
The American Cherimoya Genome Reveals Insights into the Intra-Specific Divergence, the Evolution of Magnoliales, and a Putative Gene Cluster for Acetogenin Biosynthesis

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. PacBio Sequencing and Assembly
2.3. Dovetail Hi-C Sequencing and Assembly Scaffolding with HiRise
2.4. Repeat and Noncoding RNA Annotation
2.5. Protein-Coding Gene Prediction
2.6. Orthologous Gene Clusters and Phylogenetic Analyses
2.7. GO Enrichment Analysis
2.8. Genome Synteny
2.9. Plant Resistance Gene Identification
2.10. Acetogenin (ACG) Biosynthetic Gene Cluster Identification
3. Results
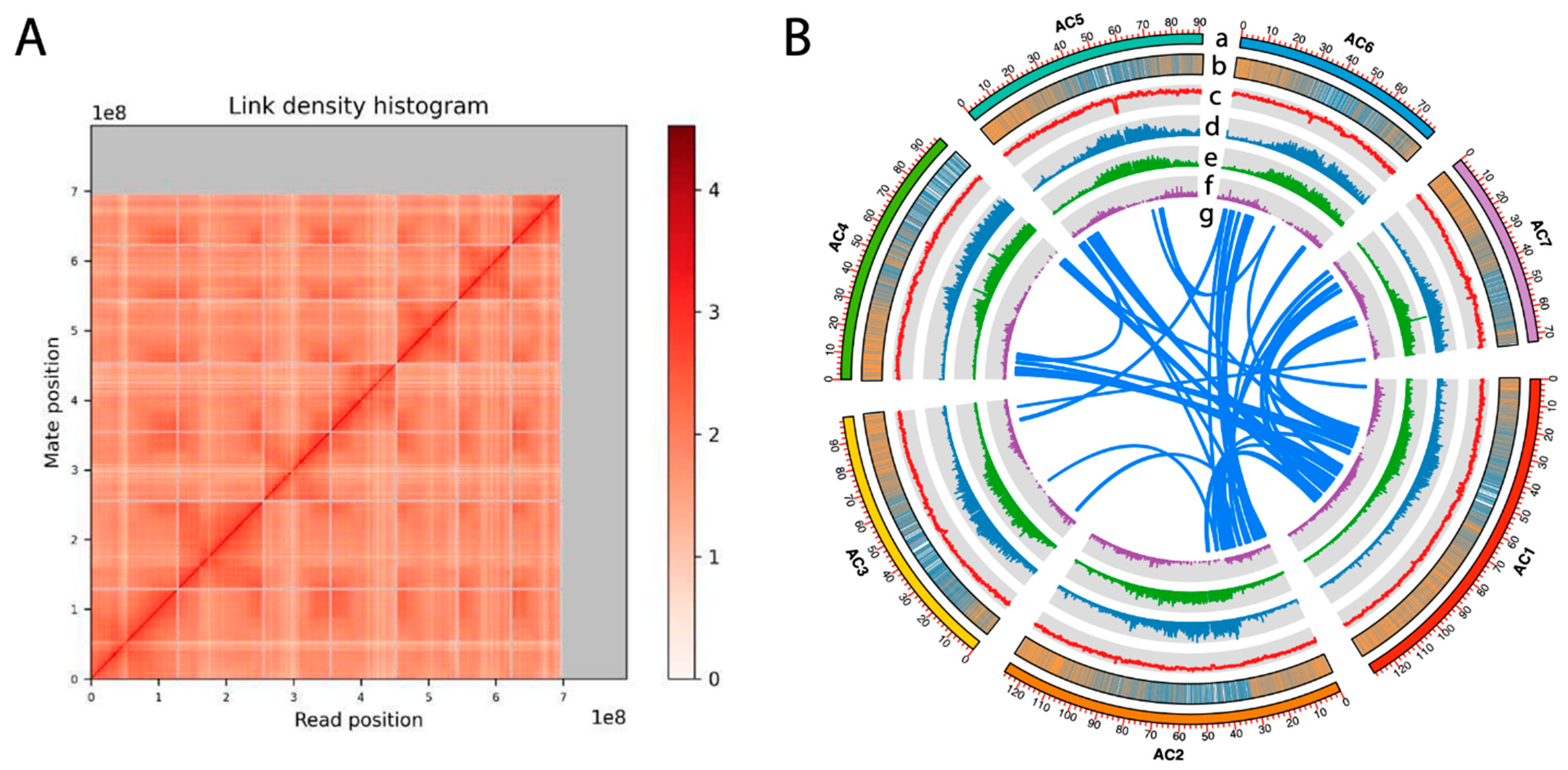
3.1. The Chromosome-Level Genome Assembly of Cherimoya “Booth”
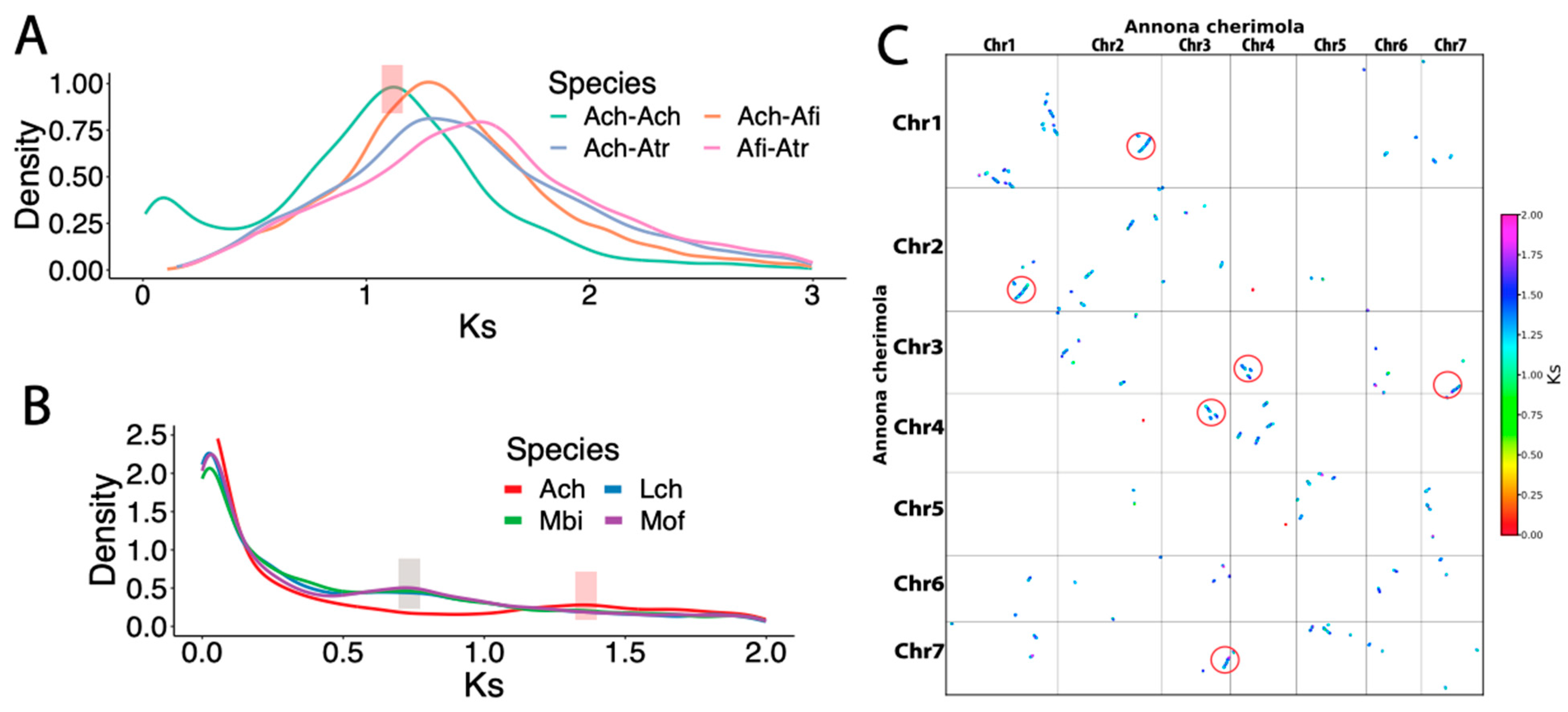
3.2. No Clear Whole Genome Duplication Event Was Found in Cherimoya
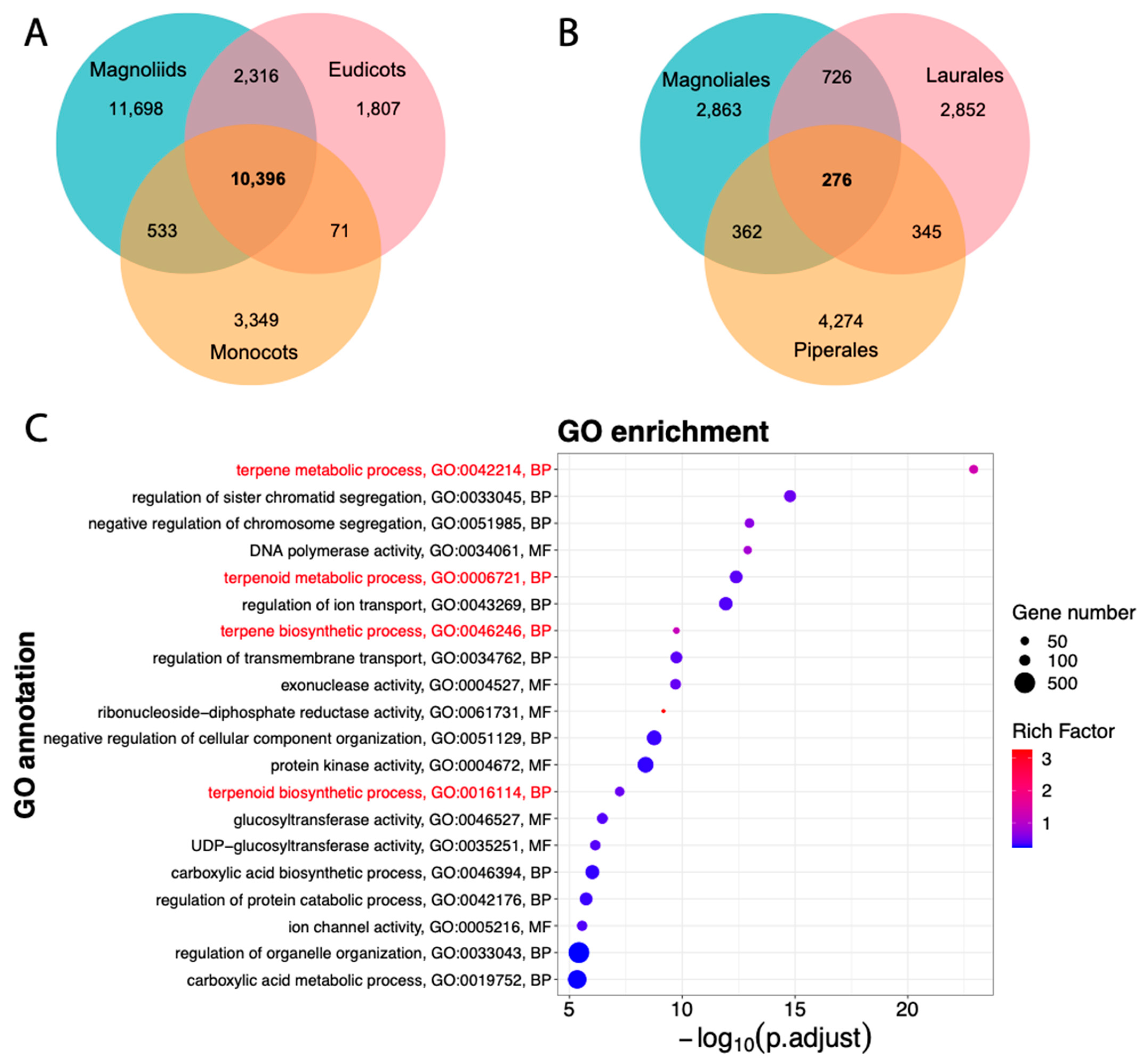
3.3. Magnoliids Are Phylogenetically Closer to Eudicots Than to Monocots
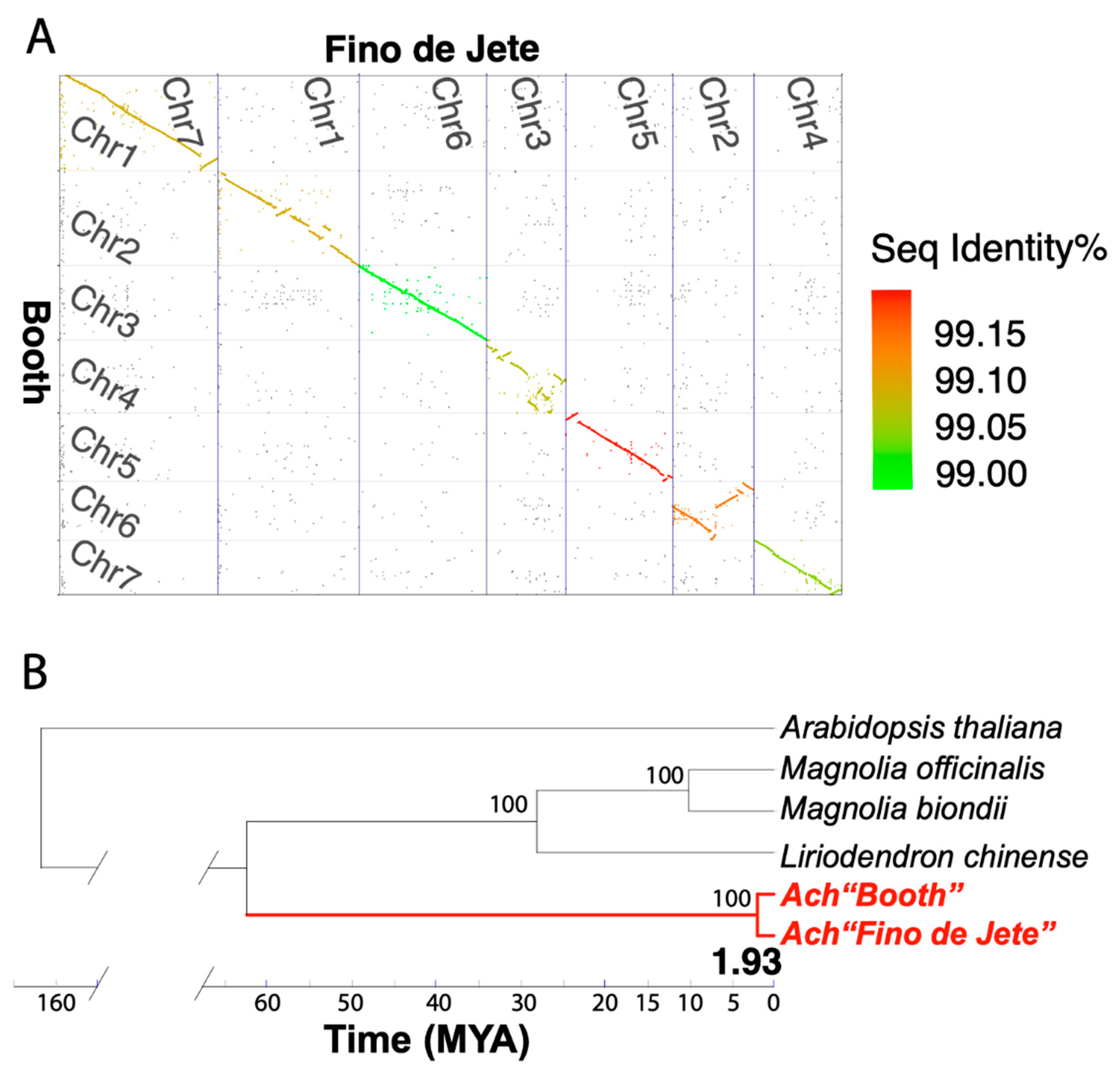
3.4. Comparative Genomics Identifies Intra-Specific Genomic Variations within Annona cherimola
3.5. Terpene/Terpenoid Metabolism Functions Are Enriched in Magnoliales
3.6. Plant Resistance TNL Genes Are Absent in Magnoliales
3.7. A Potential Acetogenin Biosynthetic Gene Cluster in A. cherimola Genome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto, A.D.Q.; Cordeiro, M.C.R.; De Andrade, S.R.M.; Ferreira, F.R.; Filgueiras, H.D.C.; Alves, R.E.; Kinpara, D.I. Annona Species; University of Southampton, International Centre for Underutilised Crops: Southampton, UK, 2005; p. 284. [Google Scholar]
