Abstract
WUSCHEL-related homeobox (WOX) genes are a class of plant-specific transcription factors, regulating the development of multiple tissues. However, the genomic characterizations and expression patterns of WOX genes have not been analyzed in lotus. In this study, 15 NnWOX genes were identified based on the well-annotated reference genome of lotus. According to the phylogenetic analysis, the NnWOX genes were clustered into three clades, i.e., ancient clade, intermediate clade, and WUS clade. Except for the conserved homeobox motif, we further found specific motifs of NnWOX genes in different clades and divergence gene structures, suggesting their distinct functions. In addition, two NnWOX genes in the ancient clade have conserved expression patterns and other NnWOX genes exhibit different expression patterns in lotus tissues, suggesting a low level of functional redundancy in lotus WOX genes. Furthermore, we constructed the gene co-expression networks for each NnWOX gene. Based on weighted gene co-expression network analysis (WGCNA), ten NnWOX genes and their co-expressed genes were assigned to the modules that were significantly related to the cotyledon and seed coat. We further performed RT-qPCR experiments, validating the expression levels of ten NnWOX genes in the co-expression networks. Our study reveals comprehensive genomic features of NnWOX genes in lotus, providing a solid basis for further function studies.
1. Introduction
WUSCHEL-related homeobox (WOX) genes are a plant-specific transcription factor family, belonging to the homeobox superfamily [1,2]. The proteins of WOX genes contain a common homeobox domain, a helix–turn–helix structure consisting of 60–66 amino acids, which was a key regulatory element recognized by specific DNA sequences and regulated target gene expression at different stages of plant development [3,4]. According to most phylogenetic analysis, WOX genes were divided into three separate clades, modern/WUS clade (WC), intermediate clade (IC), and ancient clade (AC) [5]. The gradual expansion trend of the WOX gene number was approximately matched with the continuous evolution from lower plants to higher plants. Additionally, the last common ancestors of WOX proteins were identified in green algae [6].
Recent genetics and molecular biology investigations of WOX genes have indicated the vital roles of WOX members in various physiological and developmental processes in plants [7,8,9]. Since Vollbrecht et al. first found the Knotted-1 gene (i.e., WOX gene) that contained a homeodomain contributed to the cell fate determination in a maize mutant [10], an increasing number of studies have focused on the biological functions of WOX members in different plant tissues. In particular, a total of 15 WOX genes have been identified in the model eudicot plant Arabidopsis thaliana [11,12]. Several AtWOX genes are essential for multiple processes of tissue formation, including AtWUS and PRS1/AtWOX3 in the shoot apical meristem (STM) [13,14], AtWOX2 in the egg cell and zygote [15], AtWOX6 in the ovule development [16], STIP/AtWOX9 in STM, the root and other aerial organs [17], and AtWOX13 in floral transition [18]. In a model rice plant, the interaction of OsWOX11 and SLG2 determined grain width and affected quality traits [19]. Variation in the gene expression level of OsWUS negatively affects the phenotypic traits in rice [20]. The expression patterns of OsWOX genes were significantly divergent in reproductive organs, vegetative tissues, and multiple tissue-specific expressed OsWOX genes where they were identified [21]. The genome-wide identifications of the WOX gene family in non-model plants were conducted on various crops [22,23,24,25,26]. Furthermore, due to the changing expression level, WOX genes help plants respond to abiotic stress, indicating a more complex regulatory network of WOX genes under environmental stress. For example, a complex consisting of WOX and FRUCTOSE INSENSITIVE1 proteins regulated the expression level of downstream genes where Arabidopsis responds to fructose signaling during growth and development [27]. A reduction in WOX gene expression in the roots of tobacco seedlings grown under induction of alanine–glutamine acid–asparagine acid–leucine increases the differentiation of stem cells and leads to root elongation [28]. However, only a few studies have constructed the gene regulatory network of WOX genes, which limits further exploration of the complex interactions of WOX genes in plants.
Nelumbo nucifera, also called sacred lotus, is a considerable aquatic crop widely planted in Asia. With specific purposes of artificial domestication, the cultural lotuses were divided into three taxonomic groups, i.e., rhizome, seed, and flower lotus [29]. Based on the well-assembled reference genome of lotus [30], recent transcriptomic and proteomic research on sacred lotus was carried out to reveal the molecular mechanism of regulating the development of tissues [31,32,33]. According to previous studies, a single whole-genome duplication event occurred in ancestors of the N. nucifera during the K-Pg boundary [30], similar to the basal eudicot Amborella trichopoda [34]. Notably, the Nelumbo genome database including multiple tissue RNA-seq samples provided a superb platform for lotus investigation [35]. Several transcription factor families were identified in lotus, such as MADS-box [36], WRKY [37], and YABBY [38]. Although a previous study conducted a preliminary analysis of the WOX gene family in lotus [39], the interactions of WOX genes is still largely unknown. Therefore, we describe WOX genes systematically in N. nucifera and construct the co-expressed networks to find vital interactions regarding NnWOX genes during plant growth and development. This study consists of several parts, including the identification of WOX family members in N. nucifera, phylogenetic tree analysis, chromosomal localization, gene structure, conserved domain analysis, synteny and duplicated gene analysis, transcriptomic analysis, and gene co-expression analysis.
2. Results
2.1. Identification and Characterization of NnWOX Genes
To obtain a comprehensive WOX gene family in lotus, three identification methods were performed (see Section 4). The orthologs of A. thaliana WOX genes that were downloaded from the TAIR database (https://www.arabidopsis.org/ (accessed on 1 July 2023)), genes annotated to WOX-related homeobox in the Nelumbo Genome Database (http://nelumbo.cngb.org/nelumbo/ (accessed on 1 July 2023)), and genes identified under WOX transcription factors in the PlantTFDB were merged into candidate NnWOX genes in lotus. Given the nature of conservative homeobox domain in WOX sequences, the candidate NnWOX genes were mapped to three genome databases (i.e., NCBI-CDD, SMART, and Pfam) to filter out the false-positive candidate. Finally, a total of 15 high-confidence NnWOX genes were identified and used for downstream analysis (Table 1). The length of these NnWOX proteins varied from 182 to 366 amino acids. The molecular weight gradually increased with the length of proteins. Therefore, the longest NnWOX protein has a maximum weight of 40,294.14 kDa and the shortest NnWOX protein is the lightest with a weight of 20,640.22 kDa in all NnWOXs. Also, the chemical and physical characteristics of NnWOX genes were analyzed, including theoretical PI values, instability index, aliphatic index, and grand average of hydropathicity (Table 1). Furthermore, the hydrophily and hydrophobicity of amino acids in each NnWOX gene sequence were analyzed (Figure S1). Our results indicate low levels of hydrophobicity and high levels of hydrophily in all NnWOX proteins. Except for Nn5g30993, 14 NnWOX genes have at least one N-glycosylation site (Figure S2). Interestingly, the longest Nn1g06358 does not have the most N-glycosylation sites, but the second longest Nn3g18264 does, which could result from the obvious differences in sequences.

Table 1.
Physicochemical properties of NnWOX proteins in Nelumbo nucifera.
