Abstract
Citrus is affected by many diseases, and hence, the movement of citrus propagative materials is highly regulated in the USA. Currently used regulatory pathogen detection methods include biological and laboratory-based technologies, which are time-consuming, expensive, and have many limitations. There is an urgent need to develop alternate, rapid, economical, and reliable testing methods for safe germplasm exchange. Citrus huanglongbing (HLB) has devastated citrus industries leading to an increased need for germplasm exchanges between citrus growing regions for evaluating many potentially valuable hybrids for both HLB resistance and multilocational performance. In the present study, Next-Generation Sequencing (NGS) methods were used to sequence the transcriptomes of 21 test samples, including 15 well-characterized pathogen-positive plants. A workflow was designed in the CLC Genomics Workbench software, v 21.0.5 for bioinformatics analysis of the sequence data for the detection of pathogens. NGS was rapid and found to be a valuable technique for the detection of viral and bacterial pathogens, and for the discovery of new citrus viruses, complementary to the existing array of biological and laboratory assays. Using NGS methods, we detected beet western yellows virus, a newly reported citrus virus, and a variant of the citrus yellow vein-associated virus associated with the “fatal yellows” disease.
1. Introduction
Citrus is a clonally propagated tree crop, and the economic impact of distributing plant material from infected sources can be colossal. The importance of disease-free plant material for the success of the citrus industry was realized early in California starting with the Psorosis Freedom Program in 1937, which later evolved into the Citrus Variety Improvement Program in 1958, and was renamed the Citrus Clonal Protection Program in 1977 [1]. Early research at the University of California by Wallace and others led to an understanding of how diseases like psorosis and tristeza spread through infected budwood. Growers and nurserymen from California requested the University to assume responsibility for maintaining healthy plants in primary foundation blocks to provide the citrus industry with disease-free and true-to-type budwood under the regulatory control of the California Department of Food and Agriculture (CDFA). Biological indexing was the predominant technique used in the early budwood certification programs [2].
Citrus quarantine, certification, and distribution programs are designed to deliver healthy plant materials for both researchers and growers. The effectiveness of these programs depends on the tools used for pathogen detection. The efficacy of techniques used determines the success of safe germplasm movement. Initially, the detection of psorosis and tristeza by graft inoculation on indicator plants was used for the selection of virus-free mother trees. Additional indicator plants were included for indexing as knowledge about other diseases increased. Serological and molecular methods were incorporated as essential tools as they became available. Molecular methods such as real-time PCR are valuable and amenable to high throughput assays but allow for testing of only known pathogens. A limitation of the commonly used lab-based methods is that non-target pathogens are not discovered. Because of these factors, the California budwood certification program has retained the biological indexing methods, as have many other citrus budwood certification, sanitation, and introduction programs.
In recent times, several citrus diseases have threatened the survival of the industry. Hence, there is an increased interest in screening new breeding materials in different citrus growing areas for disease resistance and horticultural performance. The development of rapid and reliable budwood testing methods will facilitate the movement of certified plant materials across state and international borders. In recent years, metagenomic approaches have been used for the detection of pathogens because of advances in Next-Generation Sequencing (NGS) technologies. NGS, also referred to as High-Throughput Sequencing (HTS) or deep sequencing, can detect multiple known pathogens as well as novel and emergent pathogens, and hence appears to be the technique of choice for budwood certification.
Both biological and laboratory-based methods of pathogen detection have several limitations. Biological indexing is labor-intensive and time-consuming, and not amenable to high throughput processing. Changes in greenhouse conditions, grafting efficiency, soil, fertilizers, light, temperature, etc., can affect the success of biological indexing [2,3]. Many viruses and/or virus populations can be non-symptomatic on standard indicator plants. Pathogen detection by serological and molecular methods is target-specific; the techniques may not detect pathogen variants that have sequence differences in the genomic regions selected for designing the detection assay. NGS methodology may be able to circumvent most of the problems encountered with both biological and laboratory-based detection methods.
NGS technology allows for the identification of most viruses in a sample without prior information about the pathogen status [4]. NGS has the potential to detect the nucleic acids of any organism present in a sample [5,6,7]. Scientists from the Animal and Plant Health Inspection Service (APHIS), the regulatory agency of USDA, collaborated with scientists from multiple countries and have developed guidelines for the use of NGS technologies to detect plant pathogens and pests [8].
Plant virus detection and discovery using NGS were described in detail [9]. NGS has been used for the detection of several new viruses [10,11,12,13]. In citrus, NGS has also been used for the discovery and characterization of various novel viruses including Citrus chlorotic dwarf-associated virus [14], citrus yellow vein clearing virus [15], citrus leprosis virus cytoplasmic type 2 [16], citrus vein enation virus [17], citrus tatter leaf virus [18], and citrus yellow vein virus [19]. NGS-based microRNA analysis was used to analyze populations of Citrus dwarfing viroid [20]. NGS data was also used to develop e-probes for multiple citrus pathogen detection [21]. Various bioinformatics pipelines have been developed for the identification of viral sequences from NGS data including VirFind [22] and Virusdetect [23]. Other pipelines available are summarized in a review by Villamor et al. [9].
In clonally propagated crops like citrus, the economic consequences of the use of infected budwood can be severe. Citrus concave gum disease has been considered a disease of unknown etiology since 1930s. No vectors are known, and the disease is believed to be transmitted mostly through infected budwood. In the absence of molecular characterization of many viruses, certification programs depended mostly on biological indexing for pathogen detection; development of symptoms for diseases like concave gum is dependent on growth conditions during indexing. Disease symptoms on inoculated indicator plants may disappear quickly leading to inconclusive results. The widespread distribution of certain citrus diseases throughout the world may be attributed to the use of infected budwood. Recently, the NGS analysis of trees with citrus concave gum has led to the discovery of two negative stranded coguviruses, citrus concave gum-associated virus (CCGaV) and citrus virus A (CiVA) [24,25].
Adaptation of NGS as a tool for the certification of citrus appears to be highly useful. NGS analysis can detect both known and novel pathogens. Proper guidelines need to be developed before NGS can be used as a certification tool. There is an effort to integrate NGS as an important tool for certification in South Africa [26,27]. In the present study, 21 plant samples, including fifteen from the pathogen-positive inventory of the USDA National Clonal Germplasm Repository for Citrus & Dates, Riverside, California, USA, were analyzed by NGS transcriptome study. RNA was used as the target for the detection of pathogens with both RNA and DNA genomes (viruses and bacteria). First, we depleted the sample of ribosomal RNAs, constructed NGS transcriptome libraries, and sequenced them on the Illumina platform. We developed a workflow in the CLC software, v 21.0.5 environment to analyze NGS transcriptome data, removed host sequences, and constructed de novo assemblies of non-host sequence reads. Three local databases were generated using: (i) all available sequences of major citrus viruses, (ii) representative genomes of all viruses, and (iii) genomes of known citrus bacterial pathogens. Local blast searches for all viruses and citrus bacterial pathogens were carried out. The results from NGS-based pathogen detections were compared to existing data developed using available biological and laboratory-based test results. We report the detection of multiple citrus viruses and bacteria from the metatranscriptomes of several samples. Raw reads from all the samples analyzed have been deposited in NCBI and are freely accessible for further analysis, as needed. Unlike other biological and laboratory detection methods, the raw data and results from NGS analysis can be made publicly available, making this an open process.
