
Function of FT in Flowering Induction in Two Camellia Species



Abstract
1. Introduction
2. Results
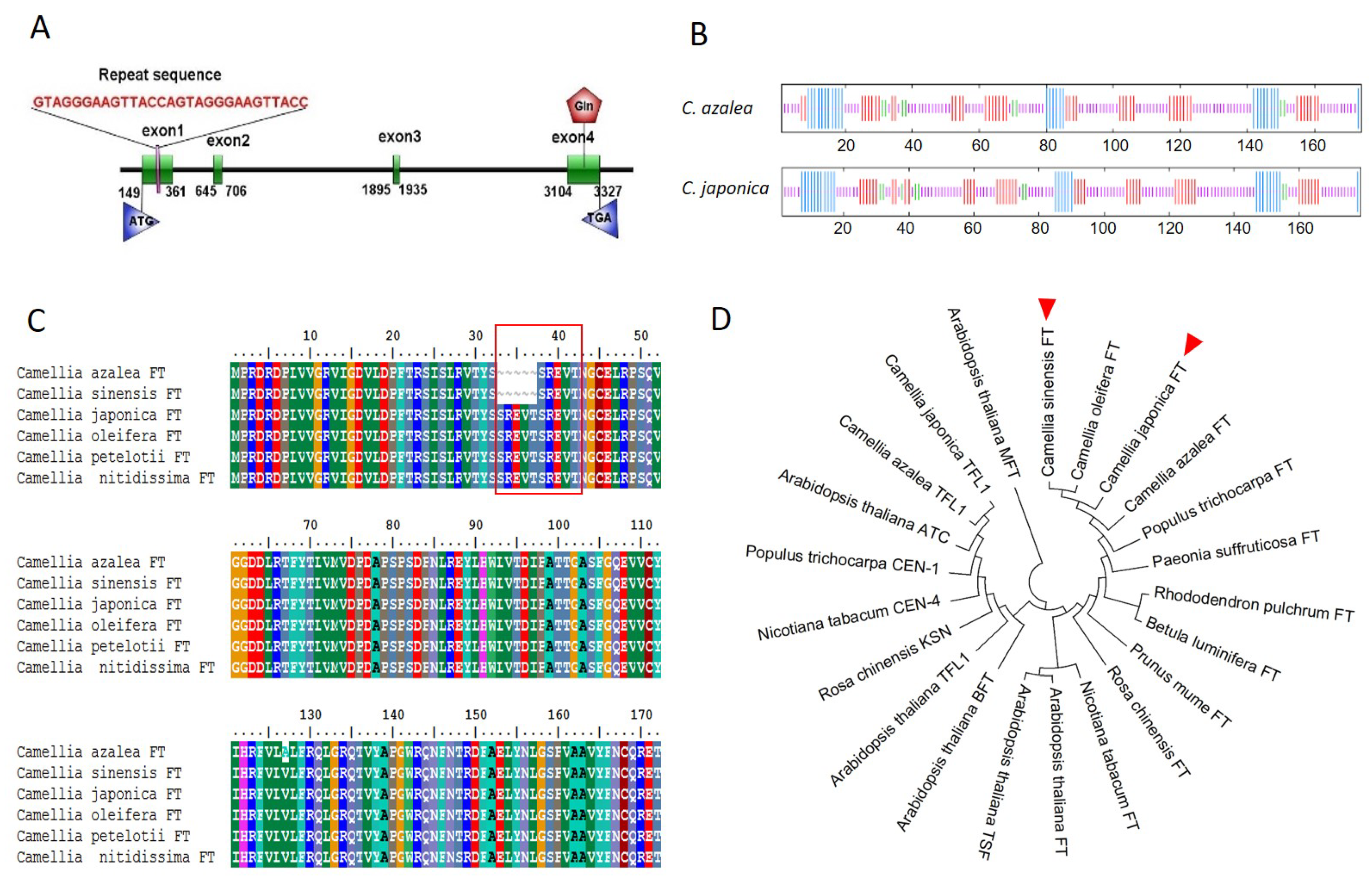
2.1. Identification and Sequence Analysis of FT Genes in C. japonica and C. azalea
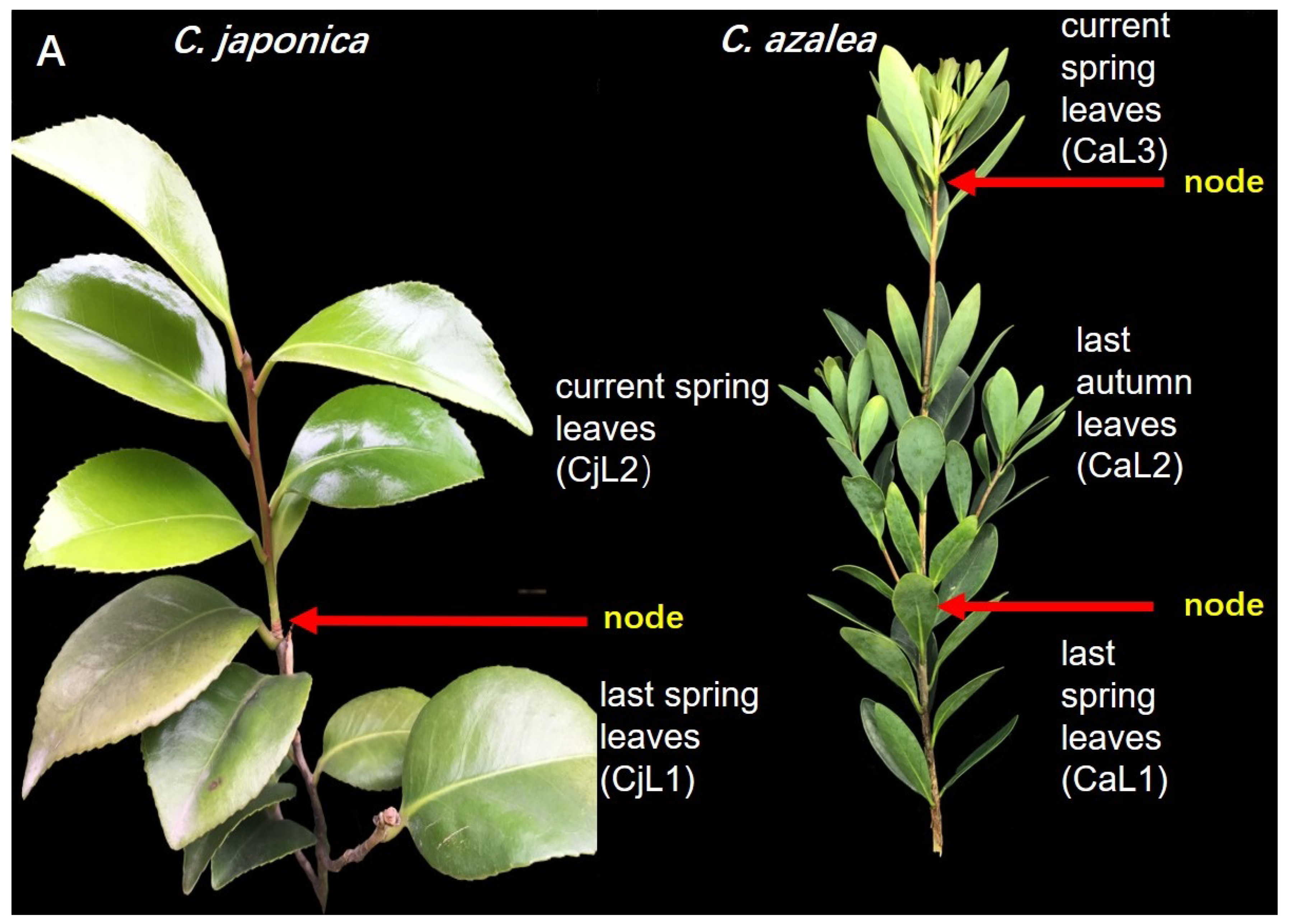
2.2. Expression Patterns of CaFT and CjFT in the Leaves and Floral Buds
2.3. Introduction of CaFT and CjFT Advanced the Flowering Time of Arabidopsis
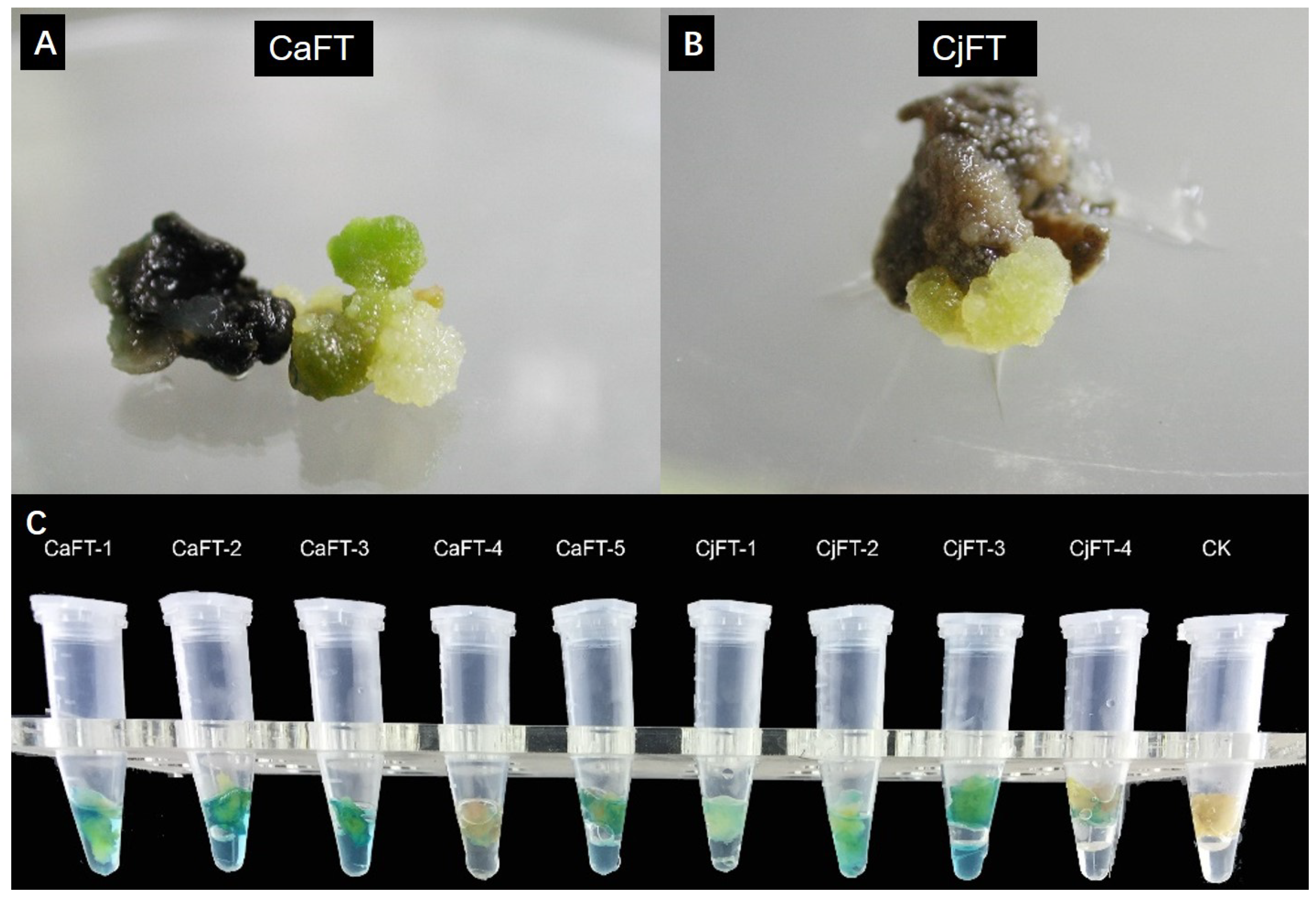
2.4. Transgenic Calli of CaFT and CjFT Were Successfully Obtained from C. azalea
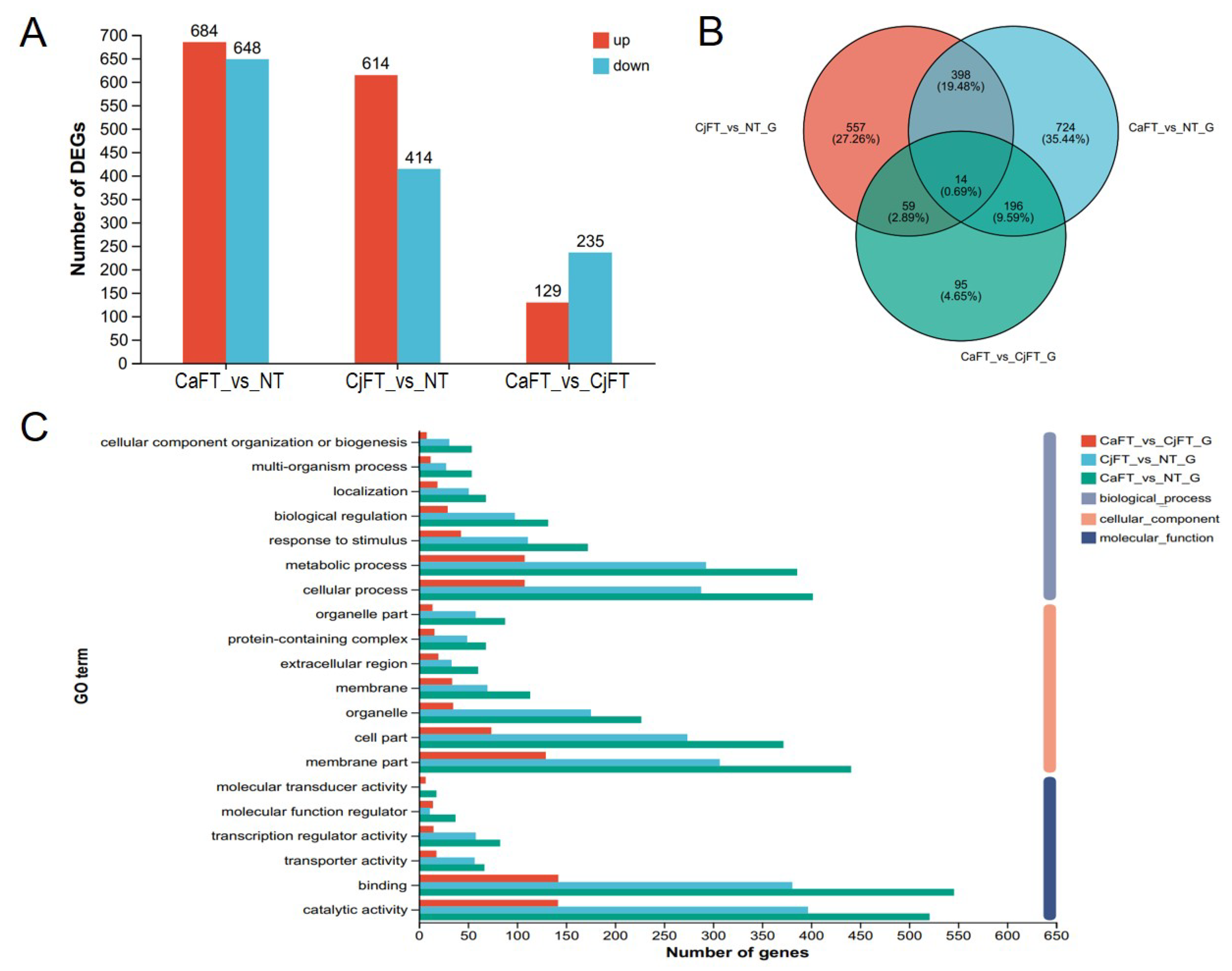
2.5. Transcriptome Analysis of the Transgenic Calli of CaFT and CjFT
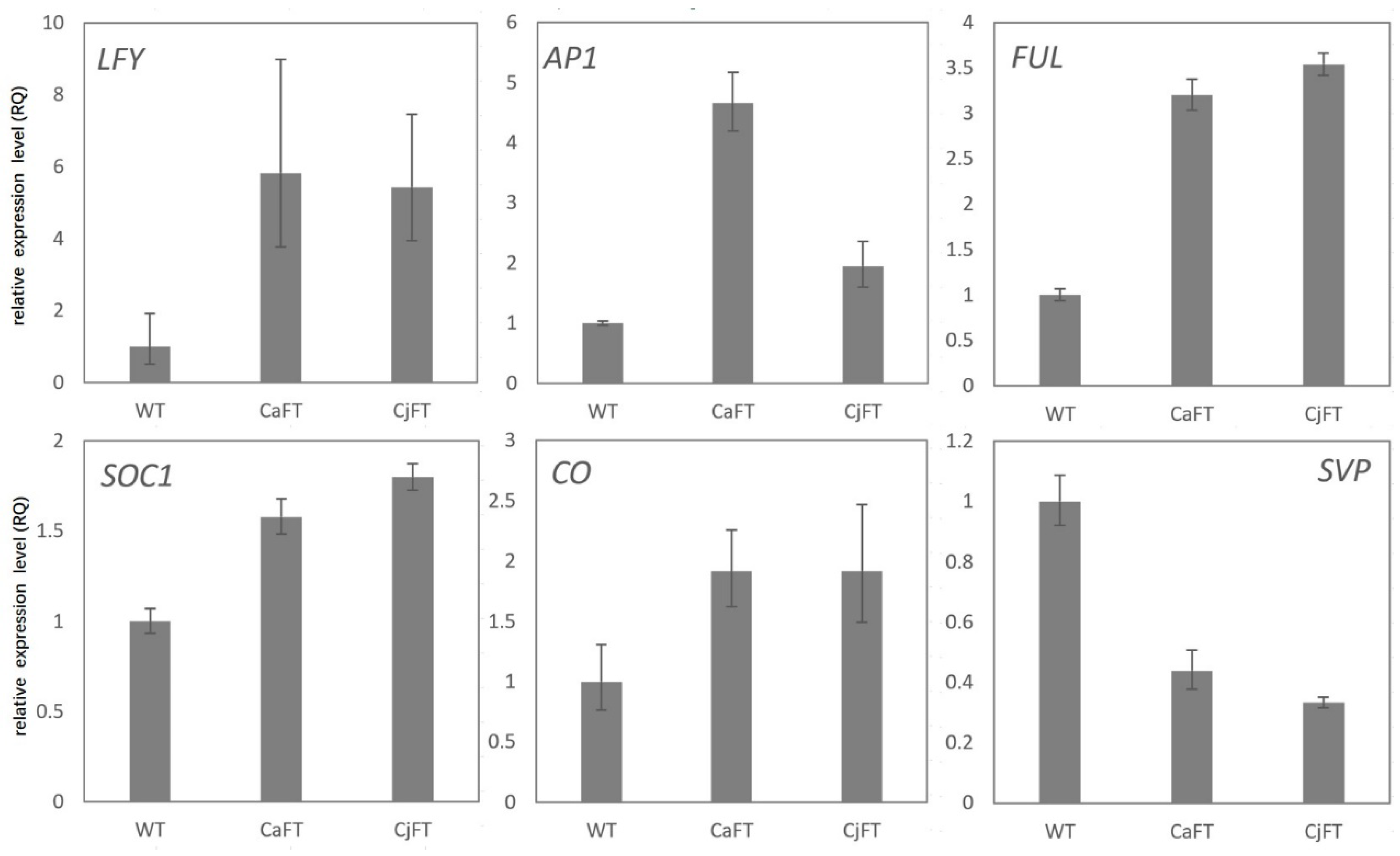
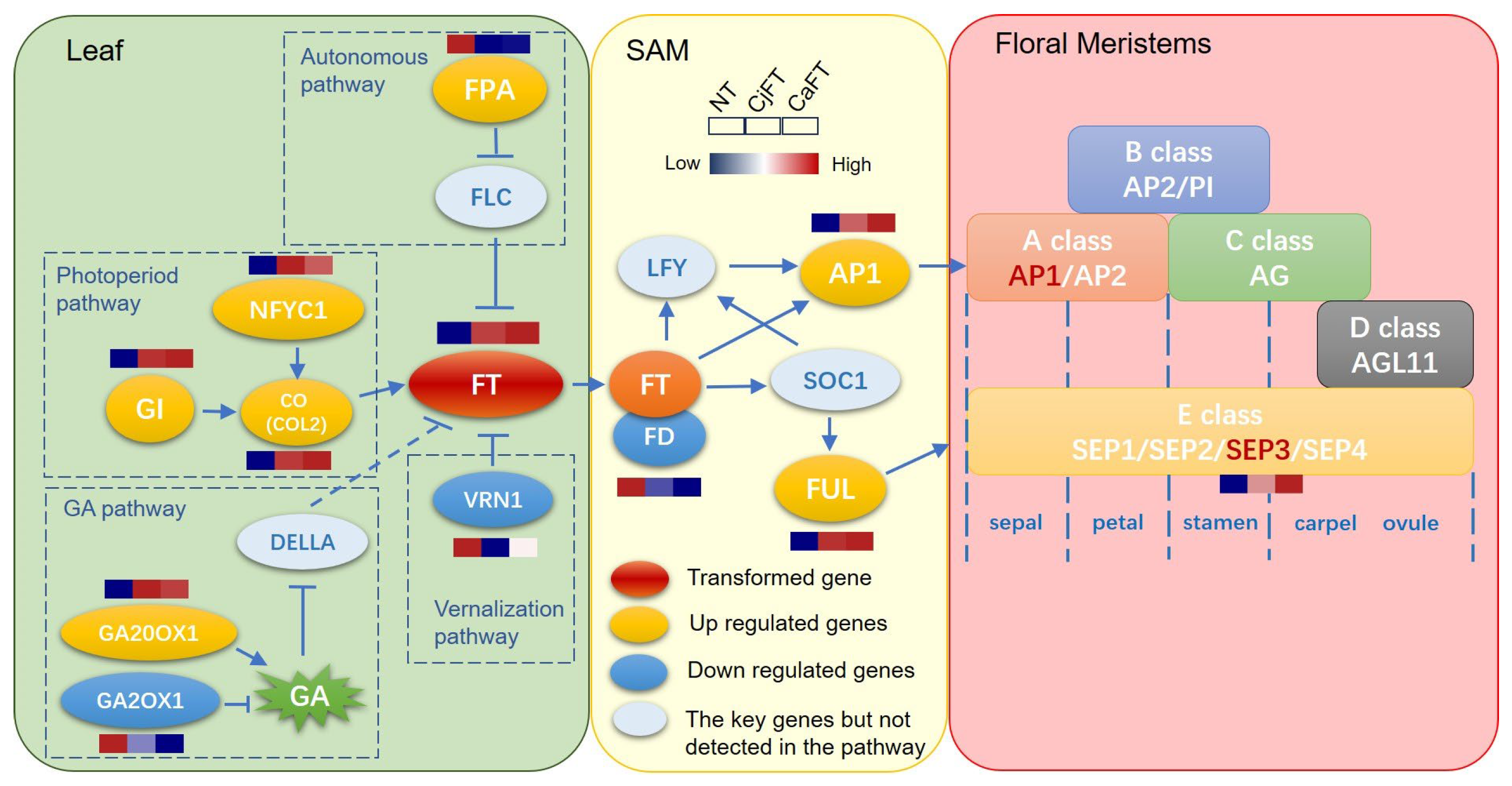
2.6. Differential Regulation of Floral Induction and Floral Organ-Related Genes by FT
2.7. Validation of the Key Genes of Transgenic Calli by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Isolation of FT Genes
4.3. Sequence Alignment and Phylogenetic Analysis
4.4. Transformation of FT Genes to Arabidopsis thaliana and C. azalea
4.5. Transcriptome Sequencing and Analysis of the Transgenic Callus of C. azalea
