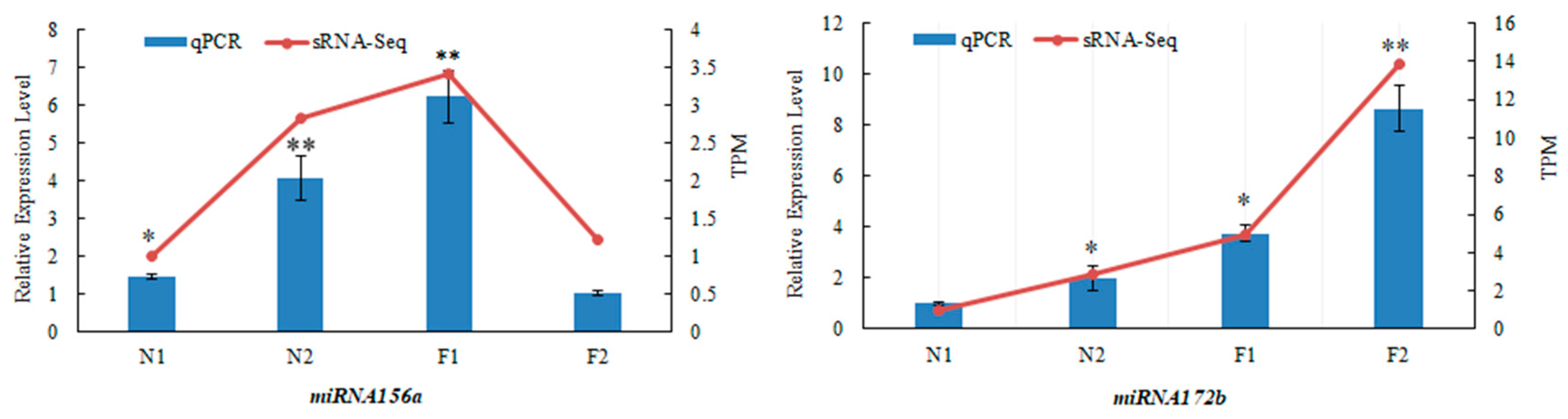
3.6. Validation of Two Known miRNAs in Age Pathway by qRT-PCR
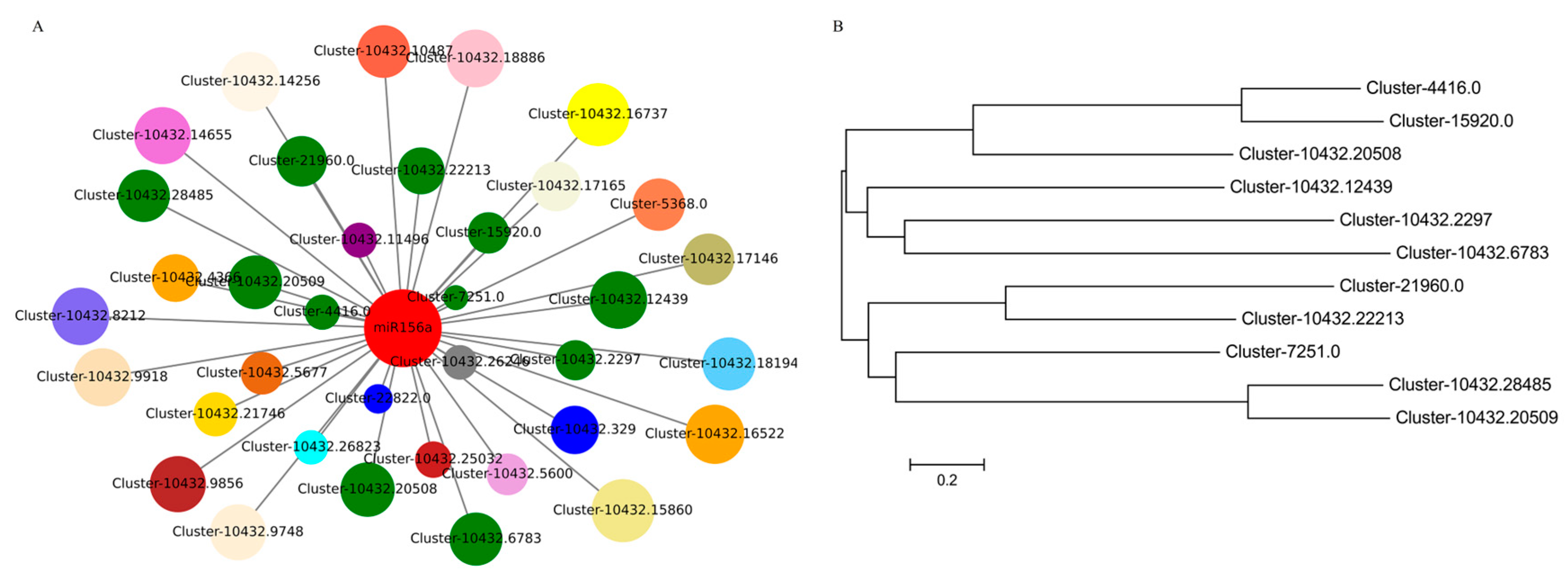
3.7. Identification of miR156a-PpSPLs Modules in P. pygmaeus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.; Kim, S.; Choi, S.J.; Joung, E.; Kwon, M.; Park, H.J.; Shim, J.S. Regulation of flowering time by environmental factors in plants. Plants 2023, 12, 3680. [Google Scholar] [CrossRef] [PubMed]
- Poethig, R.S.; Fouracre, J. Temporal regulation of vegetative phase change in plants. Dev. Cell 2024, 59, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Maple, R.; Zhu, P.; Hepworth, J.; Wang, J.-W.; Dean, C. Flowering time: From physiology, through genetics to mechanism. Plant Physiol. 2024, 195, 190–212. [Google Scholar] [CrossRef] [PubMed]
- E Colleoni, P.; van Es, S.W.; Winkelmolen, T.; Immink, R.G.H.; van Esse, G.W. Flowering time genes branching out. J. Exp. Bot. 2024, 75, 4195–4209. [Google Scholar] [CrossRef]
- Willmann, M.R.; Poethig, R.S. Conservation and evolution of miRNA regulatory programs in plant development. Curr. Opin. Plant Biol. 2007, 10, 503–511. [Google Scholar] [CrossRef]
- Dong, Q.; Hu, B.; Zhang, C. microRNAs and their roles in plant development. Front. Plant Sci. 2022, 13, 824240. [Google Scholar] [CrossRef]
- Spanudakis, E.; Jackson, S. The role of microRNAs in the control of flowering time. J. Exp. Bot. 2014, 65, 365–380. [Google Scholar] [CrossRef]
- Hong, Y.; Jackson, S. Floral induction and flower formation: The role and potential applications of miRNAs. Plant Biotechnol. J. 2015, 13, 282–292. [Google Scholar] [CrossRef]
- Waheed, S.; Zeng, L. The critical role of miRNAs in regulation of flowering time and flower development. Genes 2020, 11, 319. [Google Scholar] [CrossRef]
- Samad, A.F.A.; Sajad, M.; Nazaruddin, N.; Fauzi, I.A.; Murad, A.M.A.; Zainal, Z.; Ismail, I. MicroRNA and transcription factor: Key players in plant regulatory network. Front. Plant Sci. 2017, 8, 565. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S. Pioneer transcription factors initiating gene network changes. Annu. Rev. Genet. 2020, 54, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W. Regulation of flowering time by the miR156-mediated age pathway. J. Exp. Bot. 2014, 65, 4723–4730. [Google Scholar] [CrossRef]
- Wang, J.-W.; Czech, B.; Weigel, D. miR156-Regulated SPL Transcription Factors Define an Endogenous Flowering Pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Zhou, B.; Luo, Q.; Shen, Y.; Wei, L.; Song, X.; Liao, H.; Ni, L.; Shen, T.; Du, X.; Han, J.; et al. Coordinated regulation of vegetative phase change by brassinosteroids and the age pathway in Arabidopsis. Nat. Commun. 2023, 14, 2608. [Google Scholar] [CrossRef]