2. Materials and Methods
2.1. Plant Samples
A total of 21 plant samples were used in this study. Four plants (samples 1–4; Table 1) used for the study belonged to genera closely related to Citrus, and Poncirus trifoliata, held in quarantine at the USDA Repository. A healthy plant (C. sinensis) from the greenhouse (sample 10) and a C. sinensis plant graft inoculated with Candidatus Liberibacter asiaticus from a Biological Safety level 3 facility (sample 11) were included in the analysis. Fourteen samples were selected from the pathogen-positive inventory.

Table 1.
Details of 21 samples used for RNA-Seq analysis for identification of viral and bacterial pathogens. The description is based on information documented at the time of sample collection. Further details on most samples are available in the USDA ARS GRIN database. The inventory number is specific to the plant from which samples were collected for the study, and the accession number refers to a genotype or selection.
Sample collection and further processing were done in two sets: samples 1–12 in the first set, samples 13–21, and repeat runs of 5–7 in the second set. Three of the eight libraries (#15, 16, and 19; Table 1) were processed using combinations of samples as shown in Table 1. Further information on each sample can be found in the hyperlinks provided in Table 1. Most of the isolates in the pathogen-positive inventory were collected over the last 50–60 years and are maintained in quarantine facilities. The known disease status of all 20 plant samples used in the study is indicated in Table 1.
2.2. Sample Processing and Library Construction
Total RNA was extracted from plant tissue (Table 1) using Qiagen Plant RNeasy kits (Qiagen, Inc., Germantown, MD, USA), including on-column DNase reaction, according to the manufacturer’s protocol. Extracted RNA samples were stored at −80 °C. Libraries were prepared using the Zymo-Seq Ribofree Total RNA Library Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. Briefly, about 0.5 to 1 µg of total RNA with an A260/A280 ratio of >1.8 was used for reverse transcription. The cDNAs were processed for “Ribofree® Universal Depletion” to remove ribosomal RNAs. The cDNAs were ligated with P7 adapters, and the second strand synthesis was carried out. The double-stranded DNA from the above reaction was ligated with P5 adapters and a short PCR reaction of 12 cycles was carried out with unique dual-index primers (barcoding). The reactants between each of the above steps, and after the final step were cleaned up using “Select-a-size Magbeads”. The final elution of the library was carried out in 25 µL of DNA elution buffer and stored at −20 °C. The first set of 12 libraries was shipped for sequencing to Zymo Research, Irvine, CA, USA, and the second set of eight samples to Quick Biology Inc., Monrovia, CA, USA. The pooled libraries were sequenced to generate 100 bp paired-end (PE) sequences (#1–12) or 150 bp PE sequences (#13–20) using the HiSeq platform (Illumina, San Diego, CA, USA).
2.3. Workflow for Bioinformatics Analysis
Data processing was done using a custom workflow (Figure 1) created in CLC Genomics Workbench software (Qiagen Inc., Germantown, MD, USA). Sequence reads were trimmed to remove poor-quality sequences. The remaining high-quality sequences were mapped successively to the genomic sequences of C. clementina (GCF_000493195.1), C. sinensis (GCF_022201045.2), and chloroplast (DQ864733) and mitochondrial (NC_037463) sequences of C. sinensis. At each of the above mapping steps, only the unmapped sequences were used for the next step. The unaligned sequence reads at the end of the mapping steps were used to assemble de novo contigs.
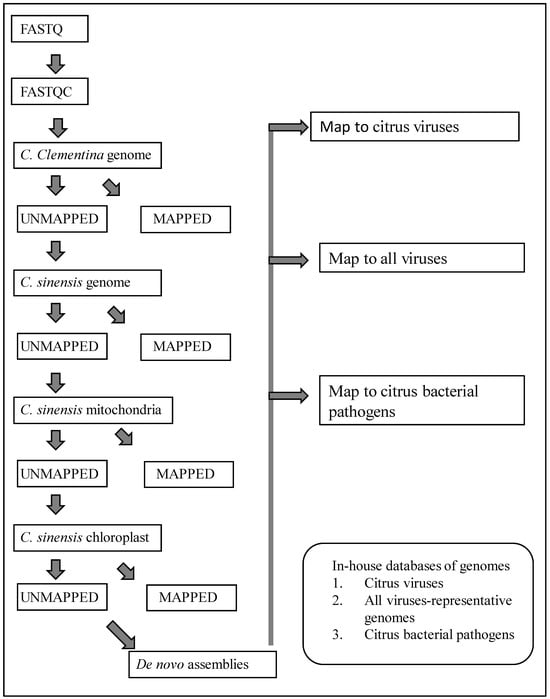
Figure 1.
Workflow in CLC Genomics Workbench software used for removal of host sequences, generating de novo contigs of unmapped sequence reads, and local blast for detection of citrus viruses and bacteria.
2.4. Pathogen Detection
Three in-house databases were created in the CLC environment as described below by importing from NCBI: (i) all available full-length genomes of citrus viruses of interest in the present study; (ii) representative genomes of all viruses from the RefSeq database; and (iii) complete genomes of all available citrus bacterial pathogens. A local blast search of de novo contigs of non-host sequence reads was conducted sequentially using all three pathogen genome databases. Where necessary, multiple contigs of some viruses were combined by re-mapping the reads to the closest viral genome as a reference. In the case of DNA pathogens (only bacterial pathogens included in this study), a blast search of multiple contigs was used to identify the pathogen.
Beet western yellows virus (BWYV) sequences from citrus were analyzed using blast search to find closely related poleroviruses. Multiple sequence alignment of nucleotide sequences was performed as described [28]. ClustalX v2.0 [29] was implemented with default parameters, and phylogenetic trees were generated using ClustalX and MEGA 11.0 [30] applying the neighbor-joining method [31]. The reliability of branches was inferred from bootstrap analysis of 1000 replicates. The final phylogenetic tree was then edited by using ITOL (http://itol.embl.de/, accessed on 12 December 2023). Recombination analysis of BWYV isolates was performed using various algorithms included in the RDP software package v4.0 [32].
Citrus yellow vein-associated virus (CYVaV) genome sequences identified in this study were compared to other genomes in the NCBI database using blast search. The 5′ and 3′ untranslated regions and two open reading frames were compared to respective regions of other viruses with similar genome organization.
The sequence of Citrus blight-associated pararetrovirus (CBaPRV) identified from the analysis of the de novo assembly from ‘Fuming Evergreen’ trifoliate (Poncirus polyandra; sample #21 in library #19) was used to do an NCBI blast search. A real-time PCR assay was developed to specifically detect the endogenous CBaPRV reportedly expressed at higher levels in roots with citrus blight, but not other citrus endogenous pararetroviruses (CEPRV) (see next section). These CEPRVs have been reported to lack the 3′ terminus of about 2000 bases of the 7 kb viral genome. Expression of CBaPRV of leaves and roots of ‘US 942′ rootstock seedlings grown from seeds received from Florida was assayed by real-time PCR. Further, over 1000 plants of various genotypes of citrus from greenhouse and field were tested by qPCR for expression of CBaPRV. Blast searches of genomes of several members of the subtribe Citreae and Clauseneae were conducted to detect CBaPRV. The negative test results from non-trifoliate accessions may also be due to sequence differences in the target region selected for real-time PCR assay (Supplementary Table S3).
