
Development and Assessment of SNP Genotyping Arrays for Citrus and Its Close Relatives


Abstract
:1. Introduction
2. Results
2.1. Variant Selection According to In Silico Analysis of Re-Sequencing Data
2.2. Axiom® Citrus HD Genotyping Array
2.3. PHR Probes on the Axiom® Citrus HD Genotyping Array AX1 and the Axiom® Citrus HD Genotyping Array AX2
2.4. Axiom® Citrus Genotyping Array
2.5. PHR Probes on the Axiom® Citrus Genotyping Array
2.6. Chromosome Distribution and Functional Annotation of the Axiom® Citrus HD Genotyping Array and the Axiom® Citrus Genotyping Array Markers
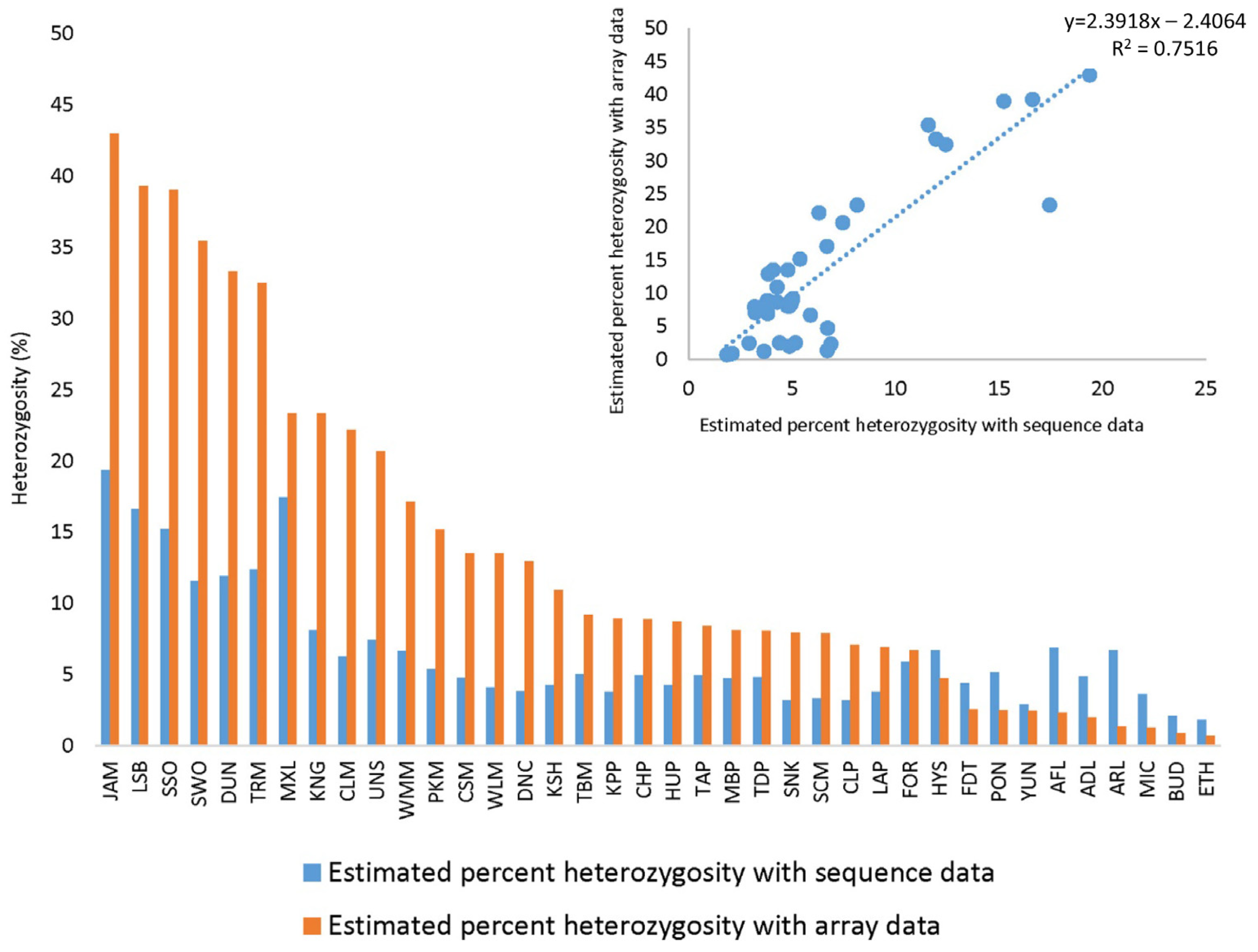
2.7. Heterozygosity and Reproducibility of Genotyping Calls between Sequencing Data and the Axiom® Citrus HD Genotyping Array
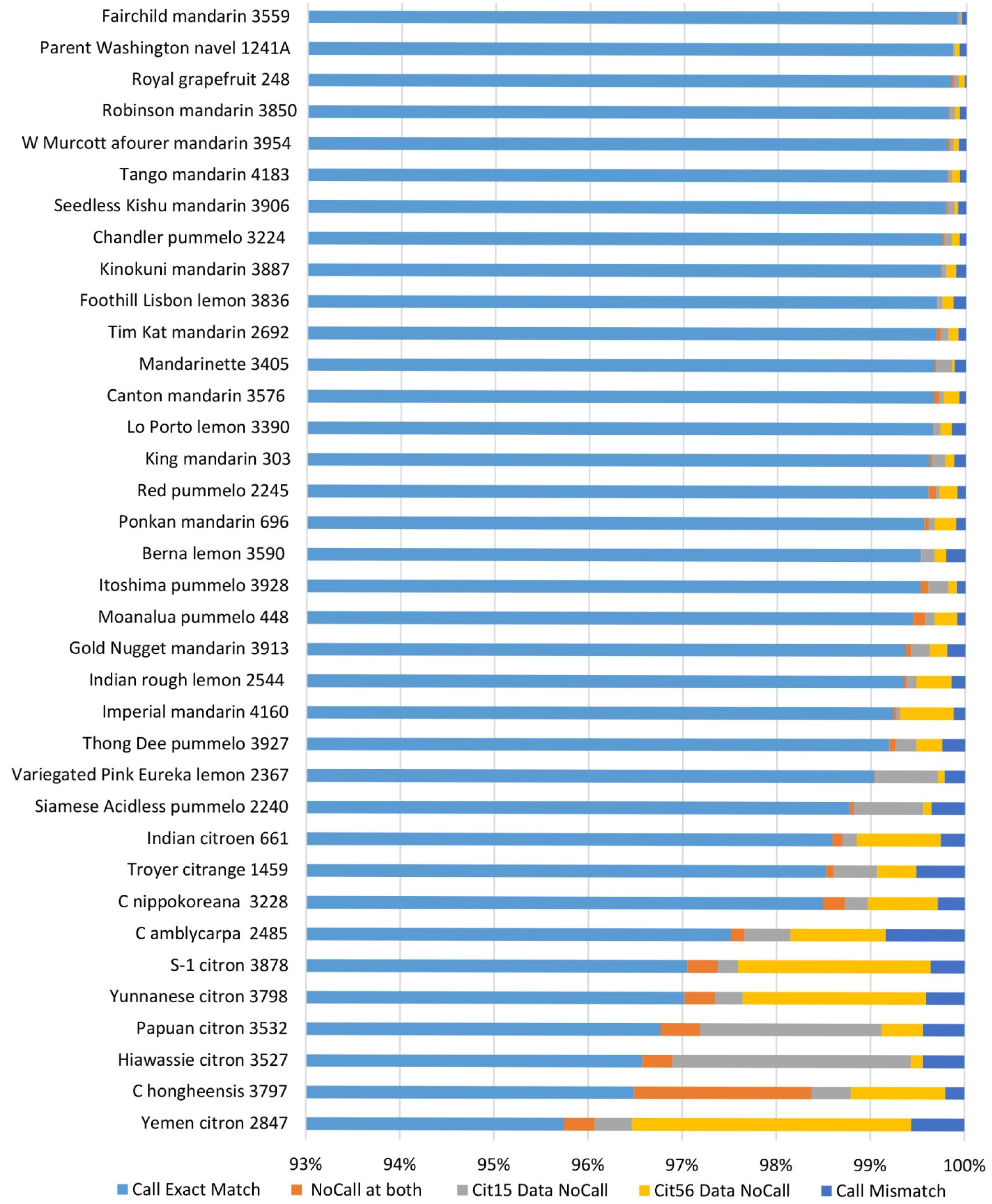
2.8. Genotyping Accuracy and Reproducibility of Genotyping Calls of Axiom® Citrus HD Genotyping Array and Axiom® Citrus Genotyping Array
2.9. Percent Identity of Accessions in Groups Composed of Clonal Accessions
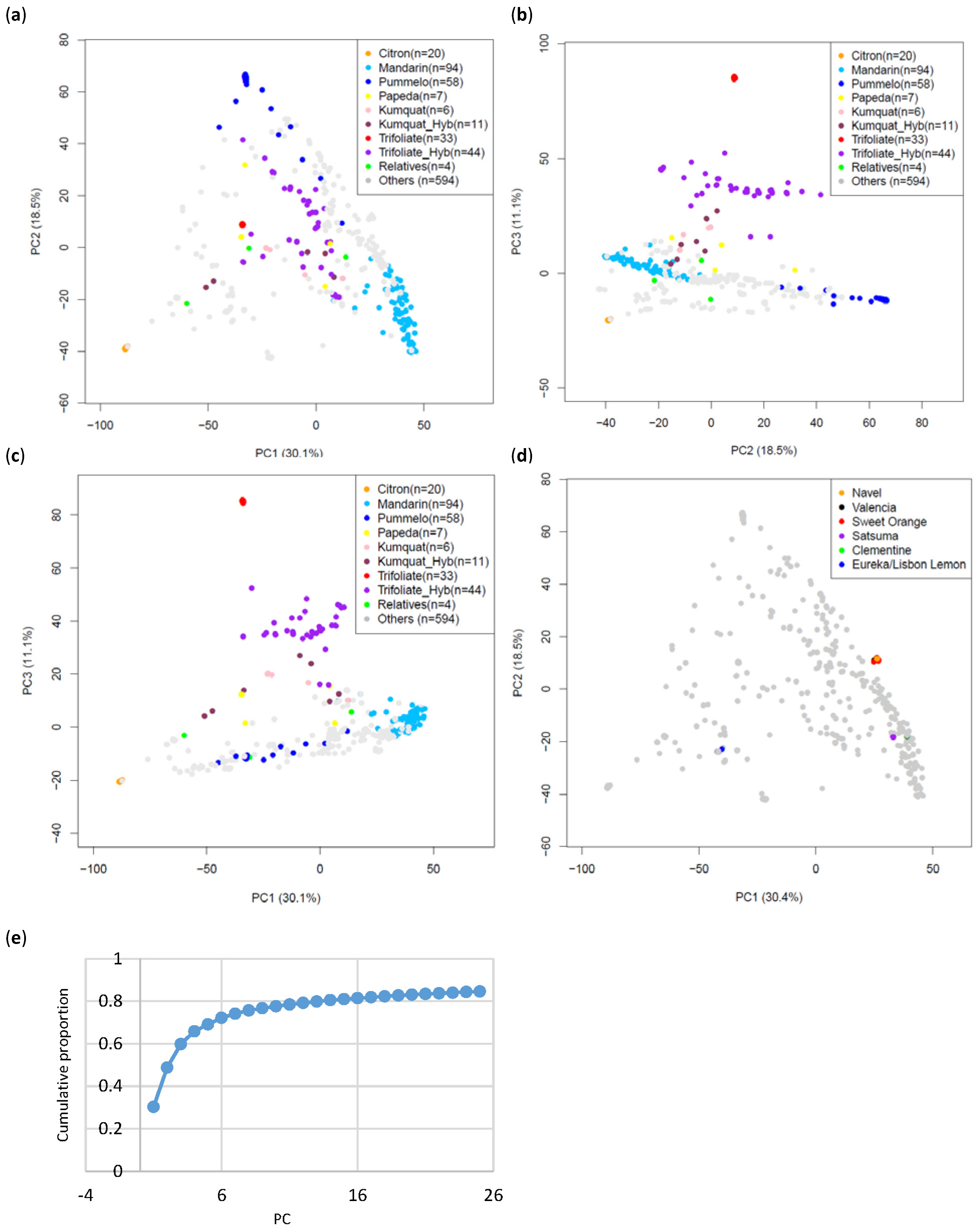
2.10. PCA
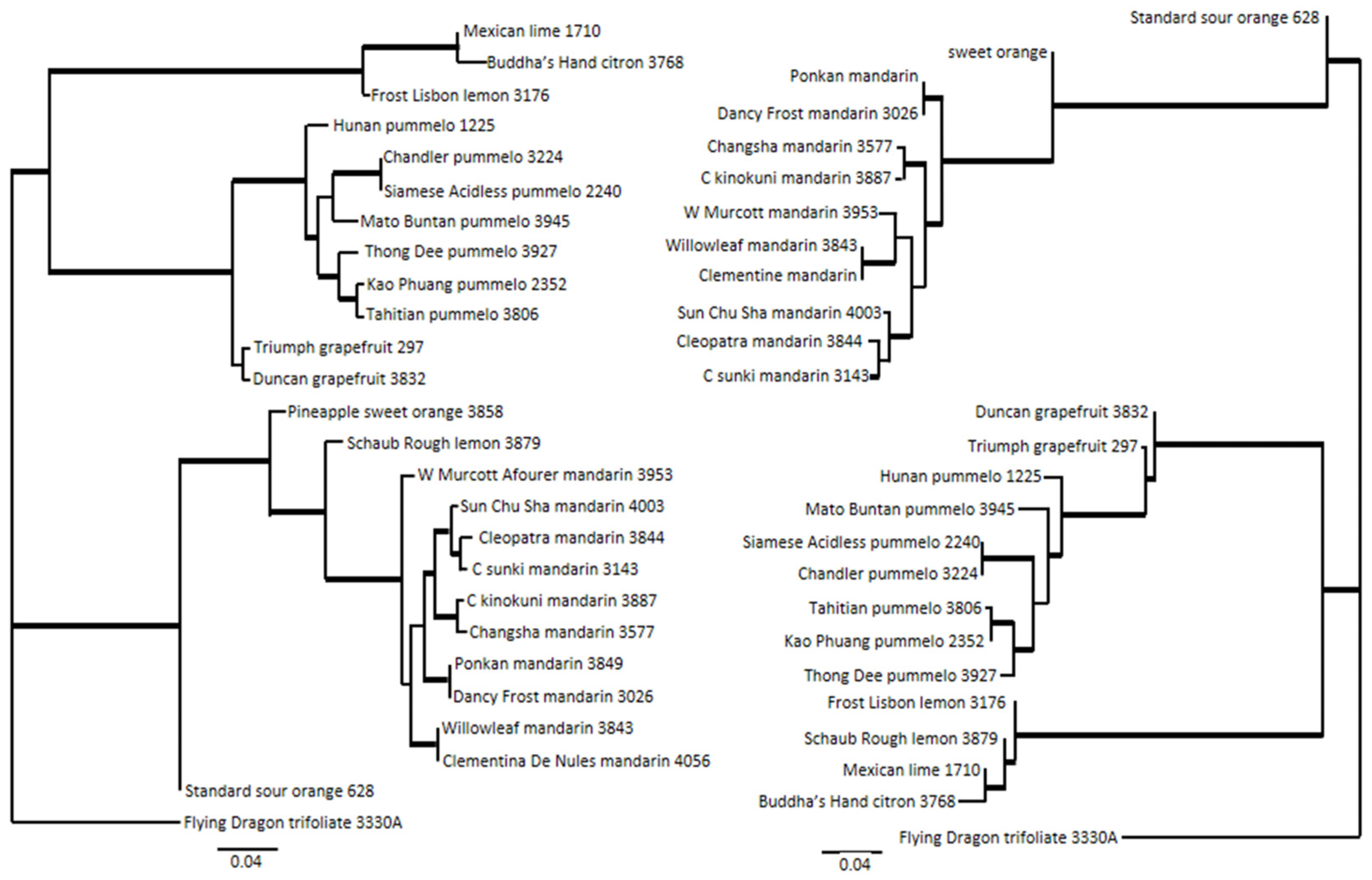
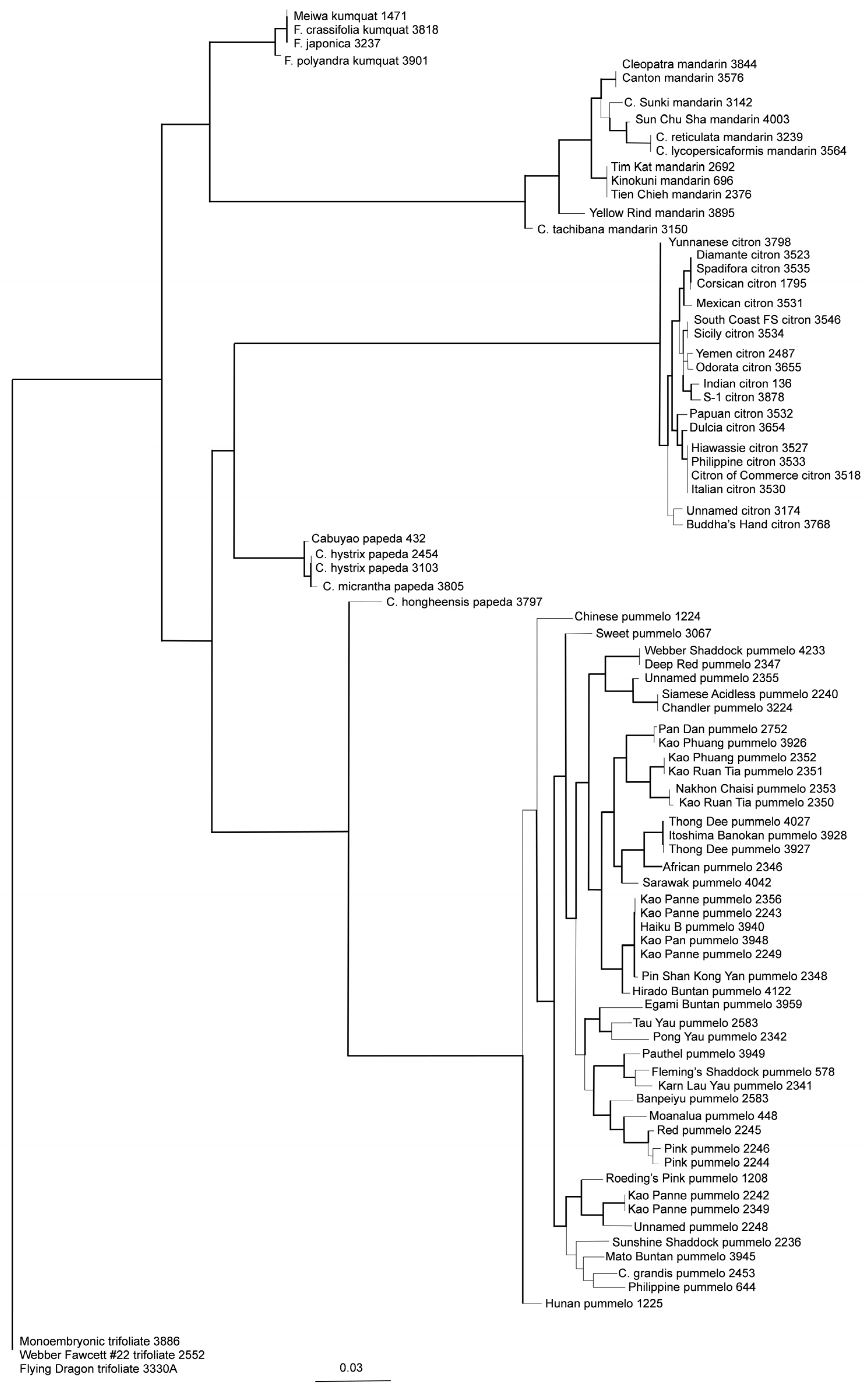
2.11. Phylogenetic Inference
3. Discussion
3.1. Accuracy of Array Data in Citrus and Citrus Relatives
3.2. Population Structure
3.3. Inference of Citrus Phylogeny with Array Data
3.4. Inference of Phylogeny of 87 Non-Admixed Accessions of Citrus and Close Relatives
4. Materials and Methods
4.1. Sample Preparation for Array Hybridization
4.2. Variant Selection According to In Silico Analysis of Re-Sequencing Data
4.3. Variant Filtering Part 1
4.3.1. In Silico Analysis of Variants
4.3.2. SNP Validation Panel
4.4. Variant Filtering Part 2
- To design an array that accurately genotypes SNPs in both Citrus and its close relatives, we performed variant filtering part 2. In analysis 1, SNP genotyping calls for 254 genomic DNA samples analyzed with Axiom® Citrus HD Genotyping Array were made using Axiom® Analysis Suite 1.1.1.66 by Affymetrix™ at Axiom® BestPractice workflow default setting (default DQC, QC, and % passing) to identify “PolyHighResolution” (PHR) probes. PHR probes are the most informative and accurate probe performance class on the Axiom® platform based on the genotype call cluster separation, cluster variance, and cluster position.
