
Genetic Diversity and Population Structure Analysis in Guar

Abstract
:1. Introduction
2. Results
2.1. Single Nucleotide Polymorphism Profiling
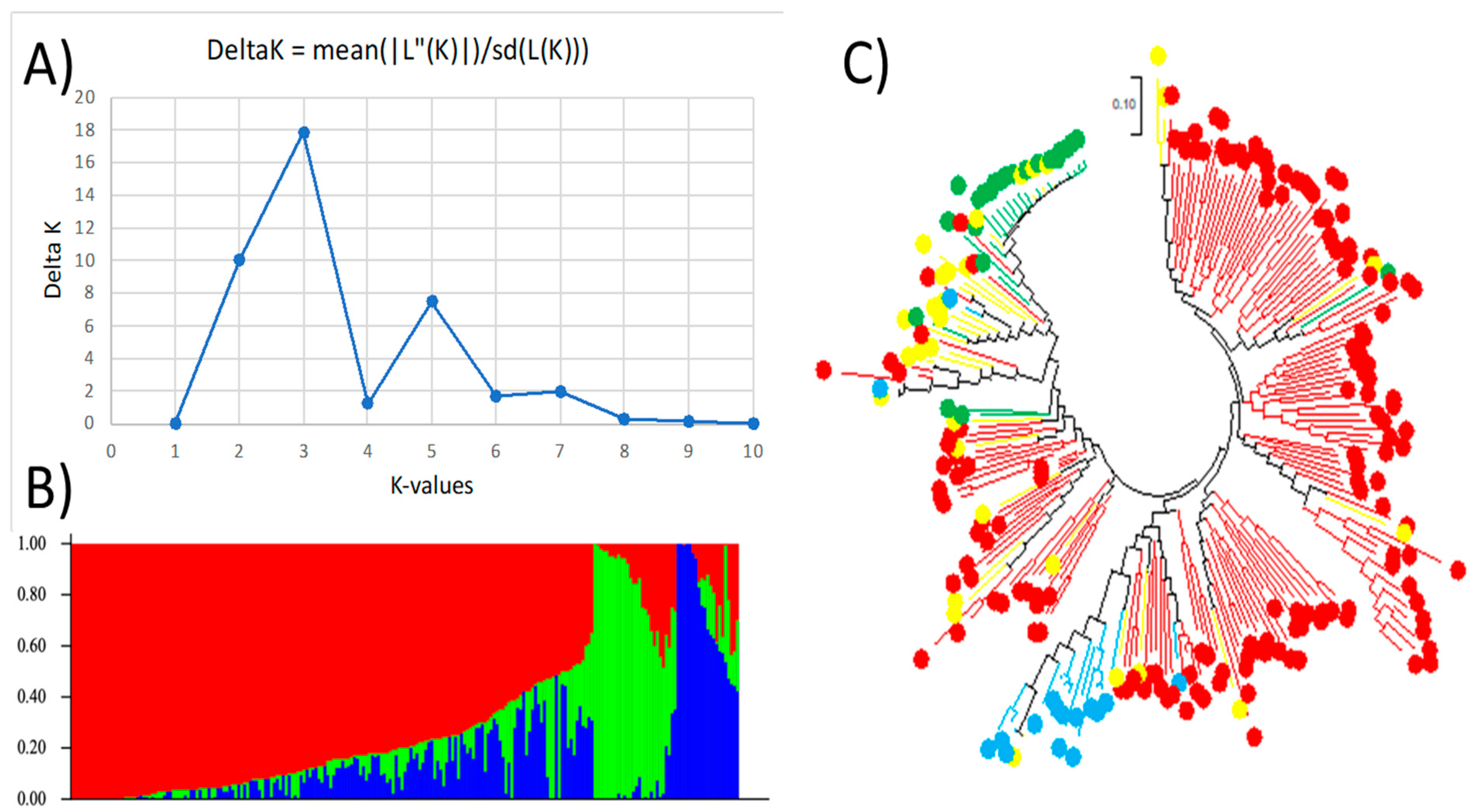
2.2. Population Structure and Genetic Diversity
2.3. Population Structure by Country of Genotype Origin
3. Discussion
4. Materials and Method
4.1. Plant Materials
4.2. DNA Extraction, Library Preparation, and Genotyping by Sequencing (GBS)
4.3. SNP Assembly, Mapping, Discovery, and Filtering
4.4. Population Structure Analysis
4.5. Population Diversity Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ravelombola, W.; Manley, A.; Adams, C.; Trostle, C.; Ale, S.; Shi, A.; Cason, J. Genetic and genomic resources in guar: A Review. Euphytica 2021, 217, 1–19. [Google Scholar] [CrossRef]
- Gresta, F.; Avola, G.; Cannavò, S.; Santonoceto, C. Morphological, biological, productive and qualitative characterization of 68 guar (Cyamopsis tetragonoloba (L.) Taub.) Genotypes. Ind. Crops Prod. 2018, 114, 98–107. [Google Scholar] [CrossRef]
- Gresta, F.; Sortino, O.; Santonoceto, C.; Issi, L.; Formantici, C.; Galante, Y.M. Effects of sowing times on seed yield, protein and galactomannans content of four varieties of guar (Cyamopsis tetragonoloba L.) in a mediterranean environment. Ind. Crops Prod. 2013, 41, 46–52. [Google Scholar] [CrossRef]
- Thapa, S.; Adams, C.B.; Trostle, C. Root nodulation in guar: Effects of soils, Rhizobium inoculants, and guar varieties in a controlled environment. Ind. Crops Prod. 2018, 120, 198–202. [Google Scholar] [CrossRef]
- Morris, J.B. Morphological and reproductive characterization of Guar (Cyamopsis tetragonoloba) genetic resources regenerated in Georgia, USA. Genet. Resour. Crop Evol. 2010, 57, 985–993. [Google Scholar] [CrossRef]
- MacMillan, J.; Adams, C.B.; Trostle, C.; Rajan, N. Testing the efficacy of existing USDA Rhizobium Germplasm Collection Accessions as inoculants for guar. Ind. Crops Prod. 2021, 161, 113205. [Google Scholar] [CrossRef]
- Lal, G.; Shekhawat, N.; Yadav, S.; Choudhary, M.K.; Bhatiya, M. Guar gum valuable for pharmacological and pharmaceutical conscious: A review. J. Pharmacogn. Phytochem. 2021, 10, 7–13. [Google Scholar]
- Hinson, P.O.; Adams, C.B. Quantifying tradeoffs in nodulation and plant productivity with nitrogen in guar. Ind. Crops Prod. 2020, 153, 112617. [Google Scholar] [CrossRef]
- Stafford, R.E.; Kirby, J.S.; Kinman, M.L.; Lewis, C.R. Registration of Kinman and Esser Guar 1 (Reg. No 4. to 5). Crop Sci. 1976, 16, 310. [Google Scholar] [CrossRef]
- Stafford, R.E.; Ray, D.T. Registration of Lewis Guar. Crop Sci. 1985, 25, 365. [Google Scholar] [CrossRef]
- Swarup, S.; Cargill, E.J.; Crosby, K.; Flagel, L.; Kniskern, J.; Glenn, K.C. Genetic diversity is indispensable for plant breeding to improve crops. Crop Sci. 2021, 61, 839–852. [Google Scholar] [CrossRef]