2.2. Phylogenetic Analysis
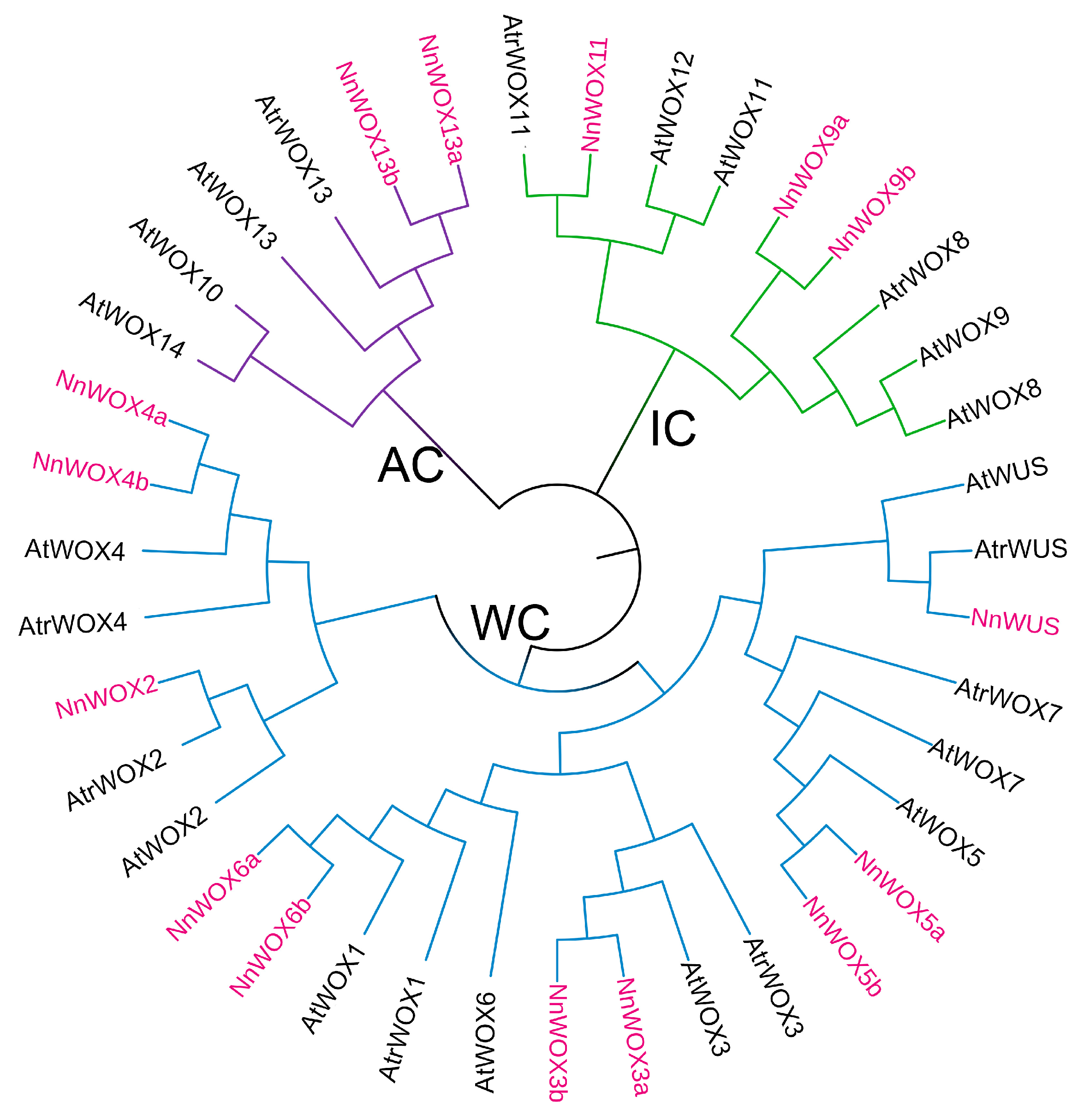
Using the same identification pipeline of the WOX gene in the lotus genome, a total of nine WOX genes were identified in a basal dicotyledon Amborella trichopoda. WOX proteins from A. thaliana and A. trichopoda were selected to construct the phylogenetic tree (Figure 1). Combining the orthologous mapping results and the clade of the phylogenetic tree, 15 NnWOX and 9 AtrWOX genes were named according to their closest orthologs to AtWOX. Similar to the previous study [6], these WOX proteins were classified into three clades, i.e., the ancient clade (AC), intermediate clade (IC), and WUS clade (WC) (Figure 1). Of which, the WC clade has the most NnWOX genes and the AC clade has only two NnWOX genes (i.e., NnWOX13a, NnWOX13b). Additionally, three NnWOX genes belong to the IC clade. Notably, lotus and A. thaliana have the same number of NnWOX genes, but some AtWOX genes have no orthologs in lotus, such as AtWOX8, AtWOX9, and AtWOX10. This could be as a result of these two species undergoing different whole-genome duplication events.

Figure 1.
Phylogenetic tree of WOX proteins in N. nucifera, A. thaliana, and A. trichopoda. This tree could be divided into three clades, including ancient clade (AC, purple), intermediate clade (IC, green), and WUS clade (WC, blue).
2.3. Gene Structure Analysis of NnWOXs
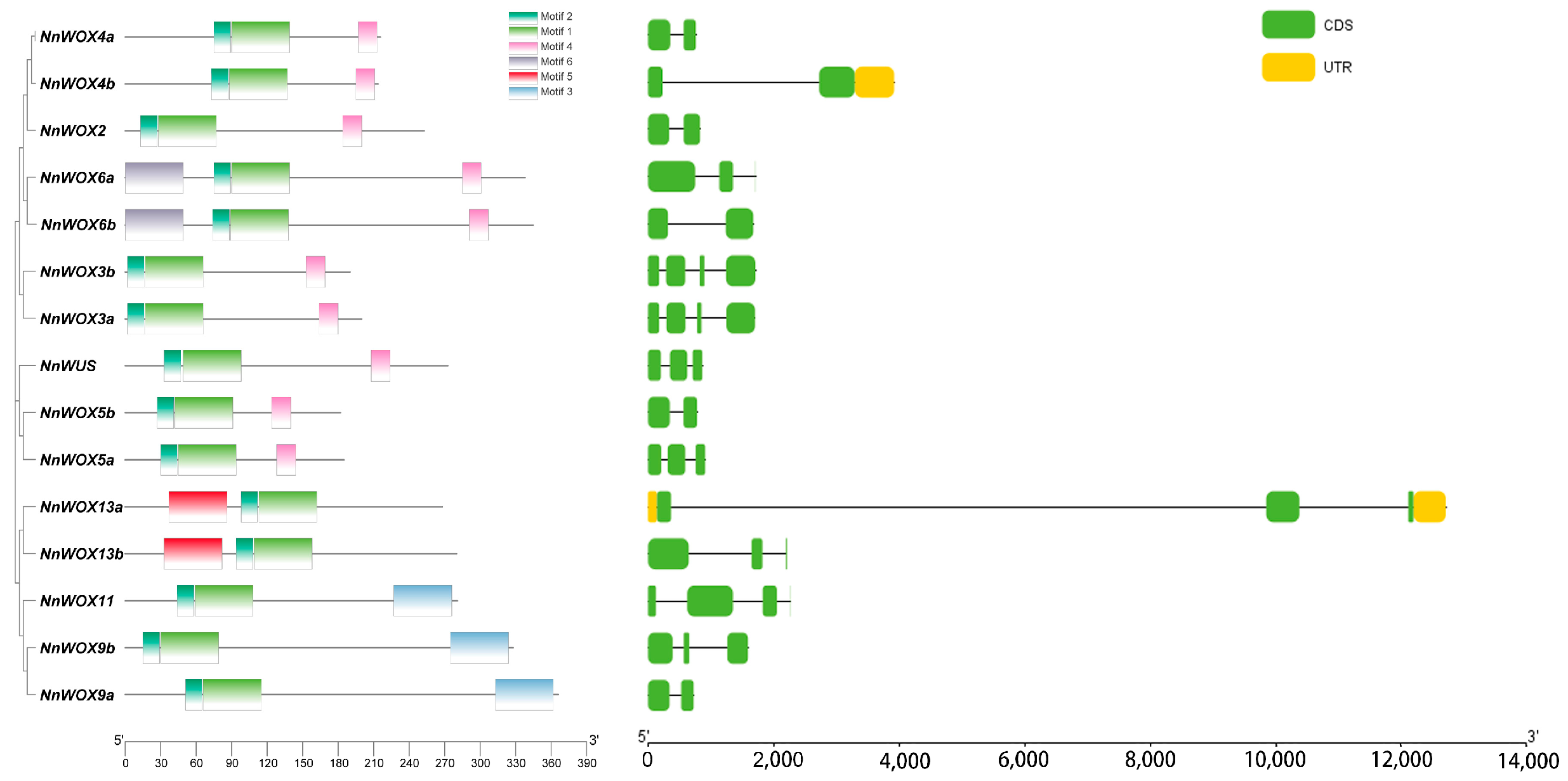
The motifs, mathematical–statistical models of specific sequences, were predicted for NnWOX proteins. Our results showed that all NnWOX members have motif1 and motif2 near the carboxy terminus, which consisted of the conserved HB domain and was consistent with the specific motif in the WOX gene family (Figure 2). Notably, we found that NnWOX genes clustered in the same clade shared the same motifs. For example, NnWOX9a, NnWOX9b, and NnWOX11 belonging to the IC clade have motif3, and motif5 was only identified in NnWOX genes from the AC clade (i.e., NnWOX13a and NnWOX13b) (Figure 2). A total of nine NnWOX genes belonging to the WC clade have the same motif4, excluding NnWOX6a and NnWOX6b that have motif6. The distinct sequence motifs in clade members were associated with their different functions.

Figure 2.
Characterizations of the identified WOXs in N. nucifera. The left panel shows the conserved motif location and the right panel shows the gene structures (black line: intron; green box: coding regions; yellow box: untranslated regions).