4.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fornara, F.; de Montaigu, A.; Coupland, G. SnapShot: Control of Flowering in Arabidopsis. Cell 2010, 141, 550–550.e2. [Google Scholar] [CrossRef]
- Wellmer, F.; Riechmann, J.L. Gene networks controlling the initiation of flower development. Trends Genet. 2010, 26, 519–527. [Google Scholar] [CrossRef]
- Turck, F.; Fornara, F.; Coupland, G. Regulation and Identity of Florigen: FLOWERING LOCUS T Moves Center Stage. Annu. Rev. Plant Biol. 2008, 59, 573–594. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Wigge, P.A. FT Protein Acts as a Long-Range Signal in Arabidopsis. Curr. Biol. 2007, 17, 1050–1054. [Google Scholar] [CrossRef]
- Lin, M.-K.; Belanger, H.; Lee, Y.-J.; Varkonyi-Gasic, E.; Taoka, K.-I.; Miura, E.; Xoconostle-Cázares, B.; Gendler, K.; Jorgensen, R.A.; Phinney, B.; et al. FLOWERING LOCUS T Protein May Act as the Long-Distance Florigenic Signal in the Cucurbits. Plant Cell 2007, 19, 1488–1506. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Warthmann, N.; Küttner, F.; Schmid, M. Export of FT Protein from Phloem Companion Cells Is Sufficient for Floral Induction in Arabidopsis. Curr. Biol. 2007, 17, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Weigel, D. Move on up, it’s time for change—Mobile signals controlling photoperiod-dependent flowering. Genes Dev. 2007, 21, 2371–2384. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, C.; Hou, X.; Xi, W.; Shen, L.; Tao, Z.; Wang, Y.; Yu, H. FTIP1 is an essential regulator required for florigen transport. PLoS Biol. 2012, 10, e1001313. [Google Scholar] [CrossRef] [PubMed]
- Corbesier, L.; Vincent, C.; Jang, S.; Fornara, F.; Fan, Q.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kaya, H.; Goto, K.; Iwabuchi, M.; Araki, T. A Pair of Related Genes with Antagonistic Roles in Mediating Flowering Signals. Science 1999, 286, 1960–1962. [Google Scholar] [CrossRef] [PubMed]
- Kardailsky, I.; Shukla, V.K.; Ahn, J.H.; Dagenais, N.; Christensen, S.K.; Nguyen, J.T.; Chory, J.; Harrison, M.J.; Weigel, D. Activation tagging of the floral inducer FT. Science 1999, 286, 1962–1965. [Google Scholar] [CrossRef]
- Tamaki, S.; Matsuo, S.; Wong, H.L.; Yokoi, S.; Shimamoto, K. Hd3a Protein Is a Mobile Flowering Signal in Rice. Science 2007, 316, 1033–1036. [Google Scholar] [CrossRef]
- Li, C.; Luo, L.; Fu, Q.; Niu, L.; Xu, Z.F. Isolation and functional characterization of JcFT, a FLOWERING LOCUS T (FT) homologous gene from the biofuel plant Jatropha curcas. BMC Plant Biol. 2014, 14, 125. [Google Scholar] [CrossRef]