- Leal, F.; Paull, R.E. The genus Annona: Botanical characteristics, horticultural requirements and uses. Crop Sci. 2023, 63, 1030–1049. [Google Scholar] [CrossRef]
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef]
- Pino, J.A. Annona Fruits. In Handbook of Fruit and Vegetable Flavors; Wiley: Hoboken, NJ, USA, 2010; pp. 229–246. [Google Scholar] [CrossRef]
- National Research Council. Lost Crops of the Incas: Little-Known Plants of the Andes with Promise for Worldwide Cultivation; National Academies Press: Washington, DC, USA, 1989. [Google Scholar] [CrossRef]
- Amoo, I.A.; Emenike, A.E.; Akpambang, V.O.E. Compositional Evaluation of Annona cherimoya (Custard Apple) Fruit. Trends Appl. Sci. Res. 2008, 3, 216–220. [Google Scholar] [CrossRef]
- Mannino, G.; Gentile, C.; Porcu, A.; Agliassa, C.; Caradonna, F.; Bertea, C.M. Chemical Profile and Biological Activity of Cherimoya (Annona cherimola Mill.) and Atemoya (Annona atemoya) Leaves. Molecules 2020, 25, 2612. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, T.G.; Santos, F.; Sanches-Silva, A.; Beatriz Oliveira, M.; Bento, A.C.; Costa, H.S. Nutritional and phytochemical composition of Annona cherimola Mill. fruits and by-products: Potential health benefits. Food Chem. 2016, 193, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Quílez, A.M.; Fernández-Arche, M.A.; García-Giménez, M.D.; De la Puerta, R. Potential therapeutic applications of the genus Annona: Local and traditional uses and pharmacology. J. Ethnopharmacol. 2018, 225, 244–270. [Google Scholar] [CrossRef] [PubMed]