2.5. Real-Time PCR Assays
Real-time PCR primers were designed for the host reference gene and four viruses of interest: Beet western yellows virus (BWYV), citrus yellow vein-associated virus (CYVaV), citrus virus A (CIVA), and citrus blight-associated pararetrovirus (CBaPRV). To specifically amplify the virus reported to be associated with citrus blight, and to avoid amplification of the other known citrus endogenous pararetroviruses, a region of about 6 kb in the genome was selected as the PCR template. For the other three viruses, primers were designed to amplify conserved regions of available genomes. As a reference, the malate dehydrogenase gene [33] was used, except that the forward primer was designed to incorporate 21 bases from the 3′ end of exon 12 and four bases from the 5′ end of exon 13 so that the amplification would take place only from the reverse transcribed RNA template, and not from genomic DNA.
Synthetic DNA (gBlocksTM) was synthesized for the above five templates by Integrated DNA Technologies, Inc., San Diego, CA, USA. Serial dilutions were made from 108 to 100 copies/µL and used for real-time PCR assays using appropriate primers and probes. Standard curves were constructed to calculate linearity and reaction efficiency.
Complementary DNA (cDNA) was synthesized using a mixture of 5 µL of RNA, 2 µL of Lunascript (New England Labs, Ipswich, MA, USA), and 3 µL of water, and incubating at 25 °C for 2 min, 55 °C for 10 min, and 95 °C for 1 min, and then cooled to room temperature. Real-time PCR mixes were prepared using 10 µL of Sso Advanced universal probes supermix (Biorad, Inc., Hercules, CA, USA), 240 nM primers and 120 nM probe for both the target virus and MDH, and water to make up the volume to 20 µL. PCR reactions were conducted in 96 well plates in a ViiA7 real-time PCR machine using the thermal profile mentioned here: 95 °C for 20 s, 40 cycles of 95 °C for 5 s, and 60 °C for 20 s. Both dilute gBlocksTM and known positive extractions served as positive controls. RNA extractions were also made from 40 seedlings of US-942. All extractions were assayed for the presence of the four viruses mentioned above by real-time PCR.
2.6. Testing Inventory of Pathogen-Positive Plants
The citrus germplasm repository has over 115 accessions of citrus pathogens, maintained in planta in quarantine with appropriate permits. While most of them were collected from within California over the last 50 to 60 years, others were received from other states or countries under appropriate permits. Real-time assays for the presence of BWYV, CIVA, CYVaV, and CIVA were conducted.
3. Results
3.1. Bioinformatics Analysis of NGS Data
Twenty RNA-Seq libraries from 21 plant samples (Table 1) constructed in the present study were checked for library quality by the TapeStation system (Agilent Technologies, Santa Clara, CA, USA). The number of reads and read lengths for each library used in the study are shown in Table 2 along with links to metadata and raw data submitted to the National Center for Biotechnology Information (NCBI) databases. With the first set of 12 samples, an average of about 30 million reads (100 bases per read) were generated per sample. In the second set of eight samples, about 120 million reads (150 bases per read) were obtained per sample (Table 1).
Table 2.
Details of host sequence removal and de novo assembly of non-host sequences obtained from CLC Genomics Workbench analysis.
The workflow used for bioinformatics analysis is shown in Figure 1. The sequence reads were trimmed to remove poor-quality sequences. Over 96% of the good-quality reads were retained in all samples. The exact number of host genome sequences removed at each step is shown in Table 2. About 81% to 98% of the sequence reads were mapped to host genomes (Table 2). The number of unmapped sequence reads varied significantly between the samples ranging from 1.17% to 18.07%. Sample 6, with 13.27% of the total sequence reads unmapped to the host genome was infested with thrips; sample 20 with 18.07% of sequence reads unmapped to the host genome was infested with mealybugs. A sample from the citrus relative Clausena spp. showed a higher percentage of sequence reads that did not map to the citrus genome probably because of sequence diversity between Citrus and Clausena (Table 2). Many unmapped sequence reads in the current study presumably originated from the associated insect species.
Local BLASTN search of de novo assemblies generated from unmapped reads was conducted against a database of citrus viruses and bacterial pathogens. The pathogens described in this study are listed in Table 3. The analysis resulted in the identification of many citrus pathogens and other citrus-associated viruses (Table 4). No citrus pathogens were identified in samples 1–4 and 10. Samples 8, 11, and 12 were infected by a single citrus pathogen while others had mixed infections with multiple pathogens. Mapping of sequence reads to the reference genomes was carried out to calculate the depth of the genome coverage of the pathogen (Table 4) which ranged from about 280,000× (for RNA3 of both CLRV and CVV in library #15) to below 10× for some viruses.
Table 3.
Names and abbreviations of citrus pathogens detected in this study according to current and earlier nomenclatures.
Table 4.
Viruses and bacteria identified through NGS in the present study. Blast search of de novo assemblies of non-host sequence reads was done against the in-house databases of viruses and bacteria as described in materials and methods.
3.2. Detection of Pathogen Sequences
The current nomenclature of viruses and bacteria is used throughout the manuscript. Previously used names mentioned in old literature, and maintained in database records are relevant. For clarity, the current nomenclature of viruses and bacteria along with alternate names used in the literature are provided in Table 3.
Table 5 compares the results of pathogen testing in the present study with previous characterization using several biological and laboratory-based assays. Beet western yellows virus (BWYV) was detected in samples 5 (libraries 5 and 17), and 16 (library 16). This is the first report of BWYV from citrus. A variant of Citrus yellow vein-associated virus was found in the plant with “fatal yellows”, a disease of unknown etiology.
Table 5.
Viruses and bacteria detected in 21 samples by NGS analysis and by other methods.
3.3. Insect Viruses and Bacteria
Transcriptome analysis also revealed a few viruses were presumably derived from insect pests prevalent in the greenhouses. A picorna-like virus (PLV; GenBank accession MW674792) was detected in sample 6; real-time PCR analysis of citrus trees from the greenhouse and field revealed its widespread presence and uneven distribution within plants and variability during different seasons. Analysis of sequence reads from sample 6 unmapped to the host revealed the DNA of a common greenhouse pest, Scirtothrips citri. Analysis of unmapped sequence reads from sample 20 revealed the presence of another greenhouse citrus pest, mealybug, Planococcus sp. Complete genomes of two endophytic bacteria of Planococcus, Candidatus Trembalya princeps (CaTP) and Candidatus Monarella endobia (CaME) were also recovered from sample 20.
3.4. Suppression of Viruses
Sample 7 (Cristacortis, IVNO 8078) was collected at two different times and two NGS libraries were constructed (#7 and #18, Table 1). Both libraries showed the presence of CBLVd, CBCVd, and CPsV. While all four viroid sequence reads were present in library #7 at about 60× to 548× depth, in library #18 sequence reads belonging to HSVd and CBCVd were either absent or very low; reads of CBLVd were present at over 4200× genome coverage (Table 4). Similarly, both CVd V and BWYV sequence reads increased significantly in library #17 compared to library #5. However, it did not appear to affect the CDVd population (Table 4).