- In analysis 2, SNP genotyping calls of 254 diploid genomic DNA samples were made for PHR probe loci identified in analysis 1 using Axiom® Analysis Suite at lowered DQC, QC, and % passing thresholds (=0) to allow Axiom® Analysis Suite to analyze all the diploid samples including citrus relatives which otherwise failed the analysis due to their divergence from the majority of the samples being analyzed. DQC and QC probes are used by the Affymetrix™ Axiom® platform as quality controls. DQC probes are 31-mer sequences that are non-polymorphic in the variant discovery panel, and QC probes are the best-performing probes selected by Affymetrix™ in silico analysis. Among the 20,000 QC probes, the number of probes polymorphic in various groups were 15,539 in Group 1 only, 786 in Group 2 only, 120 in Group 3 only, 1181 in Groups 1 and 2, 487 in Groups 1 and 3, 78 in Groups 2 and 3, and 1809 polymorphic in all three groups (Figure S13).
- R package SNPolisher (SNPolisher, v1.5.2, Affymetrix™) was used to perform the “BalleleFreq test”, which is a post-processing function to check if there is a shift in intensity in probes across samples. When samples are processed at the same time, an intensity shift in a small number of probes in some of the samples can, in fact, cause a sample to be assigned to the wrong cluster. BalleleFreq test is performed by comparing the SNP genotyping calls between analysis 1 and analysis 2 at the PHR probe loci in each individual analyzed. We then selected loci for which calls did not differ between analysis 1 and analysis 2. Analysis 1 identified 728,831 PHR probe loci that accurately genotype SNPs in Citrus and analysis 2 identified 369,269 PHR probe loci that accurately genotype SNPs in both Citrus and its relatives.
4.5. Heterozygosity and Reproducibility of Genotyping Data
4.6. PCA
4.7. Phylogenetic Inference
4.8. Simulation Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. FAO Statistical Yearbook: World Food and Agriculture; FAO: Roma, Italy, 2013. [Google Scholar]
- FAO. FAOstat. 2020. Available online: https://www.fao.org/faostat/ (accessed on 23 December 2023).
- United Nations. Food and Agriculture Organization of the United Nations (FAO). In Yearbook of the United Nations 1995 (UN); United Nations, Ed.; United Nations: New York, NY, USA, 1995; pp. 1469–1472. [Google Scholar]