We analyzed the gene structures of these candidate proteins based on the locus annotation in the lotus genome for further exploration of genomic characteristics of NnWOX proteins. The exon number of NnWOX genes varied from two to four (Figure 2). NnWOX13a has the longest genome locus, which was significantly longer than other NnWOX genes (Figure 2). The untranslated regions (UTRs) were identified in NnWOX13a and NnWOX4b (Figure 2). Interestingly, we did not find similar gene structures of NnWOX genes belonging to the same clade, indicating large variations in lotus genome structure. Based on the well-annotated functions of AtWOX genes and the homologous relationships between NnWOX and AtWOX genes, we deduced that specific motifs and gene structures had a vital role in distinguishing the functions of WOX genes.
2.4. Localization and Duplicated Gene Analysis
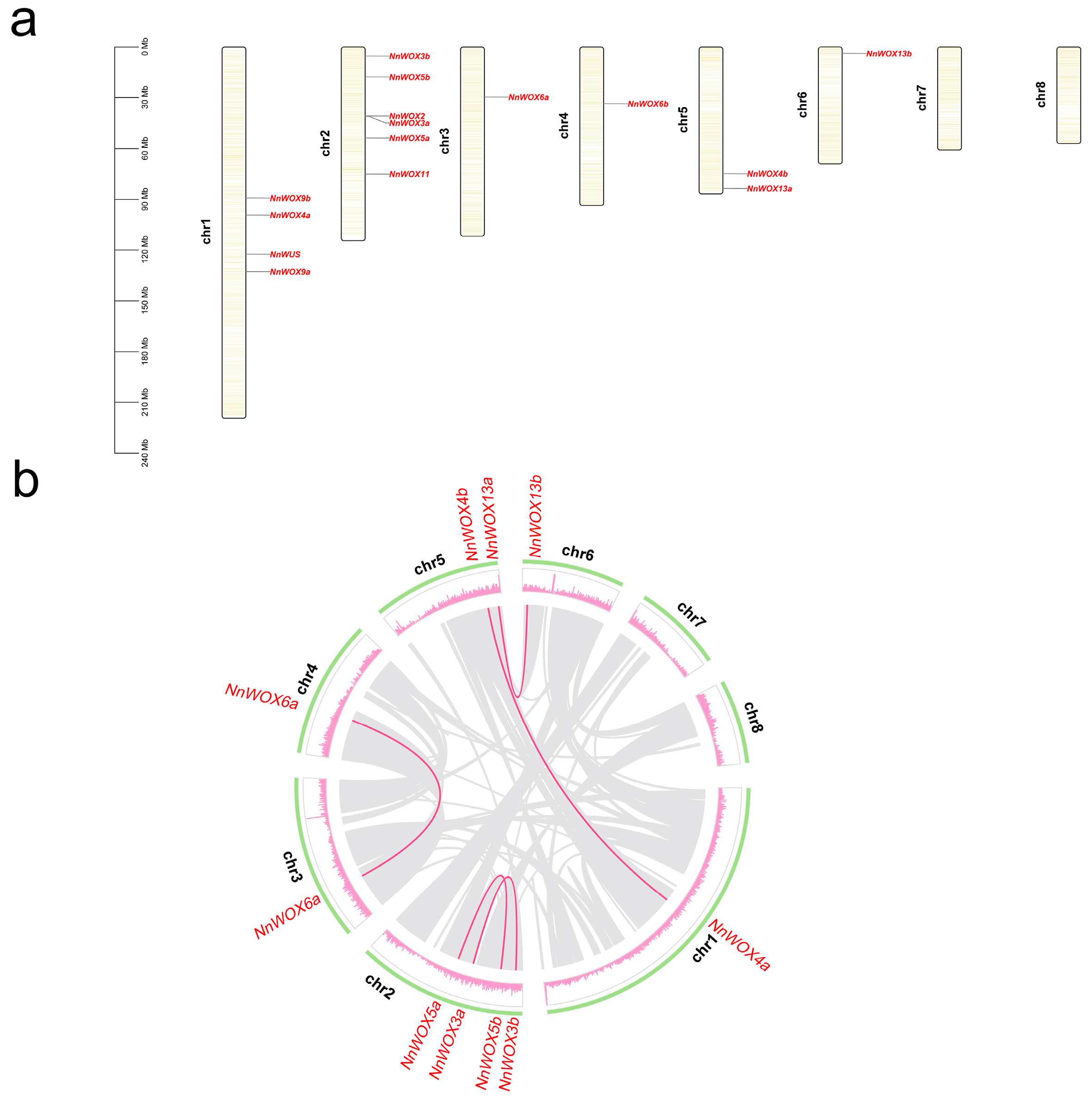
The position of NnWOX genes was annotated to the pseudochromosome in the lotus genome and visualized using TBtools (Figure 3a). The 15 NnWOX genes were located on six lotus chromosomes, except for Chr7 and Chr8 (Figure 3a). Chr2 has the most NnWOX genes (six), followed by Chr1 with four NnWOX genes. Chr3, Chr4, and Chr5 have one NnWOX gene each. The physical distance between NnWOX2 and NnWOX3a was the shortest compared to other NnWOX genes (Figure 3a). Furthermore, we found that NnWOX genes in the same clade of the phylogenetic tree are likely not located in the same chromosome.

Figure 3.
Distribution of WOXs in N. nucifera genome. (a) Chromosome distribution of NnWOXs. The scale on the left is in megabases (Mb). (b) Circle graph showing the location of genes in N. nucifera genome. The green circle represents the chromosomes, and the pink circle represents the gene density on the N. nucifera genome. Grey links show the collinearity regions in the lotus genome, and pink lines show the duplicated pairs of NnWOX.
Due to lotus occurring in one ancient whole-genome duplication event [30], we investigated the duplicated type of NnWOX genes. According to MCScanX, a total of five whole-genome duplicated pairs of NnWOX genes were identified, and all these duplications belonged to the WGD/segmental duplication type. Among these five duplications, three duplicated gene pairs were in different chromosomes, including NnWOX4a/NnWOX4b, NnWOX6a/NnWOX6b, and NnWOX13a/NnWOX13b (Figure 3b). And two duplicated gene pairs were in the same chromosome, i.e., NnWOX3a/NnWOX3b and NnWOX5a/NnWOX5b (Figure 3b). Moreover, no duplicates were identified for the other five NnWOX genes. We speculated that the duplicates were lost due to the redundancy functions or genome variations during the evolution of lotus.