For most of the viral sequences identified in the present study, full-length genome sequences were recovered. A total of 46 full-length genome sequences have been deposited in NCBI (Table 6).
Table 6.
Viral genome sequences deposited in the NCBI database from the present study.
3.5. Analysis of Samples Infected with “Fatal Yellows” Disease
The pathogen sequences identified from “fatal yellows disease” (library 14, Table 1), a disease of unknown etiology, contained sequences of CYVaV along with very low levels of CLRV and CVV (Table 4). The genomes of CYVaV from fatal yellows (Table 6; MZ330113) and citrus yellow vein (Table 6; accession MZ33089) differed significantly (Table 7) showing only about 86% nucleotide identity at the genome level, 91% at the amino acid level (between the two RNA dependent RNA polymerase), and 86% identity with the hypothetical protein.
Table 7.
A comparison of genomic regions of three different isolates of citrus yellow vein-associated virus (CYVaV) isolates. The sequence of YV920a (JX101610.1) was taken from a previous study [19], and sequences of YV920b (MZ330089.1) and FY940 (MZ30113.1) are from the present study.
3.6. Analysis of BWYV Sequences
The blast search of de novo contigs of non-host sequence reads from two plants (IVNO 4501 and 5046) revealed the presence of BWYV in these samples. The sequences of two BWYV genomes reported in this study are about 98.5% identical (Supplementary Table S1). BWYV isolates from citrus showed 97% nt identity with an isolate of BWYV reported from Coriandrum sativum from Cyprus (OM419176.1) [34]. Other BWYV genomes included in this analysis showed about 81–93% identity with the citrus isolates; 93% nt identity with SDWF16 isolate (MK307780.1) on Capsicum annuum from China, NJ22 isolate on Raphanus sativus from S. Korea (OQ625515.1), and S19 isolate reported on spinach from Japan (LC428356.1) [35]. Phylogenetic analysis of BWYV and a few closely related poleroviruses (Figure 2) indicated that the BWYV genomes from citrus clustered closely with other BWYV genomes. A similar analysis using BWYV genome sequences from different geographic regions (Supplementary Figure S1) shows that the citrus isolate of BYWV clusters closely with an isolate from coriander reported from Cyprus and some other isolates from Asia.
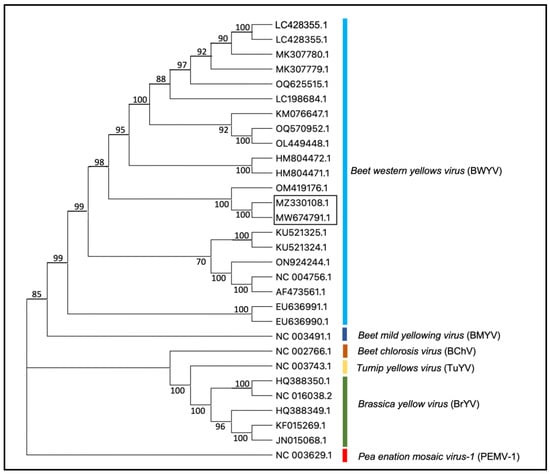
Figure 2.
Phylogenetic tree representing the relationship between BWYV isolates and other closely related viruses in the genus Polerovirus. Accessions from the citrus described in the present study are shown in the box. Pea enation mosaic virus-1 (PEMV-1, genus Enamovirus, NC_003629) is used as an outgroup. The phylogenetic tree was constructed using the neighbor-joining method and visualized with MEGA11.0. Bootstrap values shown at the nodes indicate data analyzed from 1000 replications. The names of different viruses and accession numbers of isolates used in this study are shown.
Recombination analysis using the RDP software package [32] strongly supported a recombination event at the 5′ region of the BWYV/citrus genome with an isolate of BWYV on spinach from Japan (Figure 3). These preliminary results suggest that BWYV infected citrus in Asia well before it was introduced to Western countries. However, further analysis of citrus trees from other geographic regions may help understand the origin of BWYV in citrus.

Figure 3.
Recombination analysis of Beet western yellows virus (BWYV) sequences from various host plants. Analyses were performed using various algorithms included in the RDP software package [32]. The order of the designation of the recombination events is as follows: host plant name, country of origin, NCBI accession number, and algorithm (R, RDP; G, GENECONV; C, Chimaera; M, MaxChi; B, Bootscan; S, SiScan; T, 3SEQ). Only recombination events with a p-value of <0.01 detected based on at least three different algorithms are shown.
3.7. Analysis of Citrus Blight-Associated Pararetrovirus Sequences
The genomic sequence of citrus blight-associated pararetrovirus (CBaPRV) showed 92% nucleotide identity, with genomic sequences of the CBaPRV-LC isolate reported to be associated with citrus blight. Several other endogenous pararetrovirus-like sequences also showed very high similarity, but none were of genomic length (Figure 4).

Figure 4.
A cartoon showing results of blast analysis of 7652 nucleotides long assembly (NCBI accession MZ330114.1) of sequence reads from library 19 with IVNOs 2327 and 3591. The above assembly was created by conducting a de novo assembly of sequence reads unmapped to the citrus genome (as described in Figure 1). The sequences of Citrus blight-associated pararetrovirus (CBaPRV-LC; NCBI accession MN8814438.1) showed about 92% nucleotide identity with the query sequence while five other sequences in Genbank showed about 90% identity at the nucleotide level, restricted to the 5′ 5000 bases of the viral genome. The genomic location utilized to design a real-time PCR assay is indicated (nucleotides 5852–5943).
3.8. Real-Time PCR Analysis for Detection of New Citrus Viruses
The sequences of synthetic DNA templates (gBlocksTM), primers, and probes designed for real-time PCR assay for BWYV, CYVaV, CBaPRV, CiVA, and the host reference gene, MDH, are shown in Figure 5. The primers and probe for selective amplification of MDH RNA, but not DNA was achieved by the design of the forward primers in the exon regions as shown in Supplementary Figure S2.

Figure 5.
Real-time PCR: templates, primers, and probes. Sequences of synthetic DNA templates for four viruses of interest were discovered in this study and for the reference gene, malate dehydrogenase (RNA sequence) is shown. The forward and reverse primers are highlighted yellow; the probe sequences are highlighted purple and are depicted in bold font. The direction of primers and probes is indicated by an arrow below the sequences. Further details of primer design for specific amplification of RNA, but not DNA are shown in Supplementary Figure S2.
The gBlocksTM were provided by the manufacturer at 10 ng/µL concentration. Based on the calculated molecular weight, moles/µL and copies/µL were computed. Stock solutions were prepared at 108 copies/µL, and serial dilutions from 108 to 100 copies/µL were used for generating standard curves using appropriate primers and probes. These assays conducted for all five templates (BWYV, CYVaV, CBaPRV, CIVA, and MDH) with corresponding primers and probes revealed calibration curves with a coefficient of correlation (R2) between 0.9991 to 1.000. The slope of the curve ranged from −3.38 to −3.57, and the PCR efficiencies were calculated to be 91.4% to 95.7% (Table 8). The Ct values of the three technical replicate runs for each of the five templates (dilutions from 102 to 108) were consistent (±0.35) in the replicate reactions.
Table 8.
Results of standard curve analysis of primers and probes by real-time PCR using serial dilutions of the corresponding templates constructed by synthetic gBlocksTM.