- Tripoli, E.; Guardia, M.L.; Giammanco, S.; Majo, D.D.; Giammanco, M. Citrus flavonoids: Molecular structure, biological activity and nutritional properties: A review. Food Chem. 2007, 104, 466–479. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, L.L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.B.; Hao, B.H.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Swingle, W.; Reece, P. The botany of Citrus and its wild relatives. In The Citrus Industry; Leon Dexter Batchelor, L., Webber, H., Eds.; University California Press: Berkeley, CA, USA, 1967; pp. 190–430. [Google Scholar]
- Wu, G.A.; Prochnik, S.; Jenkins, J.; Salse, J.; Hellsten, U.; Murat, F.; Perrier, X.; Ruiz, M.; Scalabrin, S.; Terol, J.; et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 2014, 32, 656–662. [Google Scholar] [CrossRef]
- Nicolosi, E. Origin and taxonomy. In Citrus Genetics, Breeding and Biotechnology; Khan, I., Ed.; CAB International: Surrey, UK, 2007; pp. 19–43. ISBN 1845931939. [Google Scholar]
- Wu, G.A.; Terol, J.; Ibanez, V.; López-García, A.; Pérez-Román, E.; Borredá, C.; Domingo, C.; Tadeo, F.R.; Carbonell-Caballero, J.; Alonso, R.; et al. Genomics of the origin and evolution of Citrus. Nature 2018, 554, 311–316. [Google Scholar] [CrossRef]
- Minamikawa, M.F.; Nonaka, K.; Kaminuma, E.; Kajiya-Kanegae, H.; Onogi, A.; Goto, S.; Yoshioka, T.; Imai, A.; Hamada, H.; Hayashi, T.; et al. Genome-wide association study and genomic prediction in citrus: Potential of genomics-assisted breeding for fruit quality traits. Sci. Rep. 2017, 7, 4721. [Google Scholar] [CrossRef]
- Frost, H.B. Nucellar embryony and juvenile characters in clonal varieties of citrus. J. Hered. 1938, 29, 423–492. [Google Scholar] [CrossRef]
- Soost, R.K.; Cameron, J.W. Citrus. In Advances in Fruit Breeding; Janick, J., Moore, J.N., Eds.; Purdue University Press: West Lafayettena, IN, USA, 1975; pp. 507–540. [Google Scholar]
- Fang, D.; Krueger, R.R.; Roose, M.L. Phylogenetic relationships among selected Citrus germplasm accessions revealed by Inter-simple Sequence Repeat (ISSR) Markers. J. Am. Soc. Hortic. Sci. 1998, 123, 612–617. [Google Scholar] [CrossRef]
- Ruiz, C.; Paz Breto, M.; Asíns, M.J. A quick methodology to identify sexual seedlings in citrus breeding programs using SSR markers. Euphytica 2000, 112, 89–94. [Google Scholar] [CrossRef]