- Li, X.; Bian, H.; Song, D.; Ma, S.; Han, N.; Wang, J.; Zhu, M. Flowering time control in ornamental gloxinia (Sinningia speciosa) by manipulation of miR159 expression. Ann. Bot. 2013, 111, 791–799. [Google Scholar] [CrossRef]
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.-B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef]
- Fahlgren, N.; Montgomery, T.A.; Howell, M.D.; Allen, E.; Dvorak, S.K.; Alexander, A.L.; Carrington, J.C. Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNAA ects Developmental Timing and Patterning in Arabidopsis. Curr. Biol. 2006, 16, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Liese, W.; Köhl, M. Bamboo: The Plant and Its Uses; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Zheng, X.; Lin, S.; Fu, H.; Wan, Y.; Ding, Y. The bamboo flowering cycle sheds light on flowering diversity. Front. Plant Sci. 2020, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, C.; Lin, S.; Wang, J.; Fan, T.; Zhao, W. The structures of floral organs and reproductive characteristics of an ornamental bamboo species, Pleioblastus pygmaeus. Hortic. Plant J. 2023, 9, 589–601. [Google Scholar] [CrossRef]
- Wu, C.; Cheng, Z.; Gao, J. Mysterious Bamboo flowering phenomenon: A literature review and new perspectives. Sci. Total Environ. 2024, 911, 168695. [Google Scholar] [CrossRef]
- Yao, W.; Li, C.; Lin, S.; Ren, L.; Wan, Y.; Zhang, L.; Ding, Y. morphological characteristics and transcriptome comparisons of the shoot buds from flowering and non-flowering Pleioblastus pygmaeus. Forests 2020, 11, 1229. [Google Scholar] [CrossRef]
- Yao, W.; Li, C.; Fu, H.; Yang, M.; Wu, H.; Ding, Y.; Li, L.; Lin, S. Genome-wide analysis of SQUAMOSA-Promoter-Binding Protein-like family in flowering Pleioblastus pygmaeus. Int. J. Mol. Sci. 2022, 23, 14035. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An Integrative microRNA Evolutionary Analysis Platform for Next-Generation Sequencing Experiments. BMC Bioinform. 2010, 13, 140. [Google Scholar] [CrossRef]
- Ye, T.; Huang, X.; Ma, T.; Li, Y.; Wang, X.; Lu, H.; Xue, H. Integrated Analysis of miRNAome and Transcriptome Identify Regulators of Elm Seed Aging. Plants 2023, 12, 1719. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ge, W.; Zhang, Y.; Cheng, Z.; Li, L.; Hou, D.; Hou, C. Identification and characterization of microRNAs at different flowering developmental stages in moso bamboo (Phyllostachys edulis) by high-throughput sequencing. Mol. Genet. Genom. 2015, 290, 2335–2353. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Ma, Y.-K.; Chen, T.; Wang, M.; Wang, X.-J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. goseq: Gene Ontology testing for RNA-seq datasets. R Bioconduct. 2012, 8, 1–25. [Google Scholar]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Prasun, B.; Sukanya, C.; Smritikana, D.; Amita, P.; Malay, D. Bamboo flowering from the perspective of comparative genomics and transcriptomics. Front. Plant Sci. 2016, 7, 1900. [Google Scholar]