2.5. Gene Expression Pattern Analysis of NnWOX Genes
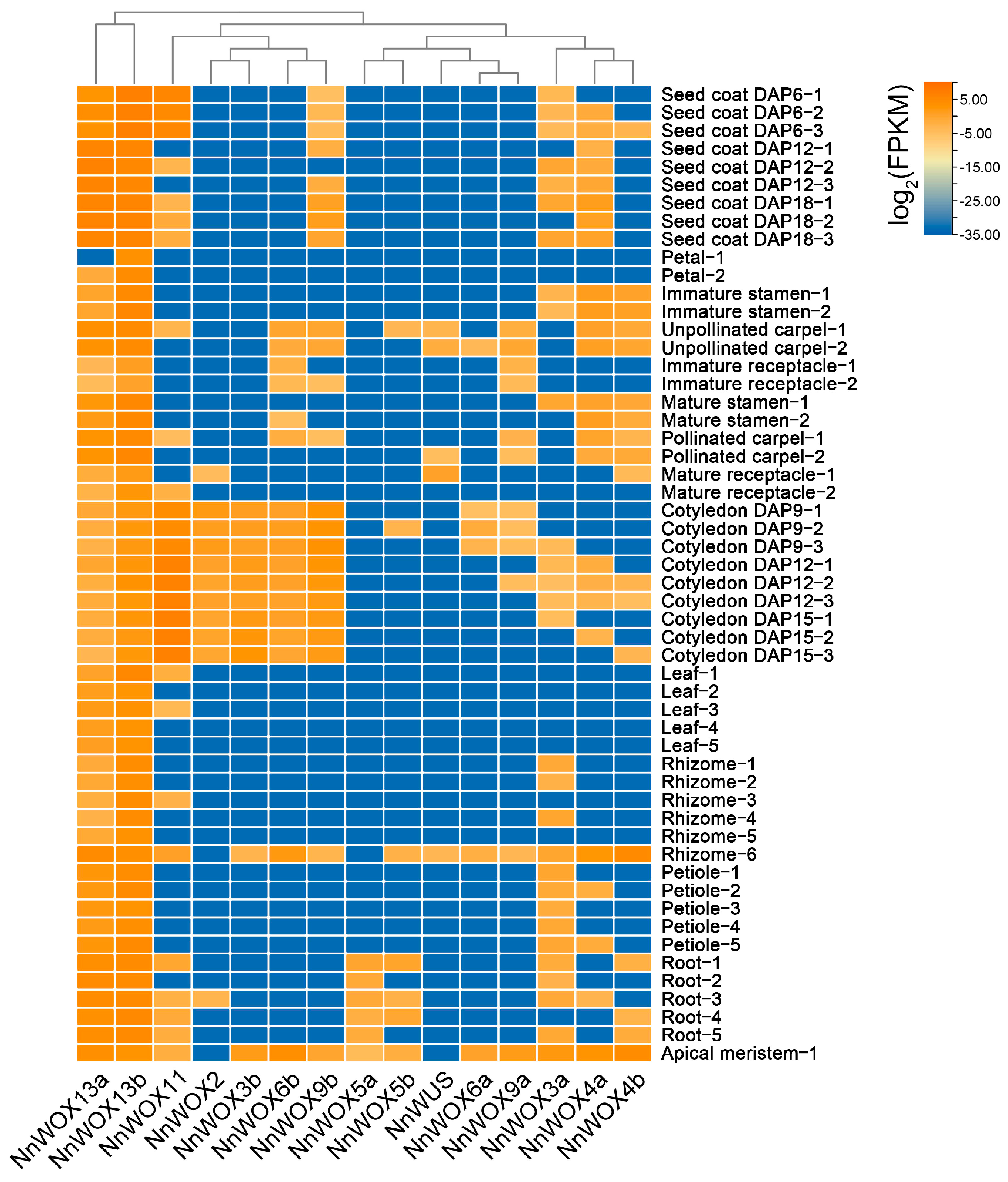
To gain insight into the functions of NnWOX genes during lotus development, we investigated their expression patterns in different tissues. The gene expression matrix of China Antique including 54 RNA-seq samples from 11 tissues was downloaded from the Nelumbo Genome Database [35]. The expression patterns of NnWOX genes are shown in Figure 4. Notably, we found that NnWOX13a and NnWOX13b have a wide tissue expression pattern, suggesting that NnWOX genes in the AC clade have multiple biological functions and play roles in different tissues. According to the heatmap cluster, the two NnWOX genes in the AC clade were clustered again, and the IC and WC members were reconstructed. This highlights the wide and vital roles of ancient WOX genes during lotus growth and development. Except for these two NnWOX genes, other NnWOX genes have tissue-specific bias expression patterns (Figure 4). For example, NnWOX2, NnWOX3b, NnWOX6b, and NnWOX9b have specifically high expression levels in cotyledon, and NnWOX5b was only expressed in root and apical meristem (Figure 4). Interestingly, few NnWOX genes were expressed in vegetative growth tissues, such as leaf, petiole, and rhizome. However multiple NnWOX genes show high expression levels in reproductive growth tissues, including stamen, carpel, and cotyledon. Therefore, NnWOX genes in IC and WC clades might contribute to the formation of reproductive tissues by targeting and interacting with other genes that regulate the reproductive processes.

Figure 4.
Expression patterns of NnWOXs in 54 tissue samples.
We found distinct expression patterns between WOX duplicates in the IC and WC clades. NnWOX3a has a wider tissue expression pattern compared to NnWOX3b, and NnWOX3b has an obvious expression bias to cotyledon (Figure 4). Compared to the duplicate NnWOX6b, NnWOX6a lacks expression in the cotyledon and immature receptacles. NnWOX9b also has a higher expression level in seed coat and cotyledon than its duplication NnWOX9a (Figure 4). Due to these obviously distinct expression patterns of duplicate WOX genes in tissues, we speculate that different functions between duplicate WOX genes were formed by the neofunctionalization during the evolution of lotus.
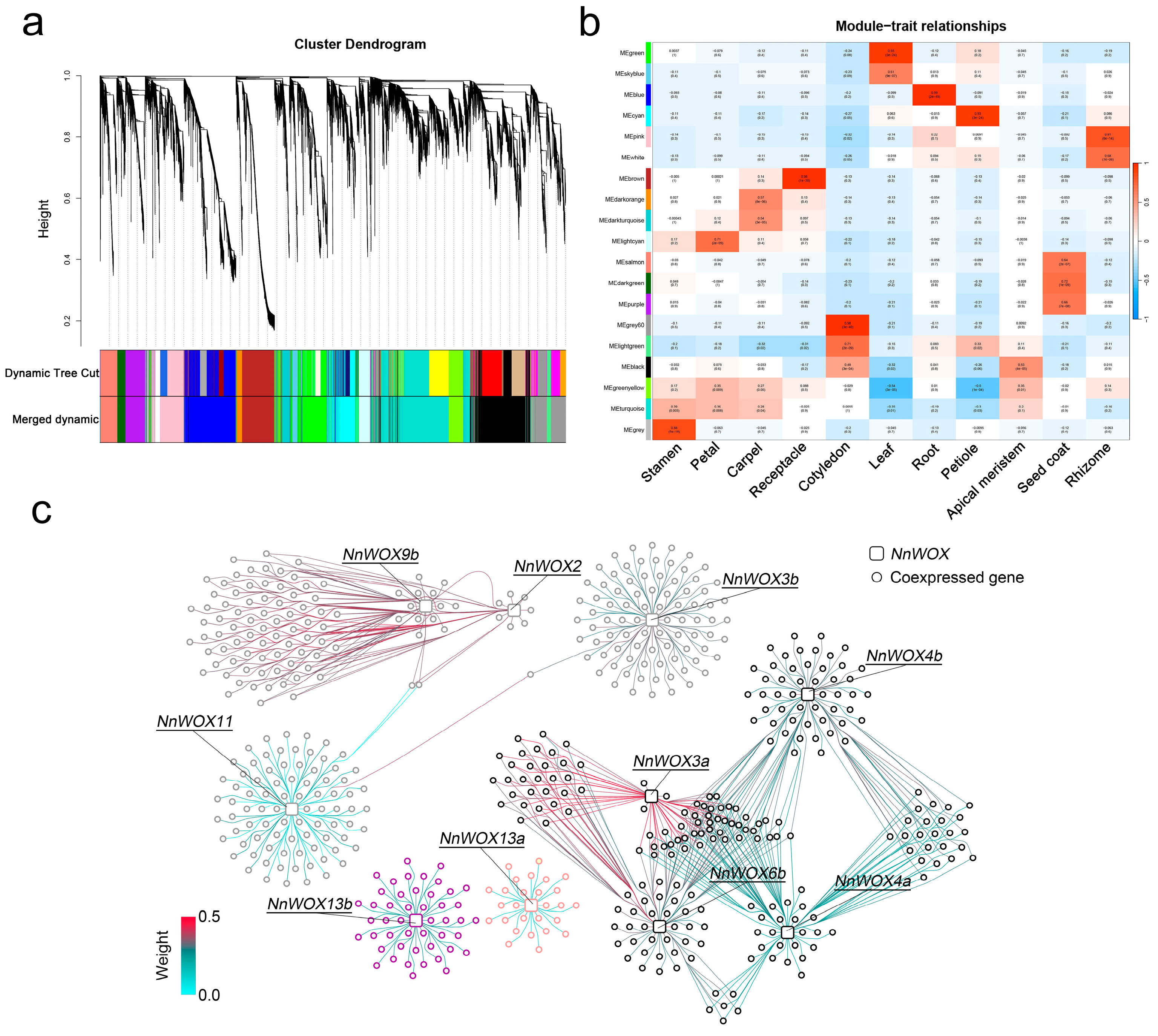
2.6. Co-Expressed Gene Networks of NnWOXs
To further explore the regulatory relationships of NnWOX genes in lotus tissues, a weight gene co-expression network analysis (WGCNA) was conducted on the filtered gene expression matrix. Due to the WGCNA gathering genes that are highly interconnected into a module, which is significantly (p < 0.001) related to the traits, the genes within similar biological processes or regulatory relationships are supposed to be identified in the same module. After excluding the silenced genes among 54 tissue samples, a total of 25,432 genes were aggregated into 19 color-coded modules (Figure 5a,b). Apart from black and turquoise modules being significantly related to two tissues, most modules were significantly related to one tissue (Figure 5b). For example, MEgrey60 was significantly related to cotyledon, and MEsalmon and MEpurple were significantly related to seed coat.

Figure 5.
Co-expression network analysis of NnWOXs. (a) Hierarchical cluster tree and color bands indicating 19 modules. (b) Heatmap showing the correlation of modules and tissues. The color in each cell represents the correlation coefficient of the module in the row and tissue in the column. The value in cells shows the p-value. (c) Co-expression networks for each NnWOX gene. Squares represent the NnWOX genes, circles represent the co-expressed genes. The colors of genes are represented in the module, and those of the lines indicate the weight value between co-expressed genes.