- Govindaraj, M.; Vetriventhan, M.; Srinivasan, M. Importance of genetic diversity assessment in crop plants and its recent advances: An overview of its analytical perspectives. Genet. Res. Int. 2015, 2015, 431487. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.A.; Prasanna, B.M. Analysis of genetic diversity in crop plants—Salient statistical tools and considerations. Crop Sci. 2003, 43, 1235–1248. [Google Scholar] [CrossRef]
- Kumara, B.N.; Gangaprasad, S.; Sridhara, S. Genetic diversity studies in guar (Cyamopsis tetragonoloba L.) genotypes. Bioscan 2015, 7, 355–359. [Google Scholar]
- Gresta, F.; Mercati, F.; Santonoceto, C.; Abenavoli, M.R.; Ceravolo, G.; Araniti, F.; Anastasi, U.; Sunseri, F. Morpho-agronomic and AFLP characterization to explore guar (Cyamopsis tetragonoloba L.) genotypes for the Mediterranean environment. Ind. Crops Prod. 2016, 86, 23–30. [Google Scholar] [CrossRef]
- Duran, C.; Appleby, N.; Vardy, M.; Imelfort, M.; Edwards, D.; Batley, J. Single nucleotide polymorphism discovery in barley using autosnpdb. Plant Biotechnol. J. 2009, 7, 326–333. [Google Scholar] [CrossRef]
- Loridon, K.; Burgarella, C.; Chantret, N.; Martins, F.; Gouzy, J.; Prospéri, J.M.; Ronfort, J. Single-nucleotide polymorphism discovery and diversity in the model legumemedicago truncatula. Mol. Ecol. Resour. 2012, 13, 84–95. [Google Scholar] [CrossRef]
- Singh, N.; Choudhury, D.R.; Singh, A.K.; Kumar, S.; Srinivasan, K.; Tyagi, R.K.; Singh, N.K.; Singh, R. Comparison of SSR and SNP markers in estimation of genetic diversity and population structure of Indian rice varieties. PLoS ONE 2013, 8, e84136. [Google Scholar] [CrossRef]
- Wang, C.; Lan, J.; Wang, J.; He, W.; Lu, W.; Lin, Y.; Luo, J. Population structure and genetic diversity in eucalyptus pellita based on SNP markers. Front. Plant Sci. 2023, 14, 1–14. [Google Scholar] [CrossRef]
- Farahani, S.; Maleki, M.; Mehrabi, R.; Kanouni, H.; Scheben, A.; Batley, J.; Talebi, R. Whole genome diversity, population structure, and linkage disequilibrium analysis of chickpea (Cicer arietinum L.) genotypes using genome-wide Dartseq-based SNP markers. Genes 2019, 10, 676. [Google Scholar] [CrossRef]
- Song, Q.; Hyten, D.L.; Jia, G.; Quigley, C.V.; Fickus, E.W.; Nelson, R.L.; Cregan, P.B. Fingerprinting soybean germplasm and its utility in Genomic Research. G3 Genes Genomes Genet. 2015, 5, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.K.; Kale, S.M.; Kumar, V.; Parupali, S.; Joshi, S.; Singh, V.; Garg, V.; Das, R.R.; Sharma, M.; Yamini, K.N.; et al. Genotyping-by-sequencing of three mapping populations for identification of candidate genomic regions for resistance to sterility mosaic disease in Pigeonpea. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2011, 30, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.C.; Sharma, A.; Rooney, W.L.; Klein, P.E.; Mullet, J.E.; Mitchell, S.E.; Kresovich, S. Genetic improvement of sorghum as a biofuel feedstock: I. Qtl for stem sugar and grain nonstructural carbohydrates. Crop Sci. 2008, 48, 2165–2179. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1996, 351, 1291–1298. [Google Scholar] [CrossRef]