- Barkley, N.A.; Roose, M.L.; Krueger, R.R.; Federici, C.T. Assessing genetic diversity and population structure in a citrus germplasm collection utilizing simple sequence repeat markers (SSRs). Theor. Appl. Genet. 2006, 112, 1519–1531. [Google Scholar] [CrossRef]
- Kepiro, J.L.; Roose, M.L. AFLP markers closely linked to a major gene essential for nucellar embryony (apomixis) in Citrus maxima × Poncirus trifoliata. Tree Genet. Genomes 2010, 6, 1–11. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Y.; Liu, Z.; He, J.; Jiang, X.; He, F.; Lu, Z.; Yang, S.; Chen, P.; Yu, H.; et al. Somatic variations led to the selection of acidic and acidless orange cultivars. Nat. Plants. 2021, 7, 954–965. [Google Scholar] [CrossRef]
- Wu, B.; Yu, Q.; Deng, Z.; Duan, Y.; Luo, F.; Gmitter, F., Jr. A chromosome-level phased genome enabling allele-level studies in sweet orange: A case study on citrus Huanglongbing tolerance. Hortic. Res. 2023, 10, uhac247. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Zhang, S.; Cao, L.; Huang, Y.; Cheng, J.; Wu, G.; Tian, S.; Chen, C.; Liu, Y.; et al. Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 2017, 49, 765–772. [Google Scholar] [CrossRef]
- Yu, H.; Yang, X.; Guo, F.; Jiang, X.; Deng, X.; Xu, Q. Genetic diversity and population structure of pummelo (Citrus maxima) germplasm in China. Tree Genet. Genomes 2017, 13, 58. [Google Scholar] [CrossRef]
- Vonholdt, B.M.; Pollinger, J.P.; Lohmueller, K.E.; Han, E.; Parker, H.G.; Quignon, P.; Degenhardt, J.D.; Boyko, A.R.; Earl, D.A.; Auton, A.; et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 2010, 64, 898–902. [Google Scholar] [CrossRef]
- Hu, J.; Diao, Y.; Zhou, Y.; Lin, D.; Bi, Y.; Pang, Z.; Trout Fryxell, R.; Liu, X.; Lamour, K. Loss of heterozygosity drives clonal diversity of Phytophthora capsici in China. PLoS ONE 2013, 8, e82691. [Google Scholar] [CrossRef] [PubMed]
- Verde, I.; Bassil, N.; Scalabrin, S.; Gilmore, B.; Lawley, C.T.; Gasic, K.; Micheletti, D.; Rosyara, U.R.; Cattonaro, F.; Vendramin, E.; et al. Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS ONE 2012, 7, e35668. [Google Scholar] [CrossRef]
- Bianco, L.; Cestaro, A.; Sargent, D.J.; Banchi, E.; Derdak, S.; Di Guardo, M.; Salvi, S.; Jansen, J.; Viola, R.; Gut, I.; et al. Development and validation of a 20K single nucleotide polymorphism (SNP) whole genome genotyping array for apple (Malus × domestica Borkh). PLoS ONE 2014, 9, e110377. [Google Scholar] [CrossRef]