Real-time PCR analysis was conducted using plant extracts from the pathogen-positive plant inventory. Plant samples originated from the quarantine facility of the USDA Germplasm Repository; extractions and PCR were carried out to detect four viruses, BWYV, CYVaV, CIVA, and CBaPRV, along with MDH as a reference gene. BWYV was detected in six of the 103 accessions tested. Four of the accessions were collected originally from California, and one each was from Florida and Spain (Table 9). CYVaV was detected in only two accessions, both collected from California. CIVA was identified in six accessions, five originating in California and one from Florida. CBaPRV was not detected in any of the 103 accessions in the positive inventory plants.
Table 9.
Testing plants from the positive plant inventory for four viruses of interest by real-time PCR assays. A total of 115 accessions were tested. Only plants positive for BWYV, CYVaV, and CiVA are shown.
DNA and RNA extractions from both leaves and roots of 40 seedlings of rootstock variety ‘US 942’ were positive for CBaPRV. Further analysis of over 1000 plants of various genotypes collected from protected facilities and fields was carried out. Higher levels of expression of CBaPRV appeared to be restricted to trifoliate and trifoliate hybrids (Table 10).
Table 10.
Testing citrus germplasm accessions for the citrus blight-associated pararetrovirus gene expression by qPCR.
3.9. Multiple Copies of CBaPRV Integrated into Citrus Genomes
A blast search of genomes of several members of Aurantioideae revealed that several copies of the full-length genomes of CBaPRV are endogenous in all the nine chromosomes in members of the tribe Citreae analyzed in the study (Supplementary Table S2), but not in the tribe Clauseneae (Murraya paniculata and Bergera koenigii). The target sequence used for real-time PCR assay from the sequence of CBaPRV (accession MZ330114.1) was 100% identical to the sequence reported to be associated with citrus blight (MN814438.1). However, the target sequences in the five genomes of the members of Aurantioideae showed several nucleotide differences (Supplementary Figure S3).
4. Discussion
A major challenge of the currently used biological and laboratory techniques is the difficulty encountered when comparing results from different laboratories. Often this results in duplicating all or many of the biological and laboratory tests in both donor and receiving industries resulting in extended waiting time for both researchers and growers to receive the germplasm of their interest. Novel pathogens and strains are often missed by the currently used testing methodologies. Often disease symptoms may not be expressed on all indicator hosts at all times; it is vital to detect all pathogens in imported propagation materials.
4.1. NGS Is Becoming a Method of Choice for Pathogen Detection in Multiple Crop Plants
NGS is a useful technology as an additional tool for the pathogen testing process and may be able to resolve some of the ambiguities that arise with the standard methods. NGS can be a powerful tool since it can detect asymptomatic infections, novel pathogens, and variant populations [3]. NGS can be applied to solve problems in a range of areas involving viral diseases, in determining the etiology of viral diseases, in virus characterization, taxonomy, viral population genetics, and in diagnostics of plant viruses [6]. Raw data on NGS analysis of test plants can be made available to others via public databases so that the data can be re-analyzed by interested groups for detection of the pathogen sequences. This makes the procedure of budwood certification an open process.
A major goal of the present study was to compare the results of NGS-based pathogen detection with the currently used biological and laboratory-based detection methods. The long-term goal is to utilize NGS as an additional tool for pathogen testing so that the process can be quick and more efficient. The USDA Repository in Riverside maintains, under appropriate regulatory permits, over 100 accessions in its pathogen-positive inventory. Many of the isolates were collected by scientists in California over the last 50 to 60 years and their pathogen status has been well studied using both biological and laboratory-based assays. They appeared to be excellent samples for the current study. Since HLB and certain other exotic disease agents are not maintained in this collection, a plant sample infected with HLB-associated CLas was obtained from an approved containment facility.
Viral metagenomics has revolutionized virus discovery using a wide variety of clinical samples [36]. Thousands of novel viruses have been discovered over the last decade through viral metagenomic studies [37]; these methods have been extensively used in the discovery of plant viruses for about 15 years [12,38,39]. Several novel citrus viruses have been discovered through NGS [14,15,16,17,19].
4.2. NGS Facilitates the Genome Characterization of Viruses and the Discovery of Both Viral and Bacterial Pathogens including New Citrus Viruses
A comparison of two sets of libraries, one with about 20–30 million reads per sample (libraries 1–12; Table 1) and another with about 120 million reads per sample (libraries 13–20; Table 1) suggested that the data from libraries 1–12 may be adequate for identification of pathogens; increased sequence depth may facilitate recovery of full-length genome sequences and increase the sensitivity of the analysis through detection of pathogens present at low titers.
A comparison of results from the present study on NGS analysis and previous test results (Table 5) shows that by utilizing NGS, we were able to find sequences of all pathogens found previously by biological, serological, and molecular methods. In addition, the method also identified new viruses associated with citrus: for example, the detection of BWYV and a variant of CYVaV. The significance, if any, of BWYV in citrus is currently not known.
Sequencing at higher depths resulted in the identification of some viruses occurring at low levels (Table 4, compare libraries 5 and 17). Except for a small number of viruses with low genome coverage (Table 4), full-length genomes were recovered from the NGS data for most of the viruses detected. A total of 46 of these full genome sequences have been deposited in NCBI (Table 6). The present study also revealed sequences of bacterial transcripts (CLas and SC; Table 3). NGS, as deployed here, should be able to detect all pathogen genomes with RNA in their life cycle, provided that the bioinformatics analysis includes the target genomes in the database for blast search. Accordingly, the present study was directed at finding all viruses, and known bacterial pathogens of citrus. Most of the pathogens found in this study also confirmed the results of previous studies (Table 5).
4.3. NGS Analysis Discovered New Citrus Viruses
BWYV, not previously reported from citrus, was detected (as six isolates) from plants that originated from various geographic locations. While the two genomic sequences of BWYV from citrus showed about 98.4% similarity, the other closest genome identified was from coriander from Cyprus with about 97% identity at the nucleotide level (Supplementary Table S1). Phylogenetic analyses (Figure 2 and Supplementary Figure S1) of the genomic sequences confirmed the identity as BWYV. Its close relationship with several isolates from Asia and its presence in isolates collected from different geographic regions suggests that BWYV may not be a new introduction to citrus. Recombination analysis suggests that the BWYV/citrus isolate may have resulted from recombination with a genomic fragment from a spinach isolate of BWYV, but the significance of this is not clear. Pathogenicity and interaction with other viruses need to be studied.
Fatal yellows was reported as a disease of lemons on Alemow rootstock [40]. Typical symptoms of yellowing of both veins and lamina are shown in Figure 6. Corky veins and rubbery stems can also be seen. An isolate has been maintained at the quarantine greenhouses since 1980 as a disease of unknown etiology. Analysis of samples from the plant showing “fatal yellows” (FY) symptoms showed the presence of a variant of CYVaV, which had been reported earlier to be associated with citrus yellow vein disease [19]. Even though the sequences of the FY-associated virus genome varied from the Citrus yellow vein-associated virus by about 14% (Table 7), they are considered the same species because of similar genome organization. The fatal yellows isolate had very low levels of CLRV and CVV, and the yellow vein sample had low levels of CVV and several other viruses (Table 4).

Figure 6.