- Fu, Y.-B. Understanding crop genetic diversity under modern plant breeding. Theor. Appl. Genet. 2015, 128, 2131–2142. [Google Scholar] [CrossRef]
- Smith, S.; Bubeck, D.; Nelson, B.; Stanek, J.; Greke, J. Genetic Diversity and Modern Plant Breeding. In Sustainable Development and Biodiversity; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Thakur, O.; Randhawa, G.S. Identification and characterization of SSR, SNP and Indel molecular markers from RNA-seq data of Guar (Cyamopsis tetragonoloba, L. Taub.) roots. BMC Genom. 2018, 19, 1–14. [Google Scholar] [CrossRef]
- Grigoreva, E.; Barbitoff, Y.; Changalidi, A.; Karzhaev, D.; Volkov, V.; Shadrina, V.; Safronycheva, E.; Ben, C.; Gentzbittel, L.; Potokina, E. Development of SNP set for the marker-assisted selection of guar (Cyamopsis tetragonoloba (L.) Taub.) based on a custom reference genome assembly. Plants 2021, 10, 2063. [Google Scholar] [CrossRef]
- Kisha, T.J.; Sneller, C.H.; Diers, B.W. Relationship between genetic distance among parents and genetic variance in populations of soybean. Crop Sci. 1997, 37, 1317–1325. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Bastien, M.; Humira, S.; François, B. Genome wide association mapping of Sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach. Plant Genome 2014, 20, 1–9. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; VonHoldt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Shi, A.; Buckley, B.; Mou, B.; Motes, D.; Morris, J.B.; Ma, J.; Xiong, H.; Qin, J.; Yang, W.; Chitwood, J.; et al. Association analysis of cowpea bacterial blight resistance in USDA cowpea germplasm. Euphytica 2016, 208, 143–155. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| India | U.S. | Pakistan | |
|---|---|---|---|
| Q1 | 56.44% | 5.33% | 6.67% |
| Q2 | 10.22% | 0% | 0% |
| Q3 | 7.11% | 0% | 0% |
| Admixture | 12.89% | 0% | 0% |
| India | U.S. | Pakistan | |
|---|---|---|---|
| Q1 | 74.67% | 5.33% | 6.67% |
| Q2 | 10.22% | 0% | 0% |
| Admixture | 1.78% | 0% | 0% |
| Chromosome | Ne | uh | %P |
|---|---|---|---|
| 1 | 3.49 | 0.624 | 71.2 |
| 2 | 2.32 | 0.587 | 65.4 |
| 3 | 1.52 | 0.394 | 60.2 |
| 4 | 2.48 | 0.434 | 57.3 |
| 5 | 2.47 | 0.586 | 58.8 |
| 6 | 1.51 | 0.341 | 51.8 |
| 7 | 3.45 | 0.683 | 68.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malani, S.; Ravelombola, W.; Manley, A.; Pham, H. Genetic Diversity and Population Structure Analysis in Guar. Plants 2024, 13, 3183. https://doi.org/10.3390/plants13223183
Malani S, Ravelombola W, Manley A, Pham H. Genetic Diversity and Population Structure Analysis in Guar. Plants. 2024; 13(22):3183. https://doi.org/10.3390/plants13223183
Chicago/Turabian StyleMalani, Shubham, Waltram Ravelombola, Aurora Manley, and Hanh Pham. 2024. "Genetic Diversity and Population Structure Analysis in Guar" Plants 13, no. 22: 3183. https://doi.org/10.3390/plants13223183
APA StyleMalani, S., Ravelombola, W., Manley, A., & Pham, H. (2024). Genetic Diversity and Population Structure Analysis in Guar. Plants, 13(22), 3183. https://doi.org/10.3390/plants13223183