- Cortes, D.; Myint, S.H.; Dupont, B.; Davoust, D. Bioactive acetogenins from seeds of Annona cherimolia. Phytochemistry 1993, 32, 1475–1482. [Google Scholar] [CrossRef]
- Neske, A.; Ruiz Hidalgo, J.; Cabedo, N.; Cortes, D. Acetogenins from Annonaceae family. Their potential biological applications. Phytochemistry 2020, 174, 112332. [Google Scholar] [CrossRef]
- Perrone, A.; Yousefi, S.; Salami, A.; Papini, A.; Martinelli, F. Botanical, genetic, phytochemical and pharmaceutical aspects of Annona cherimola Mill. Sci. Hortic. 2022, 296, 110896. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Chang, F.-R.; Liaw, C.-C.; Wu, Y.-C.; Bastow, K.F.; Lee, K.-H. Acetogenins as Selective Inhibitors of the Human Ovarian 1A9 Tumor Cell Line. J. Med. Chem. 2003, 46, 3185–3188. [Google Scholar] [CrossRef]
- Yuan, S.-S.F.; Chang, H.-L.; Chen, H.-W.; Yeh, Y.-T.; Kao, Y.-H.; Lin, K.-H.; Wu, Y.-C.; Su, J.-H. Annonacin, a mono-tetrahydrofuran acetogenin, arrests cancer cells at the G1 phase and causes cytotoxicity in a Bax- and caspase-3-related pathway. Life Sci. 2003, 72, 2853–2861. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Ammoury, C.; Haykal, T.; Nasr, L.; Sarkis, R.; Rizk, S. The selective anti-proliferative and pro-apoptotic effect of A. cherimola on MDA-MB-231 breast cancer cell line. BMC Complement. Med. Ther. 2020, 20, 343. [Google Scholar] [CrossRef] [PubMed]