Based on the pipeline of constructing gene co-expression networks, ten NnWOX genes were filtered out and assigned to four modules, including MEgrey60, MEblack, MEsalmon, and MEpurple (Figure 5b,c). Among them, NnWOX13a and NnWOX13b, two AC clade members, were assigned to MEsalmon and MEpurple modules showing significant correlation with seed coat (Figure 5b,c). Four NnWOX genes, i.e., NnWOX2, NnWOX3b, NnWOX9b, and NnWOX11, were clustered into the MEgrey60 module and significantly associated with cotyledon (Figure 5b,c). Furthermore, NnWOX3a, NnWOX4a, NnWOX4b, and NnWOX6b were assigned to the MEblack module, which was relevant to both the cotyledon and the apical meristem (Figure 5b,c). We filtered the significantly co-expressed genes to NnWOX genes based on a strict threshold (see Section 4). The GO enrichment analysis of these co-expressed genes suggested biological functions enriched in reproductive and transcriptional regulatory processes, such as “floral development”, “DNA-binding transcription factor activity”, and “transcription regulator activity” (Figure S3). These co-expressed genes may be regulated by NnWOX and play a key role in the reproductive development of lotus.
To further explore the interaction of NnWOX and co-expressed genes, the gene co-expression networks for each NnWOX gene were graphed (Figure 5c). We found that genes in the same module have distinct co-expression patterns. For example, NnWOX9b and NnWOX2 shared most co-expressed genes, but they have two co-expressed genes correlated to NnWOX11. Notably, although NnWOX3b and NnWOX11 are clustered in the same module (i.e., MEgrey60 module), NnWOX3b has the only common co-expressed gene significantly correlated to NnWOX11 (Figure 5c). However, NnWOX genes in the MEblack module have major common co-expressed genes, suggesting these NnWOX genes might regulate similar pathways in lotus (Figure 5c). The duplicated gene pairs of NnWOX4a and NnWOX4b were assigned to the same MEblack module, but different co-expressed genes were identified for these two genes, indicating their distinct co-expression network and divergent functions, respectively.
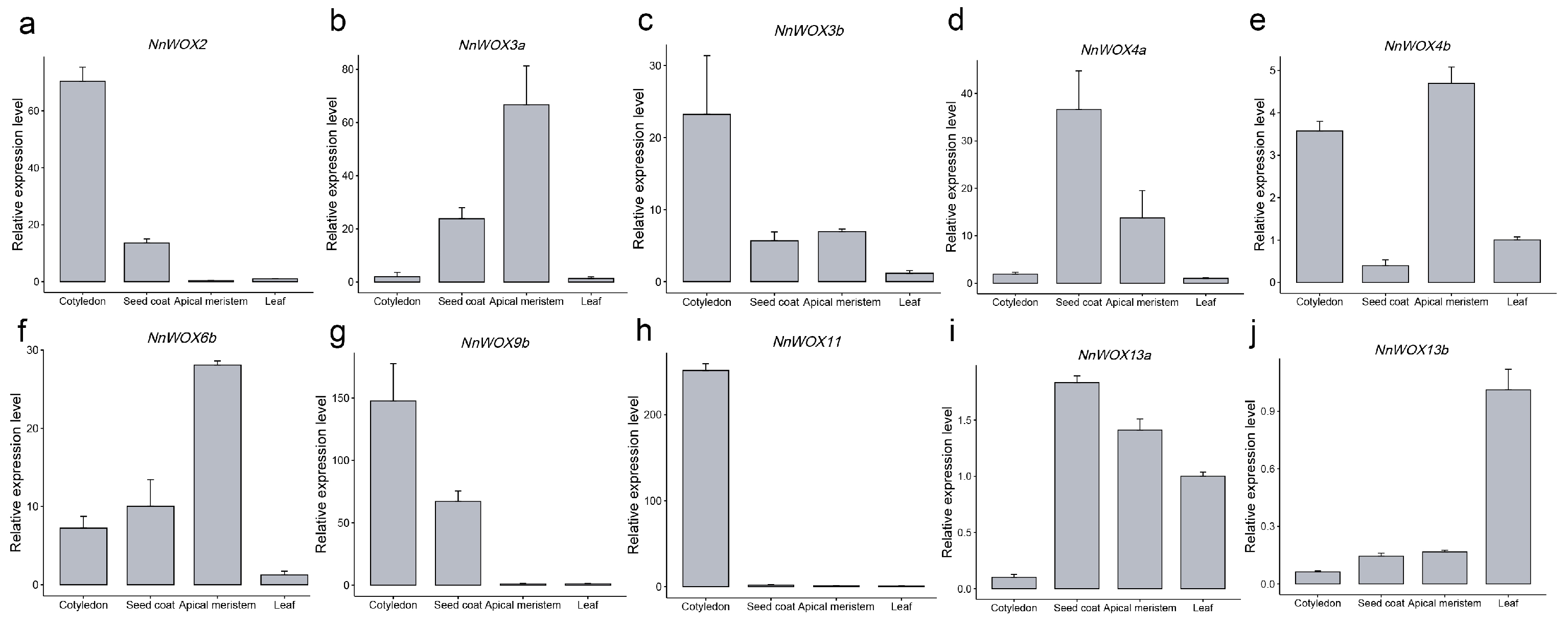
2.7. qRT-PCR Validation of NnWOX Genes in Co-Expression Network
According to the WGCNA networks and gene expression patterns of RNA-seq, the ten NnWOX genes involved in the co-expression networks exhibited a high correlation to the specific tissue. To further validate the expression level of those ten NnWOX genes in the tissues, we collected four tissues including cotyledon, seed coat, apical meristem, and leaf to perform qRT-PCR experiments. The leaf was considered the internal reference and the other three tissues represented the specifically expressed tissues of NnWOX genes. Four NnWOX genes were allocated to MEgrey60, which was significantly related to the cotyledon, exhibiting a higher expression level in the cotyledon compared to the other three tissues (Figure 6a,c,g,h). Our results showed a similar tendency for tissue expression patterns between qRT-PCR and RNA-seq data (Figure 6). These results indicated the high accuracy of RNA-seq and WGCNA networks and the tissue-specific expression patterns of NnWOX genes.

Figure 6.
qRT-PCR verified the expression levels of NnWOXs in four tissues. (a–j) Ten NnWOX genes used in WGCNA networks that were tested. Three biological replicates were performed. The error bars show the standard error of the mean.
3. Discussion
Transcription factors play important roles in regulating transcript initiation and gene expression during plant growth and development. As a plant-specific transcription factor, WOX genes play key roles in maintaining stem cell homeostasis and are involved in the growth and development of multiple tissues [40,41,42,43,44]. Considering the vital roles of WOX genes, a whole-genome analysis of the WOX family was conducted in multiple plants, including A. thaliana [12], wheat [44], cotton [45], melon [46], and loquat [47]. The number of WOX members varies in these plants. Although genome-wide identification of the WOX members in lotus were preliminarily studied [39], the regulatory interactions of NnWOXs genes need further investigation. With increasing genomics research on lotus [30,48], the Nelumbo Genome Database provided a great platform to investigate the whole-genome analysis of transcription factors [35]. In the present study, we also conduct genome-wide identification of the WOX genes in lotus according to the genomic information obtained from the published dataset.
Although lotus has the same number of WOX genes compared to A. thaliana, some orthologs of AtWOX genes have not been reported in lotus, including WOX1, WOX7, WOX8, WOX10, WOX12, and WOX14. Given the basal position of lotus in the phylogenetic tree of the dicotyledon [49], the AtWOX genes that have no orthologs to lotus are supposed to be derived from the expansion of gene families during species evolution. Despite lotus and A. trichopoda occurring once during whole-genome duplication events [30,34], more WOX genes were identified in lotus than in A. trichopoda. This could result from the distinct amplifications and losses of WOX gene family members between these two species during plant evolution. Two NnWOX genes, i.e., NnWOX13a and NnWOX13b, were identified in the ancient clade; we speculated that this WGD pair remained after the ancient whole-genome duplication in lotus. The conserved homeodomain was used as a criterion to identify members of the WOX gene family. Additionally, we found the NnWOX members in each phylogenetic clade containing specific motifs and large different genomic structures, indicating the distinctive functions of different NnWOX genes.