- Chen, H.; Xie, W.; He, H.; Yu, H.; Chen, W.; Li, J.; Yu, R.; Yao, Y.; Zhang, W.; He, Y.; et al. A high-density SNP genotyping array for rice biology and molecular breeding. Mol. Plant 2014, 7, 541–553. [Google Scholar] [CrossRef]
- Unterseer, S.; Bauer, E.; Haberer, G.; Seidel, M.; Knaak, C.; Ouzunova, M.; Meitinger, T.; Strom, T.M.; Fries, R.; Pausch, H.; et al. A powerful tool for genome analysis in maize: Development and evaluation of the high density 600 k SNP genotyping array. BMC Genom. 2014, 15, 823. [Google Scholar] [CrossRef]
- Bassil, N.V.; Davis, T.M.; Zhang, H.; Ficklin, S.; Mittmann, M.; Webster, T.; Mahoney, L.; Wood, D.; Alperin, E.S.; Rosyara, U.R.; et al. Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo-octoploid cultivated strawberry Fragaria × ananassa. BMC Genom. 2015, 16, 155. [Google Scholar] [CrossRef]
- Ganal, M.W.; Polley, A.; Graner, E.M.; Plieske, J.; Wieseke, R.; Luerssen, H.; Durstewitz, G. Large SNP arrays for genotyping in crop plants. J. Biosci. 2012, 37, 821–828. [Google Scholar] [CrossRef]
- Víquez-Zamora, M.; Vosman, B.; van de Geest, H.; Bovy, A.; Visser, R.G.; Finkers, R.; Van Heusden, A.W. Tomato breeding in the genomics era: Insights from a SNP array. BMC Genom. 2013, 14, 354. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xie, W.; Li, J.; Zhou, F.; Zhang, Q. A whole-genome SNP array (RICE6K) for genomic breeding in rice. Plant Biotechnol. J. 2014, 12, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Salhi-Hannachi, A.; Luro, F.; Vignes, H.; Mournet, P.; Ollitrault, P. Genotyping by sequencing reveals the interspecific C. maxima/C. reticulata admixture along the genomes of modern citrus varieties of mandarins, tangors, tangelos, orangelos and grapefruits. PLoS ONE 2017, 12, e0185618. [Google Scholar] [CrossRef]
- Curk, F.; Ancillo, G.; Ollitrault, F.; Perrier, X.; Jacquemoud-Collet, J.P.; Garcia-Lor, A.; Navarro, L.; Ollitrault, P. Nuclear species-diagnostic SNP markers mined from 454 amplicon sequencing reveal admixture genomic structure of modern citrus varieties. PLoS ONE 2015, 10, e0125628. [Google Scholar] [CrossRef] [PubMed]
- Scora, R.W. On the history and origin of Citrus. Bull. Torrey Bot. Club 1975, 102, 369–375. Available online: https://www.jstor.org/stable/2484763 (accessed on 23 December 2023).