- Durán, A.G.; Gutiérrez, M.T.; Mejías, F.J.R.; Molinillo, J.M.G.; Macías, F.A. An Overview of the Chemical Characteristics, Bioactivity and Achievements Regarding the Therapeutic Usage of Acetogenins from Annona cherimola Mill. Molecules 2021, 26, 2926. [Google Scholar] [CrossRef] [PubMed]
- Popenoe, W. The Native Home of the Cherimoya. J. Hered. 1921, 12, 331–336. [Google Scholar] [CrossRef]
- Larranaga, N.; Albertazzi, F.J.; Fontecha, G.; Palmieri, M.; Rainer, H.; van Zonneveld, M.; Hormaza, J.I. A Mesoamerican origin of cherimoya (Annona cherimola Mill.): Implications for the conservation of plant genetic resources. Mol. Ecol. 2017, 26, 4116–4130. [Google Scholar] [CrossRef]
- Bonavia, D.; Ochoa, C.M.; Óscar Tovar, S.; Palomino, R.C. Archaeological Evidence of Cherimoya (Annona cherimolia Mill.) and Guanabana (Annona muricata L.) in Ancient Peru. Econ. Bot. 2004, 58, 509–522. [Google Scholar] [CrossRef]
- Scheldeman, X. Distribution and Potential of Cherimoya (Annona cherimola Mill.) and Highland Papayas (Vasconcellea Spp.) in Ecuador. Ph.D. Thesis, Faculty of Agricultural and Applied Biological Sciences, Department Plant Production, Laboratory of Tropical and Subtropical Agronomy and Ethnobotany, Ghent, Belgium, 2002. [Google Scholar]
- Larranaga, N.; Albertazzi, F.J.; Hormaza, J.I. Phylogenetics of Annona cherimola (Annonaceae) and some of its closest relatives. J. Syst. Evol. 2019, 57, 211–221. [Google Scholar] [CrossRef]
- Morton, J.F. Fruits of Warm Climates; Miami, FL, USA, 1987. Available online: https://www.echopointbooks.com/agriculture/fruits-of-warm-climates (accessed on 17 January 2024).