A previous study revealed that the expression patterns of genes in lotus were different in multiple tissues [32], which enabled further investigation of the tissue expression level of the NnWOX genes. Interestingly, we only found similar tissue expression patterns between the duplicated NnWOX13a/b genes in the ancient clade. Distinctive expression patterns of NnWOX genes within the same clade and even between duplicates were identified, suggesting different roles in regulating the development of tissues at different growth stages. The AtrWUS gene played an important role in the formation of stem cells and promoted vegetative-to-embryogenic transition in A. thaliana [50,51,52]. However, the orthologous NnWUS only exhibits high expression levels in the carpel before and after pollination, in the mature receptacle, but a low expression levels in the rhizome and root. This indicated that NnWUS likely did not regulate the stem cells but might participate in the reproductive processes in lotus. A previous study also suggested that the stable expression level of AtWUS in root apical meristems was the key to maintaining the cellular organization of apical meristems [53]. However, the silent expression of the NnWUS and high expression levels of other NnWOX genes in apical meristem indicated that NnWOX genes were the real regulators of lotus apical meristem and not NnWUS. We speculated that the homeostasis of stem cells regulated by AtWUS was a neofunctionalization in the evolution of A. thaliana. Notably, we found a class of NnWOX genes showing specific high expression levels in the cotyledon of different developmental stages, including NnWOX2, NnWOX3b, NnWOX6b, NnWOX9b, and NnWOX11. Due to cotyledon being the key tissue for storing and providing nutrients during seed dormancy and germination, the development of cotyledon regulated by these NnWOX genes was supposed to be relevant to specific phenomena that lotus seed kept active after being buried underground for hundreds of years [54]. Furthermore, previous studies revealed that WOX genes contributed to the formation of somatic embryogenesis in plants, such as A. thaliana [15], cotton [45], maize [55], and wheat [56]. Therefore, our results indicated that several NnWOX genes played vital roles in the development of lotus cotyledon.
Gene co-expression networks were performed to filter gene sets involved in the same biological pathways or gene pairs interacting with each other [57,58]. Due to the minimum sample size and correlation of gene modules and tissue, weight gene co-expression network analysis (WGCNA) was widely used for identifying the co-expressed gene pairs in different plant tissues or experimental treatment samples [59,60,61]. Previous transcriptomic research on lotus has constructed a gene-level network involved in only six tissues and a transcript-level co-expression network including eleven tissues using WGCNA [32,62]. In this study, we used the same pipeline of WGCNA to construct the gene co-expression networks for each NnWOX gene (Figure 5). Most NnWOX genes in the same module shared common co-expressed genes, except for NnWOX3b and NnWOX11. Two modules (MEgrey60 and MEblack) containing eight NnWOX genes were significantly related to the cotyledon, in line with their specific expression patterns. The GO enrichment results suggested that the significantly co-expressed genes for the ten NnWOX genes used in the networks were enriched in the reproductive and transcription processes. Therefore, our results indicate that NnWOX genes and their co-expressed genes show specific high expression levels in the cotyledon, which is likely associated with the activities taking place during the long-term storage of lotus seed.
4. Materials and Methods
4.1. Plant Materials
Wild sacred lotus was cultivated in the Xiaogan (31°33’ N, 113°25’ E), Hubei province, China. Rolled leaves, roots, seed coat, and seed cotyledon were collected from the individual plant 15 days after flowering and frozen immediately in liquid nitrogen. High-quality RNA from each sample was extracted using the RNAprep pure Plant Kit (TIANGEN) for subsequent verification experiments.
4.2. Identification of WOX Genes in Nelumbo nucifera
To obtain a comprehensive WOX gene family in lotus, we carried out three identification methods. First, a total of fifteen WOX genes (AtWOX) in the model plant Arabidopsis thaliana were acquired from the TAIR database (http://www.arabidopsis.org/ (accessed on 1 July 2023)), which were further used as query sequences to perform the BALSTP search with lotus protein sequences (downloaded from http://nelumbo.biocloud.net (accessed on 1 July 2023)). Only mapped results with p-value < 1 × 10−5 were retained. Second, the gene function annotation of lotus genes was also downloaded from the Nelumbo genome database (http://nelumbo.biocloud.net (accessed on 1 July 2023)), and the genes annotated WUSCHEL-related homeobox were filtered out. Third, the protein sequences of China Antique genes were mapped to the PlantTFDB database to identify different families of transcription factors. The genes identified under the WOX transcription factor family members were filtered. We combined the filtered genes from these three methods and defined them as candidate NnWOX genes. As the WOX gene contained a conserved HB domain, the candidate NnWOX genes were mapped to three public databases, including NCBI Conserved Domain Database (CDD, https://www.ncbi.nlm.nih.gov/cdd/ (accessed on 7 July 2023)), SMART database (http://smart.embl-heidelberg.de/ (accessed on 7 July 2023)), and Pfam database (https://pfam.xfam.org/ (accessed on 7 July 2023)). The candidate genes not containing the conserved HB domain region were weeded out. According to the abovementioned identification pipeline, the WOX genes in Amborella trichopoda (AtrWOX) were also identified, and all genomic information regarding A. trichopoda was obtained from the Joint Genome Institute database (https://jgi.doe.gov/ (accessed on 7 July 2023)). Based on the mapping results, the identified NnWOX and AtrWOX genes were named according to their orthology with AtWOX genes.
4.3. Chromosome Location and Structure of NnWOX Genes
The full-length protein sequence of NnWOX, AtWOX, and AtrWOX were aligned using ClustalW. A phylogenetic neighbor-joining tree was constructed using MEGA v7.0 software with pairwise deletion and 1000 replicates in bootstrap analysis [63]. The constructed tree was drawn and optimized using the iTOL online tools (https://itol.embl.de/ (accessed on 7 July 2023)). The physical and chemical characterizations of the NnWOX protein were analyzed using the Proparam tool of ExPASy (http://weB.expasy.org/protparam/ accessed on 7 July 2023)), including the number of amino acids, molecular weights, theoretical PI values, instability index, aliphatic index, and the grand average of hydropathicity. In addition, the N-glycosylation sites of NnWOX proteins were predicted using the NetNGlyc (https://services.healthtech.dtu.dk/services/NetNGlyc-1.0/ (accessed on 7 July 2023)), and the hydrophobicity and hydrophilicity of these protein sequences were analyzed using the ProtScale tools of ExPASy (https://web.expasy.org/protscale/ (accessed on 7 July 2023)).
4.4. Chromosome Location and Structure of NnWOXs
Locus information of the physical locations of NnWOX genes in the genomes of “China Antique” was collected from the Nelumbo Genome Database (http://nelumbo.cngb.org/nelumbo/ (accessed on 1 July 2023)), and the positions were drafted to chromosomes using TBtools software [64]. Because the gene annotation from the Nelumbo Genome Database contained multiple transcripts, the longest transcript from each gene was chosen to represent the corresponding gene. The structures of the NnWOX genes were displayed using TBtools software [64] to obtain the exon composition information. To identify the sequence motifs of NnWOX genes, the protein sequences were analyzed using MEME tools (http://meme-suite.org/meme/tools/meme (accessed on 7 July 2023)). For each sequence, the maximum of identified motifs was six.
4.5. Duplication Analysis of NnWOX Genes
Given that the lotus genome only occurred once during the ancient genome duplication event [30]. MSCcanX was used to further identify the duplicated type of NnWOX genes classifying lotus genes into five groups: singletons, WGD/segmental duplications, dispersed duplications, proximal duplications, and tandem duplications. Based on the syntenic result of duplicated genes, the duplicated NnWOX gene pairs were shown in a Circos plot using an RCircos package v1.2.0.
4.6. Gene Expression Profile and Weighted Gene Co-Expression Network Analysis
The gene expression profile of fifty-four China Antique RNA-seq among 11 tissues was downloaded from the Nelumbo Genome Database (http://nelumbo.biocloud.net (accessed on 1 July 2023)). Since weighted gene co-expression network analysis (WGCNA) was effective in identifying co-expressed genes and constructing gene co-expression networks, we performed WGCNA analysis based on the gene expression profile. To obtain a high-quality gene co-expression network, among fifty-four samples, genes with average FPKM (fragments per kilobase of exon model per million mapped fragments) < 0.1 were excluded first. The gene co-expression network was built using the WGCNA R-package with min-module 500. The co-expressed genes of NnWOXs in each module were filtered, and the top 5% of co-expressed genes were defined as significantly co-expressed genes. Subsequently, the NnWOX co-expression networks were constructed using Cytoscape.
4.7. GO Enrichment Analysis
The gene functions of lotus genes were annotated using EggNOG v5.0 with the Viridiplantae database, and the GO enrichment analysis of NnWOX co-expressed genes was carried out using TBtools [64]. The GO terms with p-value < 0.01 were defined as significantly enriched.
4.8. Quantitative Real-Time PCR Experiments
The specific adapters for every NnWOX gene were designed using Primmer5 (http://www.premierbiosoft.com/primerdesign/) (accessed on 1 February 2023) (Table S1). To verify the expression level of NnWOX genes in different tissues, high-quality RNA of five lotus samples was reverse-transcribed into five cDNA samples. The qRT-PCR experiments were performed using NovoStart SYBR qPCR SuperMix plus (Novoprotein, China), and three biological replicates for each NnWOX gene were applied. Relative expression of NnWOX genes was analyzed using the 2−∆∆Ct method, with the lotus β-Actin gene used as an internal standard gene.