- Barrett, H.C.; Rhodes, A.M. A numerical taxonomic study of affinity relationships in cultivated citrus and its close relatives. Syst. Bot. 1976, 1, 105–136. [Google Scholar] [CrossRef]
- Robinsons, O.; Dylus, D.; Dessimoz, C. The Japanese Society for the Promotion of Science. Mol. Biol. Evol. 2016, 33, 2163–2166. [Google Scholar] [CrossRef]
- Ganal, M.W.; Durstewitz, G.; Polley, A.; Bérard, A.; Buckler, E.S.; Charcosset, A.; Clarke, J.D.; Graner, E.M.; Hansen, M.; Joets, J.; et al. A large maize (Zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 2011, 6, e28334. [Google Scholar] [CrossRef]
- Chagné, D.; Crowhurst, R.N.; Troggio, M.; Davey, M.W.; Gilmore, B.; Lawley, C.; Vanderzande, S.; Hellens, R.P.; Kumar, S.; Cestaro, A.; et al. Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS ONE 2012, 7, e31745. [Google Scholar] [CrossRef]
- Muñoz-Amatriaín, M.; Mirebrahim, H.; Xu, P.; Wanamaker, S.I.; Luo, M.; Alhakami, H.; Alpert, M.; Atokple, I.; Batieno, B.J.; Boukar, O.; et al. Genome resources for climate-resilient cowpea, an essential crop for food security. Plant J. 2017, 89, 1042–1054. [Google Scholar] [CrossRef]
- Sim, S.C.; Durstewitz, G.; Plieske, J.; Wieseke, R.; Ganal, M.W.; Van Deynze, A.; Hamilton, J.P.; Buell, C.R.; Causse, M.; Wijeratne, S.; et al. Development of a large SNP genotyping array and generation of high-density genetic maps in tomato. PLoS ONE 2017, 7, e40563. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef]
- Li, Y.H.; Li, W.; Zhang, C.; Yang, L.; Chang, R.Z.; Gaut, B.S.; Qiu, L.J. Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol. 2010, 188, 242–253. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Wang Ming, W.M.; Yan JianBing, Y.J.; Zhao JiuRan, Z.J.; Song Wei, S.W.; Zhang XiaoBo, Z.X.; Xiao YanNong, X.Y.; Zheng YongLian, Z.Y. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 2012, 196, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wright, M.; Kimball, J.; Eizenga, G.; McClung, A.; Kovach, M.; Tyagi, W.; Ali, M.L.; Tung, C.W.; Reynolds, A.; et al. Genomic diversity and introgression in O. sativa reveal the impact of domestication and breeding on the rice genome. PLoS ONE 2010, 5, e10780. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Godoy, M.A.; Mauri, N.; Juarez, J.; Marques, M.C.; Santiago, J.; Forment, J.; Gadea, J. A genome-wide 20 K citrus microarray for gene expression analysis. BMC Genom. 2008, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Ollitrault, P.; Terol, J.; Garcia-Lor, A.; Bérard, A.; Chauveau, A.; Froelicher, Y.; Belzile, C.; Morillon, R.; Navarro, L.; Brunel, D.; et al. SNP mining in C. clementina BAC end sequences; transferability in the Citrus genus (Rutaceae), phylogenetic inferences and perspectives for genetic mapping. BMC Genom. 2012, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Shimada, T.; Nonaka, K.; Kita, M.; Kuniga, T.; Endo, T.; Ikoma, Y.; Omura, M. High-throughput genotyping in citrus accessions using an SNP genotyping array. Tree Genet. Genomes 2013, 9, 145–153. [Google Scholar] [CrossRef]
- Shimizu, T.; Kaminuma, E.; Nonaka, K.; Yoshioka, T.; Goto, S.; Matsumoto, T.; Katayose, Y.; Mochizuki, T.; Tanizawa, Y.; Toyoda, A.; et al. A genomic approach to selecting robust and versatile SNP sets from next-generation sequencing data for genome-wide association study in citrus cultivars. in III International Symposium on Citrus Biotechnology. Int. Soc. Hortic. Sci. 2014, 1135, 23–32. [Google Scholar] [CrossRef]
- Wu, G.A.; Sugimoto, C.; Kinjo, H.; Azama, C.; Mitsube, F.; Talon, M.; Gmitter Jr, F.G.; Rokhsar, D.S. Diversification of mandarin citrus by hybrid speciation and apomixis. Nat. Commun. 2021, 12, 4377. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Tanaka, T. Species Problem in Citrus; The Japanese Society for the Promotion of Science: Tokyo, Japan, 1954; pp. 1–99. [Google Scholar]
- Federici, C.T.; Fang, D.Q.; Scora, R.W.; Roose, M.L. Phylogenetic relationships within the genus (Rutaceae) and related genera as revealed by RFLP and RAPD analysis. Theor. Appl. Genet. 1998, 96, 812–822. [Google Scholar] [CrossRef]
- Nicolosi, E.; Deng, Z.N.; Gentile, A.; La Malfa, S.; Continella, G.; Tribulato, E. Citrus phylogeny and genetic origin of important species as investigated by molecular markers. Theor. Appl. Genet. 2000, 100, 1155–1166. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tominaga, S. High chromosomal variability of mandarins (Citrus spp.) revealed by CMA banding. Euphytica 2003, 129, 267. [Google Scholar] [CrossRef]
- Uzun, A.; Yesiloglu, T.; Aka-Kacar, Y.; Tuzcu, O.; Gulsen, O. Genetic diversity and relationships within Citrus and related genera based on sequence related amplified polymorphism markers (SRAPs). Sci. Hortic. 2009, 121, 306–312. [Google Scholar] [CrossRef]
- Penjor, T.; Yamamoto, M.; Uehara, M.; Ide, M.; Matsumoto, N.; Matsumoto, R.; Nagano, Y. Phylogenetic relationships of Citrus and its relatives based on matK gene sequences. PLoS ONE 2013, 8, e62574. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [PubMed]