- Oda, A.; Narumi, T.; Li, T.; Kando, T.; Higuchi, Y.; Sumitomo, K.; Fukai, S.; Hisamatsu, T. CsFTL3, a chrysanthemum FLOWERING LOCUS T-like gene, is a key regulator of photoperiodic flowering in chrysanthemums. J. Exp. Bot. 2012, 63, 1461–1477. [Google Scholar] [CrossRef]
- Otagaki, S.; Ogawa, Y.; Hibrand-Saint Oyant, L.; Foucher, F.; Kawamura, K.; Horibe, T.; Matsumoto, S.; Peeters, T. Genotype of FLOWERING LOCUS T homologue contributes to flowering time differences in wild and cultivated roses. Plant Biol. 2015, 17, 808–815. [Google Scholar] [CrossRef]
- Xing, W.; Wang, Z.; Wang, X.; Bao, M.; Ning, G. Over-expression of an FT homolog from Prunus mume reduces juvenile phase and induces early flowering in rugosa rose. Sci. Hortic. 2014, 172, 68–72. [Google Scholar] [CrossRef]
- Liu, X.; Zhong, N.; Feng, G.; Yan, D.; Gao, J. A new advance on Camellia breeding in China studies on thirty summer-blooming hybrids from crosses between Camellia azalea and other Camellias. J. Agric. Sci. Technol. B 2013, 3, 254–264. [Google Scholar]
- Iwata, H.; Gaston, A.; Remay, A.; Thouroude, T.; Jeauffre, J.; Kawamura, K.; Oyant, L.H.S.; Araki, T.; Denoyes, B.; Foucher, F. The TFL1 homologue KSN is a regulator of continuous flowering in rose and