5. Conclusions
In this study, the WOX gene family was identified genome-wide in lotus. Based on the phylogenetic analysis, a total of 15 identified NnWOX genes were divided into three groups, which were similar to the cluster described in previous studies. Gene structure analysis showed that all NnWOX genes contained two conserved motifs, but specific motifs were identified in different clade members, indicating the conserved sequence variations in the three groups of lotus WOX genes. Gene expression analysis suggested the obvious divergence of NnWOX genes in different lotus tissues. We constructed gene co-expressed networks for each NnWOX gene and found their co-expressed genes were enriched in reproductive pathways. In summary, this work provides insight into the characteristics of NnWOX genes, which is helpful for further research on the molecular functions in lotus.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/plants13050720/s1, Figure S1: The hydrophily and hydrophobicity of NnWOX proteins, Figure S2: N-glycosylation analysis of NnWOX genes. Figure S3: GO enrichment of genes that were co-expressed with NnWOX genes. Table S1: Summary of the qRT-PCR primers.
Author Contributions
Conceptualization, J.-j.L. and C.-c.L.; formal analysis, J.-j.L. and X.-y.Q.; writing—original draft preparation, J.-j.L.; writing—review and editing, Y.-j.D., T.M.N. and C.-c.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Department of Education of Hubei Province Project (Q20202702, Q20162704) and the outstanding young and middle-aged science and technology innovation team project in colleges and universities of Hubei Province (T2022030).
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We thank Yue Zhang at Wuhan Botanical Garden for helping us analyze the gene co-expression networks.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gehring, W.J.; Affolter, M.; Bürglin, T. Homeodomain proteins. Annu. Rev. Biochem. 1994, 63, 487–526. [Google Scholar] [CrossRef]
- Lian, G.; Ding, Z.; Wang, Q.; Zhang, D.; Xu, J. Origins and evolution of WUSCHEL-related homeobox protein family in plant kingdom. Sci. World J. 2014, 2014, 534140. [Google Scholar] [CrossRef]
- van der Graaff, E.; Laux, T.; Rensing, S.A. The WUS homeobox-containing (WOX) protein family. Genome Biol. 2009, 10, 248. [Google Scholar] [CrossRef]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 2009, 21, 3493–3505. [Google Scholar] [CrossRef]
- Alvarez, J.M.; Bueno, N.; Cañas, R.A.; Avila, C.; Cánovas, F.M.; Ordás, R.J. Analysis of the WUSCHEL-RELATED HOMEOBOX gene family in Pinus pinaster: New insights into the gene family evolution. Plant Physiol. Biochem. 2018, 123, 304–318. [Google Scholar] [CrossRef]
- Mukherjee, K.; Brocchieri, L.; Bürglin, T.R. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol. Biol. Evol. 2009, 26, 2775–2794. [Google Scholar] [CrossRef]
- Dolzblasz, A.; Nardmann, J.; Clerici, E.; Causier, B.; van der Graaff, E.; Chen, J.; Davies, B.; Werr, W.; Laux, T. Stem Cell Regulation by Arabidopsis WOX Genes. Mol. Plant 2016, 9, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Breuninger, H.; Rikirsch, E.; Hermann, M.; Ueda, M.; Laux, T. Differential expression of WOX genes mediates apical-basal axis formation in the Arabidopsis embryo. Dev. Cell 2008, 14, 867–876. [Google Scholar] [CrossRef]
- Wang, J.; Tan, M.; Wang, X.; Jia, L.; Wang, M.; Huang, A.; You, L.; Li, C.; Zhang, Y.; Zhao, Y.; et al. WUS-RELATED HOMEOBOX 14 boosts de novo plant shoot regeneration. Plant Physiol. 2023, 192, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Vollbrecht, E.; Veit, B.; Sinha, N.; Hake, S. The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature 1991, 350, 241–243. [Google Scholar] [CrossRef]
- Laux, T.; Mayer, K.F.; Berger, J.; Jürgens, G. The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development 1996, 122, 87–96. [Google Scholar] [CrossRef]
- Zhang, X.; Zong, J.; Liu, J.; Yin, J.; Zhang, D. Genome-wide analysis of WOX gene family in rice, sorghum, maize, Arabidopsis and poplar. J. Integr. Plant Biol. 2010, 52, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.; Schoof, H.; Haecker, A.; Lenhard, M.; Jürgens, G.; Laux, T. Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 1998, 95, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Ji, J.; Kelsey, E.; Ohtsu, K.; Schnable, P.S.; Scanlon, M.J. Tissue specificity and evolution of meristematic WOX3 function. Plant Physiol. 2009, 149, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Haecker, A.; Gross-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Park, S.O.; Zheng, Z.; Oppenheimer, D.G.; Hauser, B.A. The PRETTY FEW SEEDS2 gene encodes an Arabidopsis homeodomain protein that regulates ovule development. Development 2005, 132, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dabi, T.; Weigel, D. Requirement of homeobox gene STIMPY/WOX9 for Arabidopsis meristem growth and maintenance. Curr. Biol. 2005, 15, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Deveaux, Y.; Toffano-Nioche, C.; Claisse, G.; Thareau, V.; Morin, H.; Laufs, P.; Moreau, H.; Kreis, M.; Lecharny, A. Genes of the most conserved WOX clade in plants affect root and flower development in Arabidopsis. BMC Evol. Biol. 2008, 8, 291. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Wang, R.; Wang, Y.; Li, Y.; Sun, G.; Yao, S. SLG2 specifically regulates grain width through WOX11-mediated cell expansion control in rice. Plant Biotechnol. J. 2023, 21, 1904–1918. [Google Scholar] [CrossRef]
- Tang, H.; Fan, J.; Wang, R.; Zhu, J.; Xiang, X.; Dong, J.; Zhou, L.; Wang, L. Changes in the expression pattern of OsWUS negatively regulate plant stature and panicle development in rice. G3 2023, 13, jkad100. [Google Scholar] [CrossRef]
- Cheng, S.; Huang, Y.; Zhu, N.; Zhao, Y. The rice WUSCHEL-related homeobox genes are involved in reproductive organ development, hormone signaling and abiotic stress response. Gene 2014, 549, 266–274. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Wolabu, T.; Wang, Z.; Liu, Y.; Tadesse, D.; Chen, N.; Xu, A.; Bi, X.; Zhang, Y.; et al. WOX family transcriptional regulators modulate cytokinin homeostasis during leaf blade development in Medicago truncatula and Nicotiana sylvestris. Plant Cell 2022, 34, 3737–3753. [Google Scholar] [CrossRef]
- He, G.; Cao, Y.; Wang, J.; Song, M.; Bi, M.; Tang, Y.; Xu, L.; Ming, J.; Yang, P. WUSCHEL-related homeobox genes cooperate with cytokinin to promote bulbil formation in Lilium lancifolium. Plant Physiol. 2022, 190, 387–402. [Google Scholar] [CrossRef]
- Ye, S.; Yan, L.; Ma, X.; Chen, Y.; Wu, L.; Ma, T.; Zhao, L.; Yi, B.; Ma, C.; Tu, J.; et al. Combined BSA-Seq based mapping and RNA-Seq profiling reveal candidate genes associated with plant architecture in Brassica napus. Int. J. Mol. Sci. 2022, 23, 2472. [Google Scholar] [CrossRef]
- Batcho, A.A.; Nwogwugwu, J.O.; Ali, M.; Jabbar, B.; Javaid, A.; Fellner, M. Identification and characterisation of blue light photoreceptor gene family and their expression in tomato (Solanum lycopersicum) under cold stress. Funct. Plant Biol. 2022, 49, 647–658. [Google Scholar] [CrossRef]
- Shi, L.; Wang, K.; Du, L.; Song, Y.; Li, H.; Ye, X. Genome-wide identification and expression profiling analysis of WOX family protein-encoded genes in Triticeae species. Int. J. Mol. Sci. 2021, 22, 9325. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.S.; Goher, F.; Hu, C.G.; Zhang, J.Z. WUSCHEL-related homeobox (WOX) transcription factors: Key regulators in combating abiotic stresses in plants. Hortic. Adv. 2024, 2, 2. [Google Scholar] [CrossRef]