- Dugrand-Judek, A.; Olry, A.; Hehn, A.; Costantino, G.; Ollitrault, P.; Froelicher, Y.; Bourgaud, F. The distribution of coumarins and furanocoumarins in Citrus species closely matches Citrus phylogeny and reflects the organization of biosynthetic pathways. PLoS ONE 2015, 10, e0142757. [Google Scholar] [CrossRef]
- Curk, F.; Ollitrault, F.; Garcia-Lor, A.; Luro, F.; Navarro, L.; Ollitrault, P. Phylogenetic origin of limes and lemons revealed by cytoplasmic and nuclear markers. Ann. Bot. 2016, 117, 565–583. [Google Scholar] [CrossRef]
- Hirai, M.; Kozaki, I.; Kajiura, I. Isozyme analysis and phylogenic relationship of citrus. Jpn. J. Breed. 1986, 36, 377–389. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kobayashi, S.; Nakamura, Y.; Yamada, Y. Phylogenic relationships of citrus revealed by RFLP analysis of mitochondrial and chloroplast DNA. Jpn. J. Breed. 1993, 43, 355–365. [Google Scholar] [CrossRef]
- de Araújo, E.F.; de Queiroz, L.P.; Machado, M.A. What is Citrus? Taxonomic implications from a study of cp-DNA evolution in the tribe Citreae (Rutaceae subfamily Aurantioideae). Org. Divers. Evol. 2003, 3, 55–62. [Google Scholar] [CrossRef]
- Yingzhi, L.; Yunjiang, C.; Nengguo, T.; and Xiuxin, D. Phylogenetic analysis of mandarin landraces, wild mandarins, and related species in China using nuclear LEAFY second intron and plastid trnL-trnF sequence. J. Am. Soc. Hortic. Sci. 2007, 132, 796–806. [Google Scholar] [CrossRef]
- Penjor, T.; Anai, T.; Nagano, Y.; Matsumoto, R.; Yamamoto, M. Phylogenetic relationships of Citrus and its relatives based on rbcL gene sequences. Tree Genet. Genomes 2010, 6, 931–939. [Google Scholar] [CrossRef]
- Garcia-Lor, A.; Curk, F.; Snoussi-Trifa, H.; Morillon, R.; Ancillo, G.; Luro, F.; Navarro, L.; Ollitrault, P. A nuclear phylogenetic analysis: SNPs, indels and SSRs deliver new insights into the relationships in the “true citrus fruit trees” group (Citrinae, Rutaceae) and the origin of cultivated species. Ann. Bot. 2012, 111, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ajit Uchoi, A.U.; Malik, S.K.; Ravish Choudhary, R.C.; Susheel Kumar, S.K.; Rohini, M.R.; Digvender Pal, D.P.; Ercİslİ, S.; Rekha Chaudhury, R.C. Inferring phylogenetic relationships of Indian citron (Citrus medica L.) based on rbcL and matK sequences of chloroplast DNA. Biochem. Genet. 2016, 54, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Butelli, E.; Garcia-Lor, A.; Licciardello, C.; Las Casas, G.; Hill, L.; Recupero, G.R.; Keremane, M.L.; Ramadugu, C.; Krueger, R.; Xu, Q.; et al. Changes in anthocyanin production during domestication of Citrus. Plant Physiol. 2017, 173, 2225–2242. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Przeworski, M. Linkage disequilibrium in humans: Models and data. Am. J. Hum. Genet. 2001, 69, 1–14. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage disequilibrium—Understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 29, 477–485. [Google Scholar] [CrossRef]
- Roose, M.L. Mapping and marker assisted selection in Citrus. In Citrus Genetics, Breeding and Biotechnology; Khan, I.A., Ed.; CAB International: Surrey, UK, 2007; pp. 275–286. ISBN 1845931939. [Google Scholar]
- Bausher, M.G.; Singh, N.D.; Lee, S.B.; Jansen, R.K.; Daniell, H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: Organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 2006, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Guo, H.; Wang, X.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 162. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP 3.2 Manual; University of California Herbarium: Berkeley, CA, USA, 1989. [Google Scholar]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2010, 27, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of genomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Variety Name | Genus and Species Name | CRC | Group |
|---|---|---|---|---|
| MXL | Mexican lime | Citrus aurantifolia Swingle | 1710 | 1 |
| JAM | Schaub rough lemon | Citrus jambhiri Lush. | 3879 | 1 |
| KSH | Mukakukishu (Kinokuni mandarin) | Citrus kinokuni Hort. ex Tanaka | 3887 | 1 |
| LSB | Frost nucellar Lisbon lemon | Citrus limon L. Burm. f. | 3176 | 1 |
| TDP | Thong Dee pummelo | Citrus maxima (Burm.) Merr | 3927 | 1 |
| CPP | Cariappa CM3 pummelo | Citrus maxima (Burm.) Merr. | RSD2013003 | 1 |
| HUP | Hunan pummelo | Citrus maxima (Burm.) Merr. | 1225 | 1 |
| KPP | Kao Phuang pummelo | Citrus maxima (Burm.) Merr. | 2352 | 1 |
| MBP | Mato Buntan pummelo | Citrus maxima (Burm.) Merr. | 3945 | 1 |
| TAP | Tahitian pummelo | Citrus maxima (Burm.) Merr. | 3806 | 1 |
| DUN | Duncan grapefruit | Citrus paradisi Macf. | 3832 | 1 |
| TRM | Triumph grapefruit | Citrus paradisi Macf. | 297 | 1 |
| CLP | Cleopatra mandarin | Citrus reshni Hort. ex Tanaka | 3844 | 1 |
| CSM | Changsha mandarin | Citrus reticulata Blanco | 3577 | 1 |
| DNC | Dancy mandarin | Citrus reticulata Blanco | 3026 | 1 |
| SCM | Sun Chu Sha Kat mandarin | Citrus reticulata Blanco | 4003 | 1 |
| SNK | Sunki mandarin | Citrus sunki Hort. ex Tanaka | 3143 | 1 |
| TBM | Tachibana orange | Citrus tachibana Tanaka | 3150 | 1 |
| ETH | Ethrog citron | Citrus medica L. | 3891 | 2 |