strawberry. Plant J. 2012, 69, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Randoux, M.; Davière, J.M.; Jeauffre, J.; Thouroude, T.; Pierre, S.; Toualbia, Y.; Perrotte, J.; Reynoird, J.P.; Jammes, M.J.; Hibrand-Saint Oyant, L.; et al. RoKSN, a floral repressor, forms protein complexes with RoFD and RoFT to regulate vegetative and reproductive development in rose. New Phytol. 2014, 202, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Li, J.; Li, X.; Wu, B.; Wang, J.; Liu, Z.; Yin, H. Genome-wide transcriptome profiling provides insights into floral bud development of summer-flowering Camellia azalea. Sci. Rep. 2015, 5, 9729. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Su, S.; Ma, L.; Wen, Y.; Wang, X. Molecular cloning and functional characterization of CoFT1, a homolog of FLOWERING LOCUS T (FT) from Camellia oleifera. Gene 2017, 626, 215–226. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, L.; Chen, Y.; Wang, R.; Peng, S.; Chen, L.; Ma, L. Observation on morphological and anatomical characteristics of the flower bud differentiation on Camellia oleifera. J. Cent. S. Univ. For. Technol. 2011, 31, 22–27. [Google Scholar]
- Ho, W.W.; Weigel, D. Structural features determining flower-promoting activity of Arabidopsis FLOWERING LOCUS T. Plant Cell 2014, 26, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Hanzawa, Y.; Money, T.; Bradley, D. A single amino acid converts a repressor to an activator of flowering. Proc. Natl. Acad. Sci. USA 2005, 102, 7748–7753. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, S.J.; Jin, S.; Susila, H.; Youn, G.; Nasim, Z.; Alavilli, H.; Chung, K.S.; Yoo, S.J.; Ahn, J.H. Genetic interactions reveal the antagonistic roles of FT/TSF and TFL1 in the determination of inflorescence meristem identity in Arabidopsis. Plant J. 2019, 99, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Komiya, R.; Ikegami, A.; Tamaki, S.; Yokoi, S.; Shimamoto, K. Hd3a and RFT1 are essential for flowering in rice. Development 2008, 135, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Komiya, R.; Yokoi, S.; Shimamoto, K. A gene network for long-day flowering activates RFT1 encoding a mobile flowering signal in rice. Development 2009, 136, 3443–3450. [Google Scholar] [CrossRef]
- Meng, X.; Muszynski, M.G.; Danilevskaya, O.N. The FT-Like ZCN8 Gene Functions as a Floral Activator and Is Involved in Photoperiod Sensitivity in Maize. Plant Cell 2011, 23, 942–960. [Google Scholar] [CrossRef]