- Soltis, D.; Soltis, P.; Endress, P.; Chase, M.W.; Manchester, S.; Judd, W.; Majure, L.; Mavrodiev, E. Phylogeny and Evolution of the Angiosperms; University of Chicago Press: Chicago, IL, USA, 2018. [Google Scholar]
- Qin, L.; Hu, Y.; Wang, J.; Wang, X.; Zhao, R.; Shan, H.; Li, K.; Xu, P.; Wu, H.; Yan, X.; et al. Insights into angiosperm evolution, floral development and chemical biosynthesis from the Aristolochia fimbriata genome. Nat. Plants 2021, 7, 1239–1253. [Google Scholar] [CrossRef]
- Hu, L.; Xu, Z.; Wang, M.; Fan, R.; Yuan, D.; Wu, B.; Wu, H.; Qin, X.; Yan, L.; Tan, L.; et al. The chromosome-scale reference genome of black pepper provides insight into piperine biosynthesis. Nat. Commun. 2019, 10, 4702. [Google Scholar] [CrossRef]
- Rendón-Anaya, M.; Ibarra-Laclette, E.; Méndez-Bravo, A.; Lan, T.; Zheng, C.; Carretero-Paulet, L.; Perez-Torres, C.A.; Chacón-López, A.; Hernandez-Guzmán, G.; Chang, T.-H.; et al. The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proc. Natl. Acad. Sci. USA 2019, 116, 17081–17089. [Google Scholar] [CrossRef]
- Yin, Y.; Peng, F.; Zhou, L.; Yin, X.; Chen, J.; Zhong, H.; Hou, F.; Xie, X.; Wang, L.; Shi, X.; et al. The chromosome-scale genome of Magnolia officinalis provides insight into the evolutionary position of magnoliids. iScience 2021, 24, 102997. [Google Scholar] [CrossRef]
- Chaw, S.-M.; Liu, Y.-C.; Wu, Y.-W.; Wang, H.-Y.; Lin, C.-Y.I.; Wu, C.-S.; Ke, H.-M.; Chang, L.-Y.; Hsu, C.-Y.; Yang, H.-T.; et al. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants 2019, 5, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Li, Z.; Zhao, Y.-X.; Gao, M.; Wang, J.-Y.; Liu, K.-W.; Wang, X.; Wu, L.-W.; Jiao, Y.-L.; Xu, Z.-L.; et al. The Litsea genome and the evolution of the laurel family. Nat. Commun. 2020, 11, 1675. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Qiu, J.; Liu, J.; Li, Z.; Zhang, W.; Wang, Q.; Fang, J.; Pan, J.; Chen, Z.; Cheng, W.; et al. The Chimonanthus salicifolius genome provides insight into magnoliid evolution and flavonoid biosynthesis. Plant J. 2020, 103, 1910–1923. [Google Scholar] [CrossRef] [PubMed]
- Talavera, A.; Fernandez-Pozo, N.; Matas, A.J.; Hormaza, J.I.; Bombarely, A. Genomics in neglected and underutilized fruit crops: A chromosome-scale genome sequence of cherimoya (Annona cherimola). Plants People Planet 2023, 5, 408–423. [Google Scholar] [CrossRef]
- Grossberger, D. The California Cherimoya Industry. Acta Hortic. 1999, 497, 119–142. [Google Scholar] [CrossRef]
- Zheng, J.; Meinhardt, L.W.; Goenaga, R.; Zhang, D.; Yin, Y. The chromosome-level genome of dragon fruit reveals whole-genome duplication and chromosomal co-localization of betacyanin biosynthetic genes. Hortic. Res. 2021, 8, 63. [Google Scholar] [CrossRef]
- Zheng, J.; Meinhardt, L.W.; Goenaga, R.; Matsumoto, T.; Zhang, D.; Yin, Y. The chromosome-level rambutan genome reveals a significant role of segmental duplication in the expansion of resistance genes. Hortic. Res. 2022, 9, uhac014. [Google Scholar] [CrossRef]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R. RepeatModeler Open-1.0; ScienceOpen, Inc.: Lexington, MA, USA, 2010. [Google Scholar]
- Ou, S.; Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef]
- Crescente, J.M.; Zavallo, D.; Helguera, M.; Vanzetti, L.S. MITE Tracker: An accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinform. 2018, 19, 348. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 17 January 2024).
- Abrusán, G.; Grundmann, N.; DeMester, L.; Makalowski, W. TEclass—A tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 2009, 25, 1329–1330. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction: Methods and Protocols; Methods Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Seemann, T. Barrnap: BAsic Rapid Ribosomal RNA Predictor. 2013. Available online: https://github.com/tseemann/barrnap (accessed on 17 January 2024).