- Guo, Z.-H.; Ma, P.-F.; Yang, G.-Q.; Hu, J.-Y.; Liu, Y.-L.; Xia, E.-H.; Zhong, M.-C.; Zhao, L.; Sun, G.-L.; Xu, Y.-X.; et al. Genome sequences provide insights into the reticulate origin and unique traits of woody bamboos. Mol. Plant 2019, 12, 1353–1365. [Google Scholar] [CrossRef]
- Ge, W.; Zhang, Y.; Cheng, Z.; Hou, D.; Li, X.; Gao, J. Main regulatory pathways, key genes, and microRNAs involved in flower formation and development of moso bamboo (Phyllostachys edulis). Plant Biotechnol. J. 2017, 15, 82–96. [Google Scholar] [CrossRef]
- Cesarino, I. Unraveling the regulatory network of bamboo lignification. Plant Physiol. 2021, 187, 673–675. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.; Zhang, S.; Lou, Y.; An, X.; Jiang, Z.; Gao, Z. Transcriptome and miRNAome analysis reveals components regulating tissue differentiation of bamboo shoots. Plant Physiol. 2022, 188, 2182–2198. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Lou, Y.; Yang, K.; Liu, Y.; Xiao, X.; Li, Z.; Guo, D.; Sun, H.; Gao, Z. Integrative analyses of morphology, physiology, and transcriptional expression profiling reveal miRNAs involved in culm color in bamboo. Front. Plant Sci. 2022, 13, 992794. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Y.; Wang, X.-Y.; Zhao, L.; Zhang, X.-M.; Chen, S.-Y.; Ma, P.-F.; Hu, X.-Y.; Li, D.-Z.; Guo, Z.-H. Investigating the MicroRNAomes of Two Developmental Phases of Dendrocalamus latiflorus (Poaceae: Bambusoideae) Inflorescences. Plant Mol. Biol. Rep. 2015, 33, 1141–1155. [Google Scholar] [CrossRef]
- Cheng, Z.; Hou, D.; Ge, W.; Li, X.; Xie, L.; Zheng, H.; Cai, M.; Liu, J.; Gao, J. Integrated mRNA, microRNA transcriptome and degradome analyses provide insights into stamen development in moso bamboo. Plant Cell Physiol. 2020, 61, 76–87. [Google Scholar] [CrossRef]
- Ahsan, M.U.; Hayward, A.; Irihimovitch, V.; Fletcher, S.; Tanurdzic, M.; Pocock, A.; Beveridge, C.A.; Mitter, N. Juvenility and vegetative phase transition in tropical/subtropical tree crops. Front. Plant Sci. 2019, 10, 729. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Chern, M.; Zhu, Y.; Zhang, L.-L.; Lu, J.-H.; Li, X.-P.; Dang, W.-Q.; Ma, X.-C.; Yang, Z.-R.; et al. Suppression of rice miR168 improves yield, flowering time and immunity. Nat. Plants 2021, 7, 129–136. [Google Scholar] [CrossRef]
- Curaba, J.; Talbot, M.; Li, Z.; Helliwell, C. Over-expression of microRNA171 affects phase transitions and floral meristem determinancy in barley. BMC Plant Biol. 2013, 13, 6. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Bao, M.-L.; Sun, Y.-Z.; Yang, Y.-J.; Xu, X.-H.; Wang, J.-H.; Han, N.; Bian, H.-W.; Zhu, M.-Y. Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol. Biol. 2011, 77, 619–629. [Google Scholar] [CrossRef]