- Fan, C.; Hu, R.; Zhang, X.; Wang, X.; Zhang, W.; Zhang, Q.; Ma, J.; Fu, Y.-F. Conserved CO-FT regulons contribute to the photoperiod flowering control in soybean. BMC Plant Biol. 2014, 14, 9. [Google Scholar] [CrossRef]
- Kong, F.; Liu, B.; Xia, Z.; Sato, S.; Kim, B.M.; Watanabe, S.; Yamada, T.; Tabata, S.; Kanazawa, A.; Harada, K.; et al. Two Coordinately Regulated Homologs of FLOWERING LOCUS T Are Involved in the Control of Photoperiodic Flowering in Soybean. Plant Physiol. 2010, 154, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Jia, Z.; Cao, D.; Jiang, B.; Wu, C.; Hou, W.; Liu, Y.; Fei, Z.; Zhao, D.; Han, T. GmFT2a, a Soybean Homolog of FLOWERING LOCUS T, Is Involved in Flowering Transition and Maintenance. PLoS ONE 2011, 6, e29238. [Google Scholar] [CrossRef]
- Zhao, C.; Takeshima, R.; Zhu, J.; Xu, M.; Sato, M.; Watanabe, S.; Kanazawa, A.; Liu, B.; Kong, F.; Yamada, T.; et al. A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog. BMC Plant Biol. 2016, 16, 20. [Google Scholar] [CrossRef]
- Voogd, C.; Brian, L.A.; Wang, T.; Allan, A.C.; Varkonyi-Gasic, E. Three FT and multiple CEN and BFT genes regulate maturity, flowering, and vegetative phenology in kiwifruit. J. Exp. Bot. 2017, 68, 1539–1553. [Google Scholar] [CrossRef]