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Sánchez Alvarado, A.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.; Steinegger, M.; Söding, J. MMseqs software suite for fast and deep clustering and searching of large protein sequence sets. Bioinformatics 2016, 32, 1323–1330. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. Author Correction: SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 352. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Jiao, B.; Yang, Y.; Shan, L.; Li, T.; Li, X.; Xi, Z.; Wang, X.; Liu, J. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 2022, 15, 1841–1851. [Google Scholar] [CrossRef]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and Collinearity in Plant Genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef]
- Zwaenepoel, A.; Van de Peer, Y. wgd-simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 2019, 35, 2153–2155. [Google Scholar] [CrossRef]
- Li, P.; Quan, X.; Jia, G.; Xiao, J.; Cloutier, S.; You, F.M. RGAugury: A pipeline for genome-wide prediction of resistance gene analogs (RGAs) in plants. BMC Genom. 2016, 17, 852. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Suarez Duran, H.G.; Blin, K.; Osbourn, A.; Medema, M.H. plantiSMASH: Automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, W55–W63. [Google Scholar] [CrossRef]
- Strijk, J.; Hinsinger, D.; Roeder, M.; Chatrou, L.; Couvreur, T.; Erkens, R.; Sauquet, H.; Pirie, M.; Thomas, D.; Cao, K. Chromosome-level reference genome of the Soursop (Annona muricata), a new resource for Magnoliid research and tropical pomology. Mol. Ecol. Resour. 2021, 21, 1608–1619. [Google Scholar] [CrossRef]
- Martin, C.; Viruel, M.A.; Lora, J.; Hormaza, J.I. Polyploidy in Fruit Tree Crops of the Genus Annona (Annonaceae). Front. Plant Sci. 2019, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Liu, M.; Liu, Y.; Chen, F.; Yang, T.; Chen, L.; Zhang, X.; Guo, X.; Fang, D.; Li, L.; et al. The genome of Magnolia biondii Pamp. provides insights into the evolution of Magnoliales and biosynthesis of terpenoids. Hortic. Res. 2021, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hao, Z.; Guang, X.; Zhao, C.; Wang, P.; Xue, L.; Zhu, Q.; Yang, L.; Sheng, Y.; Zhou, Y.; et al. Liriodendron genome sheds light on angiosperm phylogeny and species-pair differentiation. Nat. Plants 2019, 5, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O.; Dawid, I.B. Repeated Genes in Eukaryotes. Annu. Rev. Biochem. 1980, 49, 727–764. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Koshiba-Takeuchi, K. Significance of whole-genome duplications on the emergence of evolutionary novelties. Brief. Funct. Genom. 2018, 17, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Amborella Genome Project. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Cox-Georgian, D.; Ramadoss, N.; Dona, C.; Basu, C. Therapeutic and Medicinal Uses of Terpenes. In Medicinal Plants; Springer: Berlin/Heidelberg, Germany, 2019; pp. 333–359. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, R.A. Plant terpenes: Defense responses, phylogenetic analysis, regulation and clinical applications. 3 Biotech 2015, 5, 129–151. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Tarr, D.E.K.; Alexander, H.M. TIR-NBS-LRR genes are rare in monocots: Evidence from diverse monocot orders. BMC Res. Notes 2009, 2, 197. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Vernaldi, S.; Maekawa, T. Evolution and Conservation of Plant NLR Functions. Front. Immunol. 2013, 4, 297. [Google Scholar] [CrossRef] [PubMed]
- Ngou, B.P.M.; Ding, P.; Jones, J.D.G. Thirty years of resistance: Zig-zag through the plant immune system. Plant Cell 2022, 34, 1447–1478. [Google Scholar] [CrossRef] [PubMed]
- Alali, F.Q.; Liu, X.-X.; McLaughlin, J.L. Annonaceous Acetogenins: Recent Progress. J. Nat. Prod. 1999, 62, 504–540. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Scheible, W.-R. A novel calmodulin-interacting Domain of Unknown Function 506 protein represses root hair elongation in Arabidopsis. Plant Cell Environ. 2022, 45, 1796–1812. [Google Scholar] [CrossRef]
- Datiles, M.J.; Acevedo-Rodríguez, P. Annona cherimola (cherimoya). In CABI Compendium; 18/12/2014. Available online: https://doi.org/10.1079/cabicompendium.5806 (accessed on 17 January 2024).
- Kahn, T.L.; Adams, C.J.; Arpaia, M.L. Paternal and maternal effects on fruit and seed characteristics in cherimoya (Annona cherimola Mill.). Sci. Hortic. 1994, 59, 11–25. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef]
- Dick, C.W.; Pennington, R.T. History and Geography of Neotropical Tree Diversity. Annu. Rev. Ecol. Evol. Syst. 2019, 50, 279–301. [Google Scholar] [CrossRef]
- Baker, P.A.; Fritz, S.C.; Battisti, D.S.; Dick, C.W.; Vargas, O.M.; Asner, G.P.; Martin, R.E.; Wheatley, A.; Prates, I. Beyond Refugia: New Insights on Quaternary Climate Variation and the Evolution of Biotic Diversity in Tropical South America. In Neotropical Diversification: Patterns and Processes; Springer: Berlin/Heidelberg, Germany, 2020; pp. 51–70. [Google Scholar] [CrossRef]
- Larranaga, N.; Fontecha, G.; Albertazzi, F.J.; Palmieri, M.; Hormaza, J.I. Amplification of Cherimoya (Annona cherimola Mill.) with Chloroplast-Specific Markers: Geographical Implications on Diversity and Dispersion Studies. Horticulturae 2022, 8, 807. [Google Scholar] [CrossRef]


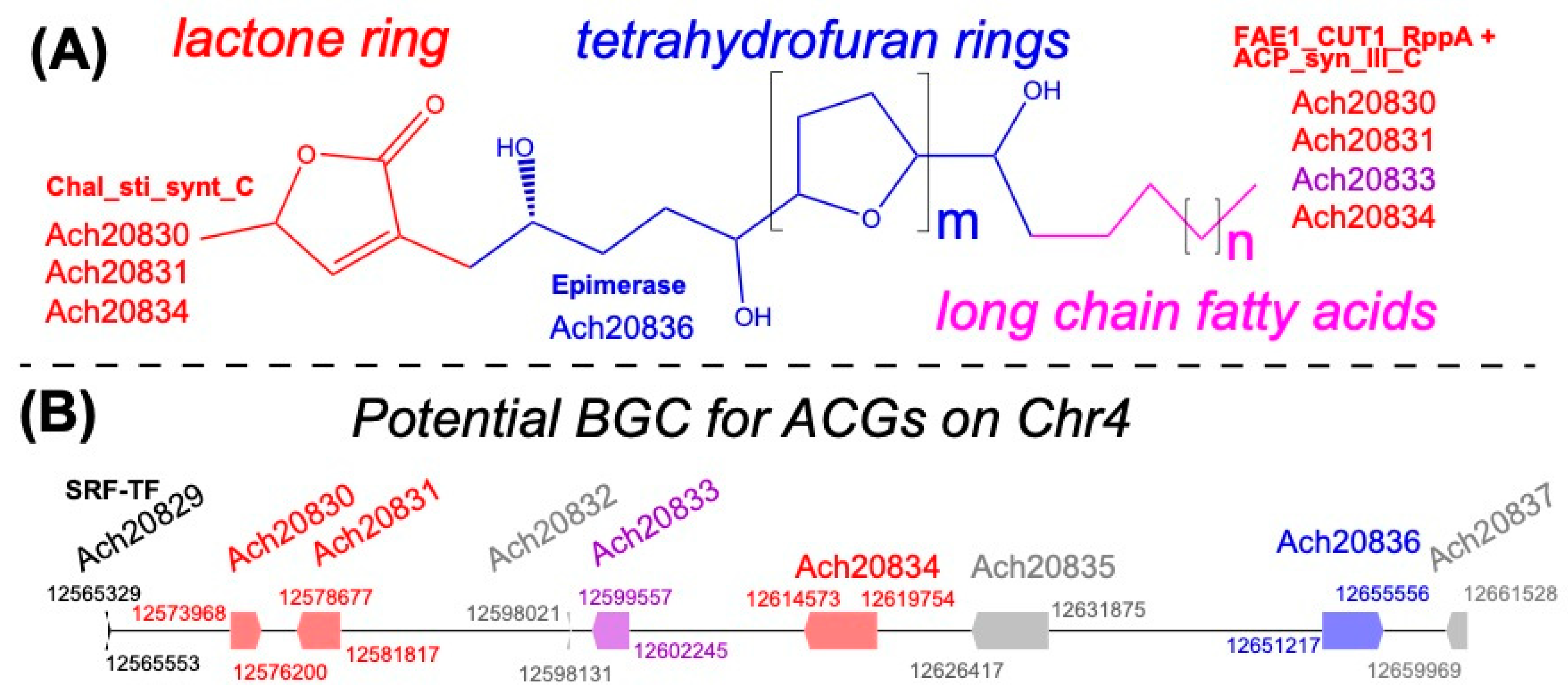

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
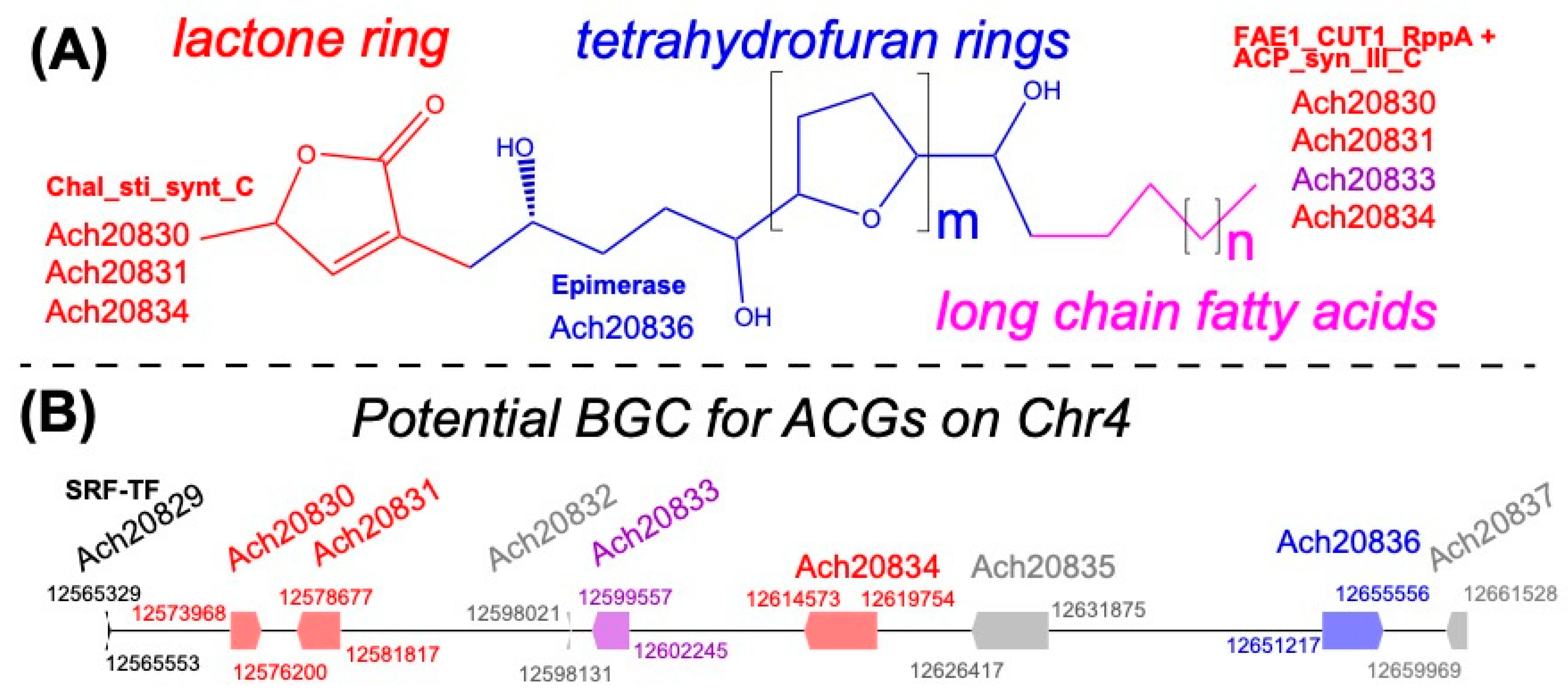{kind=link}
| Assembly | A. cherimola “Booth” | A. cherimola “Fino de Jete” * | A. muricata * |
|---|---|---|---|
| Total length (bp) | 794,023,491 | 1,137,394,475 | 656,813,740 |
| Number of scaffolds | 1377 | 2052 | 755 |
| Number of Chromosomes | 7 | 7 | 7 |