- Fedoreyeva, L.I.; Baranova, E.N.; Chaban, I.A.; Dilovarova, T.A.; Vanyushin, B.F.; Kononenko, N.V. Elongating Effect of the peptide AEDL on the root of Nicotiana tabacum under salinity. Plants 2022, 11, 1352. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, C.; Cao, D.; Damaris, R.N.; Yang, P. The latest studies on lotus (Nelumbo nucifera)-an emerging horticultural model plant. Int. J. Mol. Sci. 2019, 20, 3680. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Rahmani, R.S.; Gugger, P.F.; Wang, M.; Li, H.; Zhang, Y.; Li, Z.; Wang, Q.; Van de Peer, Y.; Marchal, K.; et al. Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol. Biol. Evol. 2020, 37, 2394–2413. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Damaris, R.N.; Shi, T.; Li, J.; Yang, P. Transcriptomic analysis identifies the key genes involved in stamen petaloid in lotus (Nelumbo nucifera). BMC Genom. 2018, 19, 554. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Van de Peer, Y.; Chen, J.; Marchal, K.; Shi, T. Evolution of isoform-level gene expression patterns across tissues during lotus species divergence. Plant J. 2022, 112, 830–846. [Google Scholar] [CrossRef]
- Cao, D.; Damaris, R.N.; Zhang, Y.; Liu, M.; Li, M.; Yang, P. Proteomic analysis showing the signaling pathways involved in the rhizome enlargement process in Nelumbo nucifera. BMC Genom. 2019, 20, 766. [Google Scholar] [CrossRef]
- Albert, V.A.; Barbazuk, W.B.; dePanphilis, C.W.; Der, J.P.; Leebens-Mack, J.; Ma, H.; Palmer, J.D.; Rounsley, S.; Sankoff, D.; Schuster, S.C.; et al. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar]
- Li, H.; Yang, X.; Zhang, Y.; Gao, Z.; Liang, Y.; Chen, J.; Shi, T. Nelumbo genome database, an integrative resource for gene expression and variants of Nelumbo nucifera. Sci. Data 2021, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Cao, D.; Damaris, R.N.; Yang, P. Genome-wide identification of MADS-box gene family in sacred lotus (Nelumbo nucifera) identifies a SEPALLATA homolog gene involved in floral development. BMC Plant Biol. 2020, 20, 497. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiong, Y.; Li, Y.; Ye, S.; Yin, Q.; Gao, S.; Yang, D.; Yang, M.; Palva, E.T.; Deng, X. Comprehensive analysis and functional studies of WRKY transcription factors in Nelumbo nucifera. Int. J. Mol. Sci. 2019, 20, 5006. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Tan, M.; Jiao, J.; Zhang, C.; Wu, P.; Feng, K.; Li, L. Identification of YABBY transcription factors and their function in ABA and salinity response in Nelumbo nucifera. Plants 2023, 12, 380. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Z.; Huang, J.; Lin, Z.C.; Wang, F.; Yang, S.M.; Jiang, X.; Ahmad, S.; Zhou, Y.Z.; Lan, S.; Liu, Z.J.; et al. Genome-wide analysis of WUSCHEL-related homeobox gene family in scared lotus (Nelumbo nucifera). Int. J. Mol. Sci. 2023, 24, 14216. [Google Scholar] [CrossRef] [PubMed]
- Tvorogova, V.E.; Krasnoperova, E.Y.; Potsenkovskaia, E.A.; Kudriashov, A.A.; Dodueva, I.E.; Lutova, L.A. What Does the WOX Say? Review of Regulators, Targets, Partners. Mol. Biol. 2021, 55, 362–391. [Google Scholar] [CrossRef]
- Vandenbussche, M.; Horstman, A.; Zethof, J.; Koes, R.; Rijpkema, A.S.; Gerats, T. Differential recruitment of WOX transcription factors for lateral development and organ fusion in Petunia and Arabidopsis. Plant Cell 2009, 21, 2269–2283. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Zhang, Q.; Yao, Y.; Cui, Y.; Huang, T. Cytosolic isocitrate dehydrogenase regulates plant stem cell maintenance in response to nutrient deficiency. Plant Physiol. 2023, 192, 3069–3087. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, Y.; Yue, Y.; Chen, Z.; Zhou, D.X.; Zhao, Y. Transcription factor WOX11 regulates protein translation via ribosome protein acetylation in rice roots. Plant Physiol. 2023, 191, 2224–2228. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, D.; Xia, Y.; Li, Z.; Jing, D.; Du, J.; Niu, N.; Ma, S.; Wang, J.; Song, Y.; et al. Identification of the WUSCHEL-related homeobox (WOX) gene family, and interaction and functional analysis of TaWOX9 and TaWUS in wheat. Int. J. Mol. Sci. 2020, 21, 1581. [Google Scholar] [CrossRef]
- Sun, R.; Zhang, X.; Ma, D.; Liu, C. Identification and evolutionary analysis of cotton (Gossypium hirsutum) WOX family genes and their potential function in somatic embryogenesis. Int. J. Mol. Sci. 2023, 24, 11077. [Google Scholar] [CrossRef]
- Tang, L.; He, Y.; Liu, B.; Xu, Y.; Zhao, G. Genome-wide identification and characterization analysis of WUSCHEL-related homeobox family in melon (Cucumis melo L.). Int. J. Mol. Sci. 2023, 24, 12326. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, M.; Liu, X.; Xia, Y.; Hu, R.; Xia, Q.; Jing, D.; Guo, Q. Genome-wide analysis of the WOX gene family and the role of EjWUSa in regulating flowering in loquat (Eriobotrya japonica). Front. Plant Sci. 2022, 13, 1024515. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, H.; Zhou, J.; Jiang, S.; Wang, Y.; Kuang, J.; Ji, Q.; Peng, J.; Wang, J.; Gao, L.; et al. Resequencing of 296 cultivated and wild lotus accessions unravels its evolution and breeding history. Plant J. 2020, 104, 1673–1684. [Google Scholar] [CrossRef]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.; Jürgens, G.; Laux, T. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.K.; Luijten, M.; Miyashima, S.; Lenhard, M.; Hashimoto, T.; Nakajima, K.; Scheres, B.; Heidstra, R.; Laux, T. Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature 2007, 446, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Niu, Q.W.; Frugis, G.; Chua, N.H. The WUSCHEL gene promotes vegetative-to-embryonic transition in Arabidopsis. Plant J. 2002, 30, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Kaya, H.; Shibahara, K.I.; Taoka, K.I.; Iwabuchi, M.; Stillman, B.; Araki, T. FASCIATA genes for chromatin assembly factor-1 in Arabidopsis maintain the cellular organization of apical meristems. Cell 2001, 104, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Q. Lotus Flower Cultivars in China; China Forestry Publishing House: Beijing, China, 2005. [Google Scholar]
- Kang, M.; Lee, K.; Ji, Q.; Grosic, S.; Wang, K. Enhancing maize transformation and targeted mutagenesis through the assistance of non-integrating Wus2 vector. Plants 2023, 12, 2799. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Shi, L.; Liang, X.; Zhao, P.; Wang, W.; Liu, J.; Chang, Y.; Hiei, Y.; Yanagihara, C.; Du, L.; et al. The gene TaWOX5 overcomes genotype dependency in wheat genetic transformation. Nat. Plants 2022, 8, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Wei, H.; Milne, J.; Page, G.P.; Somerville, C.R. Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc. Natl. Acad. Sci. USA 2005, 102, 8633–8638. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; López-Vidriero, I.; Carrasco, J.L.; Godoy, M.; Vera, P.; Solano, R. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. USA 2014, 111, 2367–2372. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, H.; Qi, Y.; Zhao, Y.; Duan, C.; Wang, Y.; Meng, Z.; Zhang, Q. Genome-wide identification of PYL/PYR-PP2C (A)-SnRK2 genes in Eutrema and their co-expression analysis in response to ABA and abiotic stresses. Int. J. Mol. Sci. 2023, 253, 126701. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Cao, J.; Wang, K.; Qin, L.; Sun, Y.; Ju, W.; Qu, C.; Miao, J. Extreme environmental adaptation mechanisms of Antarctic bryophytes are mainly the activation of antioxidants, secondary metabolites and photosynthetic pathways. BMC Plant Biol. 2023, 23, 399. [Google Scholar] [CrossRef]
- Sharma, N.; Madan, B.; Khan, M.S.; Sandhu, K.S.; Raghuram, N. Weighted gene co-expression network analysis of nitrogen (N)-responsive genes and the putative role of G-quadruplexes in N use efficiency (NUE) in rice. Front. Plant Sci. 2023, 14, 1135675. [Google Scholar] [CrossRef]
- Zhang, Y.; Rahmani, R.S.; Yang, X.; Chen, J.; Shi, T. Integrative expression network analysis of microRNA and gene isoforms in sacred lotus. BMC Genom. 2020, 21, 429. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).