Symptoms of “Fatal yellows” disease on citron (C. medica) plants in the greenhouse.
Cristacortis disease was described in 1968 from Corsica on sweet orange trees on sour orange rootstock [41]. Concave gum-like symptoms were observed on both scion and rootstock. It is not clear how to interpret the results from the analysis of plant samples with cristacortis, a disease with unknown etiology. The disease causes vertical depressions in the wood with corresponding pegs on the bark of sour orange and other citrus accessions. This isolate was received from Corsica in 1997 and has been maintained in sweet orange plants. The isolate was associated with four different citrus viroids and CPsV by both NGS analysis and qPCR (Table 4 and Table 5). The populations of HSVd and CDVd varied significantly in samples collected at different times. Since concave gum-associated viruses were not present, and none of the associated pathogens are known to cause cristacortis symptoms, we cannot draw conclusions at this time. The association of viruses in multiple samples with cristacortis may be useful for understanding its etiology.
Real-time PCR (qPCR) tests were developed for four selected pathogens of special interest discovered in this study. A qPCR analysis of over 100 samples was useful in confirming the presence of CPsV and CiVA in the positive pathogen inventory at the USDA Repository. Analysis by qPCR using CYVaV-specific primers and probes detected its presence in only two isolates in the positive inventory (Table 9).
4.4. Relevance of Citrus Blight-Associated Pararetrovirus Genome Sequence
The genomic sequence of CBaPRV, which has been reported to be highly expressed in trees with citrus blight disease [42], was detected in one of the samples studied (#21 = RRUT 178 = IVNO 3591). This was of serious concern since blight is a disease that causes significant damage in Florida [43] and several other locations [44,45] but has not yet been reported in California. A real-time PCR assay was developed in this study to selectively detect only CBaPRV, but not other CEPRVs (Figure 4). Greenhouse-grown seedlings of the trifoliate hybrid rootstock ‘US 942’, propagated from seeds received from Florida, were found to have high levels of CBaPRV expression. Assuming that the expression of CBaPRV was associated with citrus blight [42,46], these results were of serious concern. A detailed analysis of the expression of CBaPRV was studied in over 1000 accessions belonging to various genotypes. The high expression level was limited to trifoliates and trifoliate hybrids (Table 10). These results suggest that CBaPRV is constitutively expressed in trifoliate genotypes.
A search for full-length genomic sequence of CBaPRV in several genomes of Auratioideae, a subfamily of Rutaceae to which citrus belongs, revealed that multiple copies of CBaPRV are present in almost all chromosomes of the genomes of members of the tribe Citreae analyzed in the study (Table S2), but not in members of the tribe Clausineae. Multiple copies of full-length or near full-length genomes of CBaPRV were found in most genomes analyzed. The presence of CBaPRV in citrus may not be diagnostic of citrus blight.
4.5. Detection of Viruses and Bacteria from Insect Pests
Library #6 (P215) yielded unexpected results since an unknown picorna virus (MW674792) was found. Similar results were reported in metagenomes of honeybees collected from citrus-growing regions in Australia (MG995731 and MG995724). Identical results were obtained upon repeating the sequencing of the same library, but not when a new library (#17) from the same plant sample was sequenced at a different time. Real-time PCR analysis of several samples showed the presence of the picorna virus in greenhouse-grown seedlings and several other plants in both the greenhouse and field. The results strongly suggested that the virus probably originated from an insect host. Several picorna-like sequences have been recently reported in soybean thrips [47]. Revisiting 674 non-host de novo assemblies showed the presence of a small number of contigs from thrips. These results suggest that the picornavirus sequences originated from thrips, one of the most difficult pests to control in greenhouses.
Two large contigs representing two endosymbionts of mealybug, Planococcus citri, were found in library #20. Endogenous bacteria of mealybugs were not identified by the workflow described above. Large de novo contigs that did not align with either host sequences, or with the three in-house databases were identified by NCBI blast search as endogenous bacteria of mealybugs.
4.6. Pathogen Status of Citrus Relatives Held in Quarantine
Four citrus relatives (samples 1–4; Table 1) included in the present study have been maintained in the USDA Repository greenhouses for many years. Our NGS analysis as well as previous laboratory tests did not find any citrus viruses or known bacterial pathogens in all four accessions tested (Table 5). Since many citrus relatives are not graft-compatible with common indicator plants used in biological indexing, there is a need to develop alternate methods of testing before the release and distribution of germplasm. Some genera closely related to Citrus, such as Microcitrus and Eremocitrus, have been shown to have excellent tolerance/resistance to HLB [48], and have been used in breeding for HLB resistance [49]. There is a need to screen the hybrids for resistance in multiple geographic regions to assess their usefulness to the industry. Such germplasm exchanges would require approved, rapid, pathogen testing methods. We propose NGS transcriptome analysis as described here as an alternate method for releasing Rutaceous germplasm from quarantine as well as for exchanging breeders’ materials between industries. This would have to be approved by regulatory agencies, potentially at different levels (Federal and State in the USA).
4.7. Requirements for the Use of NGS as a Regulatory Test for Detection of Pathogens
For NGS methods to be used routinely, certain guidelines need to be developed and implemented. Protocols for nucleic acid extractions, guidelines for sequencing technologies recommended, minimum depth of sequencing, pipelines for different testing needs, etc. need to be standardized. The methods proposed should be user-friendly so that involved research groups can participate without requiring significant resources. The participating labs should be able to carry out the testing methods. Training sessions may be offered either by the regulatory agencies or laboratories that have successfully used the methods. NGS can serve as the main tool for testing germplasm in combination with other lab-based methods and biological indexing as optional confirmatory tests when needed. At the very least, NGS methods can be used along with the standard testing methods until proper evaluations are done.
4.8. Conclusions
Next-generation sequencing is a powerful technology that can be used as an additional tool for testing citrus germplasm. The movement of clean propagative materials across different regions requires quick and thorough testing of the germplasm. NGS would not only complement the currently used biological and laboratory assays but would also facilitate the detection of new pathogens as demonstrated in our present study. The introduction of budwood-carrying pathogens can be detrimental to the importing citrus industries if the pathogen spreads in the new region through insect vectors, farm tools, or other modes of dissemination. The addition of NGS methods to the routine disease testing regimen will complement the currently used testing methods and is beneficial for citrus germplasm certification and movement across state and international borders. Biological assays using standard citrus indicators are not amenable for testing non-citrus Rutaceous germplasm because of graft incompatibility. These citrus relatives are gaining importance because of their possible usefulness in developing resistance to HLB. NGS can be used as a reliable method for pathogen testing.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/plants13030411/s1, Figure S1: Phylogeny of Beet western yellows virus (BWYV). Genomes from different geographic locations in different host plants were analyzed. Pea enation mosaic virus-1 (PEMV-1, NC_003629) was used as an outgroup sequence. Accession numbers of different BWYV genomes, plant host information, and geographic locations are shown. The phylogenetic tree was constructed using the neighbor-joining method and visualized with MEGA11.0. Bootstrap values shown at the nodes indicate the percentage of 1000 replications supporting the clades; Figure S2: Alignment of DNA and RNA of the target area selected for designing primers and probe for real-time PCR amplification of reverse-transcribed RNA. The RNA sequence shown constitutes the 3’ terminus of exon 12 and 5’ end of exon 13 of the malate dehydrogenase gene. The forward primer was designed to align mostly to the 3’ end of exon 12 and three bases at the 5’ terminus of exon 13 so that the amplification is restricted only to reverse-transcribed RNA. The sequences of the probe and the primers as well as the direction are shown. Intron sequences are indicated by dots in the corresponding regions; Figure S3: Alignment of the target region of Citrus blight-associated pararetrovirus (MZ330114) selected for real-time PCR assay. CBaPRV reported to be associated with citrus blight (MN14438.1), and from the genomes of Poncirus trifoliata, Citrus sinensis, C. clementina, Eremocitrus glauca and Atalantia buxifolia. The primers and probe region are highlighted, and their directions of synthesis are shown. Multiple copies of full-length or near full-length genomes of CBaPRV are in all the genomes shown in the diagram; Table S1: Percent sequence identity matrix between Beet western yellows virus isolates from citrus and other closely related viruses. The sequences of 20 isolates of BWYV were downloaded from NCBI Genbank. The sequences were aligned using ClustalW (reference) to generate the phylogenetic tree (Figure 2); Table S2: Location of full-length genome sequence of CBaPRV in different chromosomes of the five selected taxa of the tribe Citreae, subfamily Aurantioideae. Only one copy with the highest nucleotide identity is shown for each chromosome. Multiple copies were found in almost all chromosomes. Query coverage and percent identity with the CBaPRV (MZ330114) identified in the present study are shown.