| YUN | Yunnanese citron | Citrus medica L. | 3798 | 2 |
| BUD | Buddha’s Hand citron (fingered citron) | Citrus medica var. sarcodactylis (Hoola van Nooten) Swingle | 3768 | 2 |
| QIN | Persistent Stigma—OPS citron | Citrus medica L. | RSD2012014 | 2 |
| HYS | Mauritius papeda | Citrus hystrix D. C. | 3103 | 3 |
| MIC | Small-fruited papeda | Citrus micrantha var.microcarpa Wester | 3605 | 3 |
| ADL | Australian desert lime | Eremocitrus glauca (Lindl.) Swingle | 3463 | 3 |
| FOR | Nagami kumquat | Fortunella margarita (Lour.) Swingle | 3877 | 3 |
| AFL | Red pulp finger lime | Microcitrus australasica var. sanguinia (F. Muell.) Swingle | 1484 | 3 |
| ARL | Australian round lime | Microcitrus australis (A. Cunn. Ex Mudie) Swingle | 3673 | 3 |
| FDT | Flying Dragon trifoliate | Poncirus trifoliata (L.) Raf. | 3330A | 3 |
| PON | Pomeroy trifoliate | Poncirus trifoliata (L.) Raf. | 1717 | 3 |
| Genotype ID | Variety Name | Genus Species Name | Group |
|---|---|---|---|
| BAN * | Banpeiyu | Citrus maxima (Burm.) Merr. | 1 |
| CHP | Chandler pummelo | Citrus maxima (Burm.) Merr. | 1 |
| CLM | Clementine mandarin | Citrus clementina hort. ex Tanaka | 1 |
| KNG * | King mandarin | Citrus nobilis Lour. | 1 |
| LAP | Low acid pummelo | Citrus maxima (Burm.) Merr. | 1 |
| PKM | Ponkan mandarin | Citrus reticulata Blanco | 1 |
| SSO | Sour orange | Citrus aurantium L. | 1 |
| SWO | Sweet orange | Citrus sinensis (L.) Osbeck | 1 |
| UNS * | Satsuma mandarin | Citrus unshiu Marcovitch | 1 |
| WLM | Willowleaf mandarin | Citrus reticulata Blanco | 1 |
| WMM | W Murcott mandarin | Citrus reticulata Blanco | 1 |
| (a) | |||
| Probe Count | |||
| Linkage Group | Size (Mb) | Axiom® Citrus HD Genotyping Array | Axiom® Citrus Genotyping Array |
| 1 | 28.94 | 137,783 | 6289 |
| 2 | 36.38 | 171,732 | 7773 |
| 3 | 51.05 | 232,654 | 10,572 |
| 4 | 25.65 | 132,515 | 6259 |
| 5 | 43.3 | 152,556 | 6665 |
| 6 | 25.61 | 120,276 | 5446 |
| 7 | 21.13 | 115,841 | 4961 |
| 8 | 25.11 | 107,769 | 4608 |
| 9 | 31.41 | 125,607 | 5360 |
| Chloroplast | 0.16 | 1002 | 500 |
| (b) | |||
| SNP Count | |||
| Phytozome Annotation | Axiom® Citrus HD Genotyping Array | Axiom® Citrus Genotyping Array | |
| Overlap with START Codons | 336 | 8 | |
| Overlap with STOP Codons | 38 | 24 | |
| Overlap with Splice Sites | 4815 | 236 | |
| Intergenic | 665,912 | 20,144 | |
| Gene Body | 630,821 | 38,289 | |
| Introns | 245,120 | 14,787 | |
| Exons | 385,701 | 23,502 | |
| mRNA | 630,821 | 38,289 | |
| CDS | 291,849 | 19,550 | |
| UTR | 93,852 | 3952 | |
| Transcription Start Site | 275 | 9 | |
| Total Probes | Autosomal SNPs | Chloroplast SNPs | SNP Density (SNP/kb) | PHR | |
|---|---|---|---|---|---|
| Axiom® Citrus HD Genotyping Array | 1,404,442 | 1,296,733 | 1002 | 3.9 | 728,831 |
| Axiom® Citrus Genotyping Array | 58,433 | 57,933 | 500 | 0.16 | 51,296 |
| Species | CVC Type | Axiom® Citrus HD Genotyping Array | Axiom® Citrus Genotyping Array |
|---|---|---|---|
| C. sinensis (Hyb) | Blood orange | 3 | 17 |
| C. sinensis (Hyb) | Navel orange | 6 | 73 |
| C. sinensis (Hyb) | Sweet orange | 3 | 71 |
| C. sinensis (Hyb) | Valencia orange | 4 | 26 |
| C. medica | Citron | 12 | 20 |
| (C. reticulata × C. medica) | Rough lemon | 6 | 15 |
| (C. micrantha × C. medica) | Lime | 5 | 26 |
| (C. reticulata × C. medica) | Rangpur lime | 3 | 17 |
| Fortunella spp. | Kumquat | 2 | 6 |
| (Hyb) | Calamondin | 1 | 3 |
| C. limon (C. aurantium × C. medica) | Lemon | 12 | 49 |
| C. limon | Eureka lemon | 2 | 14 |
| C. limon | Lisbon lemon | 3 | 13 |
| C. reticulata | Mandarin | 35 | 94 |
| C. clementina (Hyb) | Clementine | 3 | 16 |
| C. unshiu (Hyb) | Satsuma | 3 | 45 |
| (Hyb) | Tangelo | 3 | 29 |
| (Hyb) | Tangor | 5 | 18 |
| C. ichangensis, C. micrantha, etc. | Papeda | 3 | 7 |
| C. maxima | Pummelo | 25 | 58 |
| (C. maxima × C. sinensis) | Grapefruit | 6 | 34 |
| C. aurantium (C. maxima × C. reticulata) | Sour orange | 5 | 26 |
| P. trifoliata | Trifoliate | 6 | 33 |
| (C. sinensis × P. trifoliata) | Citrangelo | 2 | 24 |
| C. paradisi × P. trifoliata) | Citrumelo | 1 | 8 |
| (Hyb) | Hybrids | 7 | 76 |
| Unknown | 30 | 55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiraoka, Y.; Ferrante, S.P.; Wu, G.A.; Federici, C.T.; Roose, M.L. Development and Assessment of SNP Genotyping Arrays for Citrus and Its Close Relatives. Plants 2024, 13, 691. https://doi.org/10.3390/plants13050691
Hiraoka Y, Ferrante SP, Wu GA, Federici CT, Roose ML. Development and Assessment of SNP Genotyping Arrays for Citrus and Its Close Relatives. Plants. 2024; 13(5):691. https://doi.org/10.3390/plants13050691
Chicago/Turabian StyleHiraoka, Yoko, Sergio Pietro Ferrante, Guohong Albert Wu, Claire T. Federici, and Mikeal L. Roose. 2024. "Development and Assessment of SNP Genotyping Arrays for Citrus and Its Close Relatives" Plants 13, no. 5: 691. https://doi.org/10.3390/plants13050691
APA StyleHiraoka, Y., Ferrante, S. P., Wu, G. A., Federici, C. T., & Roose, M. L. (2024). Development and Assessment of SNP Genotyping Arrays for Citrus and Its Close Relatives. Plants, 13(5), 691. https://doi.org/10.3390/plants13050691