- Bernardi, Y.; Ponso, M.A.; Belén, F.; Vegetti, A.C.; Dotto, M.C. MicroRNA miR394 regulates flowering time in Arabidopsis thaliana. Plant Cell Rep. 2022, 41, 1375–1388. [Google Scholar] [CrossRef]
- Feng, Y.-Z.; Zhou, Y.-F.; Yang, Y.-W.; Lei, M.-Q.; Lian, J.-P.; He, H.; Zhang, Y.-C.; Huang, W.; Chen, Y.-Q. A natural variant of miR397 mediates a feedback loop in circadian rhythm. Plant Physiol. 2020, 182, 204–214. [Google Scholar] [CrossRef]
- Kim, W.; Ahn, H.J.; Chiou, T.-J.; Ahn, J.H. The role of the miR399-PHO2 module in the regulation of flowering time in response to different ambient temperatures in Arabidopsis thaliana. Mol. Cells 2011, 32, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Li, P.; Mei, H.; Wang, D.; Sun, J.; Yang, C.; Hao, L.; Cao, S.; Chu, C.; Hu, S.; et al. Fine-tuning of miR528 accumulation Modulates flowering time in rice. Mol. Plant 2019, 12, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Laufs, P. MicroRNA regulation of the CUC genes is required for boundary size control in Arabidopsis meristems. Development 2004, 131, 4311–4322. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nagpal, P.; Villarino, G.; Trinidad, B.; Bird, L.; Huang, Y.; Reed, J.W. miR167 limits anther growth to potentiate anther dehiscence. Development 2019, 146, dev.174375. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Le, N.T.; Koizumi, K.; Villar-Briones, A.; Nonomura, K.-I.; Endo, M.; Inoue, H.; Saze, H.; Komiya, R. miR2118-dependent U-rich phasiRNA production in rice anther wall development. Nat. Commun. 2020, 11, 3115. [Google Scholar] [CrossRef]
- Liebsch, D.; Palatnik, J.F. MicroRNA miR396, GRF transcription factors and GIF co-regulators: A conserved plant growth regulatory module with potential for breeding and biotechnology. Curr. Opin. Plant Biol. 2020, 53, 31–42. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, W.; Shen, P.; Yang, M.; Meng, Q.; Zhou, R.; Li, L.; Lin, S. Integrated Analysis of microRNAs and Transcription Factor Targets in Floral Transition of Pleioblastus pygmaeus. Plants 2024, 13, 3033. https://doi.org/10.3390/plants13213033
Yao W, Shen P, Yang M, Meng Q, Zhou R, Li L, Lin S. Integrated Analysis of microRNAs and Transcription Factor Targets in Floral Transition of Pleioblastus pygmaeus. Plants. 2024; 13(21):3033. https://doi.org/10.3390/plants13213033
Chicago/Turabian StyleYao, Wenjing, Peng Shen, Meng Yang, Qianyu Meng, Rui Zhou, Long Li, and Shuyan Lin. 2024. "Integrated Analysis of microRNAs and Transcription Factor Targets in Floral Transition of Pleioblastus pygmaeus" Plants 13, no. 21: 3033. https://doi.org/10.3390/plants13213033
APA StyleYao, W., Shen, P., Yang, M., Meng, Q., Zhou, R., Li, L., & Lin, S. (2024). Integrated Analysis of microRNAs and Transcription Factor Targets in Floral Transition of Pleioblastus pygmaeus. Plants, 13(21), 3033. https://doi.org/10.3390/plants13213033