Author Contributions
M.K., R.R.K. and T.H.S. conceptualized and selected plants for this study. The methodology was developed, and libraries were constructed by M.K. and C.R., bioinformatics analysis was carried out by M.K. and K.S. A draft of the article was prepared by M.K. and edited by K.S., C.R., R.R.K. and T.H.S. Funding from USDA NIFA was acquired by CR. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by USDA ARS and a grant from USDA NIFA #2020-70029-33201 awarded to C.R.
Data Availability Statement
Information on accessions used in this study is available from the National Plant Germplasm System, and raw sequence reads can be accessed by accessing the relevant Bioproject sample in Table 1 on NCBI. Total sequence reads, and depth of coverage of different viral genomes from 20 plant samples are shown in Table 4, and 46 full-length genomes of viruses (Table 6) are available from NCBI.
Acknowledgments
Technical assistance by Brittany Moreland with maintaining all the test plants used in the study and Bethany Preciado with processing plant samples and conducting real-time PCR assays is greatly expressed.
Conflicts of Interest
The authors declare no conflict of interest.
Disclaimer
The use of trade, firm, or corporation names in this publication is for the information and convenience of the reader. Such use does not constitute an official endorsement or approval by the United States Department of Agriculture or the Agricultural Research Service of any product or service to the exclusion of others that may be suitable. USDA is an equal-opportunity employer.
References
- Reuther, W. The Citrus Clonal Protection Program. Calif. Agric. 1981, 35, 30–32. [Google Scholar]
- Roistacher, C.N. Graft Transmissible Diseases of Citrus. Handbook for Detection and Diagnosis; F.A.O.: Rome, Italy, 1991; p. 283. [Google Scholar]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of Next-Generation Sequencing Versus Biological Indexing for the Optimal Detection of Viral Pathogens in Grapevine. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Maclot, F.; Candresse, T.; Filloux, D.; Malmstrom, C.M.; Roumagnac, P.; van der Vlugt, R.; Massart, S. Illuminating an Ecological Blackbox: Using High Throughput Sequencing to Characterize the Plant Virome Across Scales. Front. Microbiol. 2020, 11, 578064. [Google Scholar] [CrossRef] [PubMed]
- Lebas, B.; Adams, I.; Al Rwahnih, M.; Baeyen, S.; Bilodeau, G.J.; Blouin, A.G.; Boonham, N.; Candresse, T.; Chandelier, A.; De Jonghe, K.; et al. Facilitating the adoption of high-throughput sequencing technologies as a plant pest diagnostic test in laboratories: A step-by-step description. EPPO Bull. 2022, 52, 394–418. [Google Scholar] [CrossRef]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, A.; Flores, R.; Candresse, T.; Barba, M. Next-Generation Sequencing and Genome Editing in Plant Virology. Front. Microbiol. 2016, 7, 1325. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Adams, I.; Al Rwahnih, M.; Baeyen, S.; Bilodeau, G.J.; Blouin, A.G.; Boonham, N.; Candresse, T.; Chandellier, A.; De Jonghe, K.; et al. Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pests. Peer Community J. 2022, 2, e62. [Google Scholar] [CrossRef]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High Throughput Sequencing For Plant Virus Detection and Discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Faure, C.; Mustafayev, E.; Barone, M.; Alioto, D.; Candresse, T. Characterization by Deep Sequencing of Prunus virus T, a Novel Tepovirus Infecting Prunus Species. Phytopathology 2015, 105, 135–140. [Google Scholar] [CrossRef]
- Rott, M.E.; Kesanakurti, P.; Berwarth, C.; Rast, H.; Boyes, I.; Phelan, J.; Jelkmann, W. Discovery of Negative-Sense RNA Viruses in Trees Infected with Apple Rubbery Wood Disease by Next-Generation Sequencing. Plant Dis. 2018, 102, 1254–1263. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef]
- Villamor, D.E.; Mekuria, T.A.; Pillai, S.S.; Eastwell, K.C. High-Throughput Sequencing Identifies Novel Viruses in Nectarine: Insights to the Etiology of Stem-Pitting Disease. Phytopathology 2016, 106, 519–527. [Google Scholar] [CrossRef]
- Loconsole, G.; Saldarelli, P.; Doddapaneni, H.; Savino, V.; Martelli, G.P.; Saponari, M. Identification of a single-stranded DNA virus associated with citrus chlorotic dwarf disease, a new member in the family Geminiviridae. Virology 2012, 432, 162–172. [Google Scholar] [CrossRef]
- Loconsole, G.; Onelge, N.; Potere, O.; Giampetruzzi, A.; Bozan, O.; Satar, S.; De Stradis, A.; Savino, V.; Yokomi, R.K.; Saponari, M. Identification and characterization of citrus yellow vein clearing virus, a putative new member of the genus Mandarivirus. Phytopathology 2012, 102, 1168–1175. [Google Scholar] [CrossRef]
- Roy, A.; Choudhary, N.; Guillermo, L.M.; Shao, J.; Govindarajulu, A.; Achor, D.; Wei, G.; Picton, D.D.; Levy, L.; Nakhla, M.K.; et al. A novel virus of the genus Cilevirus causing symptoms similar to citrus leprosis. Phytopathology 2013, 103, 488–500. [Google Scholar] [CrossRef]
- Vives, M.C.; Velázquez, K.; Pina, J.A.; Moreno, P.; Guerri, J.; Navarro, L. Identification of a new enamovirus associated with citrus vein enation disease by deep sequencing of small RNAs. Phytopathology 2013, 103, 1077–1086. [Google Scholar] [CrossRef]
- Tan, S.H.; Osman, F.; Bodaghi, S.; Dang, T.; Greer, G.; Huang, A.; Hammado, S.; Abu-Hajar, S.; Campos, R.; Vidalakis, G. Full genome characterization of 12 citrus tatter leaf virus isolates for the development of a detection assay. PLoS ONE 2019, 14, e0223958. [Google Scholar] [CrossRef]