| Longest scaffold (bp) | 128,576,476 | 212,253,197 | 122,620,176 |
| Scaffolds N50 (bp) | 97,591,913 | 170,859,109 | 93,205,713 |
| GC content % | 35.25 | 34.69 | 40.07 |
| Complete BUSCOs % | 98.43 | 93.0 | - |
| Annotation | |||
| Repeat sequences % | 68.23 | 64.96 | 54.87 |
| Number of protein-coding genes | 45,272 (≥30 aa) # | 41,413 (≥50 aa) | 23,375 (≥100 aa) |
| Number of genes with annotation | 32,377 | - | 22,769 |
| Complete BUSCOs % | 92.9 | 90.9 | 92.14 |
| Species | CNL | CN | NL | TNL | TN | TX | NBS | Other |
|---|---|---|---|---|---|---|---|---|
| Mof | 125 | 28 | 97 | 0 | 1 | 6 | 18 | 0 |
| Mbi | 124 | 40 | 113 | 0 | 1 | 4 | 66 | 0 |
| Lch | 93 | 67 | 90 | 0 | 3 | 9 | 68 | 2 |
| Ach | 183 | 22 | 171 | 0 | 2 | 4 | 57 | 0 |
| Lcu | 64 | 20 | 51 | 3 | 5 | 9 | 29 | 0 |
| Cka | 174 | 32 | 128 | 2 | 3 | 9 | 31 | 0 |
| Pam | 6 | 4 | 22 | 1 | 0 | 6 | 13 | 0 |
| Csa | 81 | 23 | 53 | 0 | 1 | 6 | 25 | 4 |
| Afi | 15 | 1 | 12 | 0 | 2 | 3 | 2 | 0 |
| Pni | 115 | 43 | 168 | 0 | 7 | 2 | 203 | 0 |
| Cca | 267 | 43 | 330 | 3 | 2 | 4 | 76 | 2 |
| Ath | 39 | 1 | 21 | 75 | 14 | 34 | 5 | 15 |
| Vvi | 213 | 50 | 120 | 71 | 15 | 27 | 72 | 14 |
| Nnu | 71 | 11 | 40 | 21 | 3 | 6 | 16 | 5 |
| Sbi | 110 | 17 | 115 | 0 | 3 | 1 | 26 | 0 |
| Osa | 190 | 44 | 143 | 0 | 2 | 3 | 51 | 0 |
| Mac | 51 | 5 | 30 | 0 | 3 | 5 | 12 | 0 |
| Peq | 17 | 2 | 20 | 0 | 2 | 3 | 9 | 0 |
| Atr | 17 | 16 | 37 | 13 | 7 | 7 | 36 | 3 |
| Gbi | 33 | 2 | 67 | 91 | 36 | 16 | 9 | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zheng, J.; Nousias, O.; Yan, Y.; Meinhardt, L.W.; Goenaga, R.; Zhang, D.; Yin, Y. The American Cherimoya Genome Reveals Insights into the Intra-Specific Divergence, the Evolution of Magnoliales, and a Putative Gene Cluster for Acetogenin Biosynthesis. Plants 2024, 13, 636. https://doi.org/10.3390/plants13050636
Li T, Zheng J, Nousias O, Yan Y, Meinhardt LW, Goenaga R, Zhang D, Yin Y. The American Cherimoya Genome Reveals Insights into the Intra-Specific Divergence, the Evolution of Magnoliales, and a Putative Gene Cluster for Acetogenin Biosynthesis. Plants. 2024; 13(5):636. https://doi.org/10.3390/plants13050636
Chicago/Turabian StyleLi, Tang, Jinfang Zheng, Orestis Nousias, Yuchen Yan, Lyndel W. Meinhardt, Ricardo Goenaga, Dapeng Zhang, and Yanbin Yin. 2024. "The American Cherimoya Genome Reveals Insights into the Intra-Specific Divergence, the Evolution of Magnoliales, and a Putative Gene Cluster for Acetogenin Biosynthesis" Plants 13, no. 5: 636. https://doi.org/10.3390/plants13050636
APA StyleLi, T., Zheng, J., Nousias, O., Yan, Y., Meinhardt, L. W., Goenaga, R., Zhang, D., & Yin, Y. (2024). The American Cherimoya Genome Reveals Insights into the Intra-Specific Divergence, the Evolution of Magnoliales, and a Putative Gene Cluster for Acetogenin Biosynthesis. Plants, 13(5), 636. https://doi.org/10.3390/plants13050636