- Kwon, S.J.; Bodaghi, S.; Dang, T.; Gadhave, K.R.; Ho, T.; Osman, F.; Al Rwahnih, M.; Tzanetakis, I.E.; Simon, A.E.; Vidalakis, G. Complete Nucleotide Sequence, Genome Organization, and Comparative Genomic Analyses of Citrus Yellow-Vein Associated Virus (CYVaV). Front. Microbiol. 2021, 12, 683130. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Lavagi-Craddock, I.; Bodaghi, S.; Vidalakis, G. Next-Generation Sequencing Identification and Characterization of MicroRNAs in Dwarfed Citrus Trees Infected with Citrus Dwarfing Viroid in High-Density Plantings. Front. Microbiol. 2021, 12, 646273. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Wang, H.; Espindola, A.; Habiger, J.; Vidalakis, G.; Cardwell, K. Development and statistical validation of e-probe diagnostic nucleic acid analysis (EDNA) detection assays for the detection of citrus pathogens from raw high throughput sequencing data. PhytoFrontiers 2022, 3, 113–123. [Google Scholar] [CrossRef]
- Ho, T.; Tzanetakis, I.E. Development of a virus detection and discovery pipeline using next generation sequencing. Virology 2014, 471–473, 54–60. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Minutolo, M.; De Stradis, A.; Palmisano, F.; Alioto, D.; Di Serio, F. The first phlebo-like virus infecting plants: A case study on the adaptation of negative-stranded RNA viruses to new hosts. Mol. Plant Pathol. 2018, 19, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Zicca, S.; Minutolo, M.; Saponari, M.; Alioto, D.; Di Serio, F. A Negative-Stranded RNA Virus Infecting Citrus Trees: The Second Member of a New Genus within the Order Bunyavirales. Front. Microbiol. 2018, 9, 2340. [Google Scholar] [CrossRef] [PubMed]
- Bester, R.; Steyn, C.; Breytenbach, J.H.J.; de Bruyn, R.; Cook, G.; Maree, H.J. Reproducibility and Sensitivity of High-Throughput Sequencing (HTS)-Based Detection of Citrus Tristeza Virus and Three Citrus Viroids. Plants 2022, 11, 1939. [Google Scholar] [CrossRef] [PubMed]
- Jooste, T.L.; Visser, M.; Cook, G.; Burger, J.T.; Maree, H.J. In Silico Probe-Based Detection of Citrus Viruses in NGS Data. Phytopathology 2017, 107, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Zouhar, M.; Mazakova, J.; Rysanek, P. Genome wide identification of the immunophilin gene family in Leptosphaeria maculans: A causal agent of Blackleg disease in Oilseed Rape (Brassica napus). Omics 2014, 18, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Keremane, M.L.; McCollum, T.G.; Roose, M.L.; Lee, R.F.; Ramadugu, C. An Improved Reference Gene for Detection of “Candidatus Liberibacter asiaticus” Associated with Citrus Huanglongbing by qPCR and Digital Droplet PCR Assays. Plants 2021, 10, 2111. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Gibbs, A.J.; Fowkes, A.R.; Pufal, H.; McGreig, S.; Jones, R.A.C.; Boonham, N.; Adams, I.P. Enhanced Apiaceous Potyvirus Phylogeny, Novel Viruses, and New Country and Host Records from Sequencing Apiaceae Samples. Plants 2022, 11, 1951. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Tamada, T. Host range and molecular analysis of Beet leaf yellowing virus, Beet western yellows virus-JP and Brassica yellows virus in Japan. Plant Pathol. 2019, 68, 1045–1058. [Google Scholar] [CrossRef]
- Slavov, S.N. Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine. Viruses 2022, 14, 2448. [Google Scholar] [CrossRef] [PubMed]
- Moubset, O.; François, S.; Maclot, F.; Palanga, E.; Julian, C.; Claude, L.; Fernandez, E.; Rott, P.; Daugrois, J.H.; Antoine-Lorquin, A.; et al. Virion-Associated Nucleic Acid-Based Metagenomics: A Decade of Advances in Molecular Characterization of Plant Viruses. Phytopathology 2022, 112, 2253–2272. [Google Scholar] [CrossRef] [PubMed]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef]
- Schneider, H. Host Range of the Fatal Yellows Disease. In Proceedings of the International Organization of Citrus Virologists Conference Proceedings, Puerto Iguaçu, Argentina, 9–13 May 1983; pp. 222–228. [Google Scholar]
- Vogel, R.; Bove, J.M. Cristacortis, a Virus Disease Inducing Stem Pitting on Sour Orange and Other Citrus Species. Int. Organ. Citrus Virol. Conf. Proc. 1968, 4, 221–228. [Google Scholar] [CrossRef]
- Fu, S.; Shao, J.; Roy, A.; Brlansky, R.H.; Zhou, C.; Hartung, J.S. Transcriptomic analyses reveal physiological changes in sweet orange roots affected by citrus blight. BMC Genom. 2019, 20, 969. [Google Scholar] [CrossRef]
- Derrick, K.S.; Timmer, L.W. Citrus Blight and Other Diseases of Recalcitrant Etiology. Annu. Rev. Phytopathol. 2000, 38, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, W.; Moreira, L.; Derrick, K.S.; Beretta, M.J.G.; Rivera, C. First Report of Citrus Blight in Costa Rica. Plant Dis. 2005, 89, 108. [Google Scholar] [CrossRef] [PubMed]
- Beretta, M.J.G.; Barthe, G.A.; Ceccardi, T.L.; Lee, R.F.; Derrick, K.S. A Survey for Strains of Xylella fastidiosa in Citrus Affected by Citrus Variegated Chlorosis and Citrus Blight in Brazil. Plant Dis. 1997, 81, 1196–1198. [Google Scholar] [CrossRef]
- Roy, A.; Shao, J.; Schneider, W.L.; Hartung, J.S.; Brlansky, R.H. Population of endogenous pararetrovirus genomes in carrizo citrange. Genome Announc. 2014, 2, e01063-13. [Google Scholar] [CrossRef]
- Thekke-Veetil, T.; Lagos-Kutz, D.; McCoppin, N.K.; Hartman, G.L.; Ju, H.-K.; Lim, H.-S.; Domier, L.L. Soybean Thrips (Thysanoptera: Thripidae) Harbor Highly Diverse Populations of Arthropod, Fungal and Plant Viruses. Viruses 2020, 12, 1376. [Google Scholar] [CrossRef]
- Ramadugu, C.; Keremane, M.L.; Halbert, S.E.; Duan, Y.P.; Roose, M.L.; Stover, E.; Lee, R.F. Long-Term Field Evaluation Reveals Huanglongbing Resistance in Citrus Relatives. Plant Dis. 2016, 100, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Ramadugu, C.; Keremane, M.; Lee, R.F.; Hall, D.G.; McCollum, T.G.; Roose, M.L. Novel Citrus Hybrids with HLB Resistance. Citrograph 2019, 10, 60–64. Available online: https://citrus-research-board-static.sfo2.digitaloceanspaces.com/citrograph/pdf/CRB-Citrograph-Mag-Q2-Spring-2019-Web.pdf (accessed on 23 December 2023).




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).