- Reig, C.; Gil-Muñoz, F.; Vera-Sirera, F.; García-Lorca, A.; Martínez-Fuentes, A.; Mesejo, C.; Pérez-Amador, M.A.; Agustí, M. Bud sprouting and floral induction and expression of FT in loquat [Eriobotrya japonica (Thunb.) Lindl.]. Planta 2017, 246, 915–925. [Google Scholar] [CrossRef]
- Hamama, L.; Bosselut, J.; Voisine, L.; Chameau, J.; Foucrier, S.; Pierre, S.; Jeauffre, J.; Ogé, L.; Thouroude, T.; Foucher, F.; et al. Study of rose RoFT transgenic plants. Acta Hortic. 2023, 1362, 29–36. [Google Scholar] [CrossRef]
- Xi, W.; Yu, H. An expanding list: Another flowering time gene, FLOWERING LOCUS T, regulates flower development. Plant Signal. Behav. 2009, 4, 1142–1144. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Chen, C.; Xu, Y.; Hu, S.; Wang, L.; Sun, K.; Chen, X.; Li, X. Optimization of Agrobacterium tumefaciens-mediated transformation systems in tea plant (Camellia sinensis). Hortic. Plant J. 2017, 3, 105–109. [Google Scholar] [CrossRef]
- Liu, G.; Huang, K.; Ke, J.; Chen, C.; Bao, G.H.; Wan, X. Novel Camellia sinensis O-methyltransferase regulated by CsMADSL1 specifically methylates EGCG in Cultivar “GZMe4”. J. Agric. Food Chem. 2023, 71, 6706–6716. [Google Scholar] [CrossRef]
- Mandel, M.A.; Yanofsky, M.F. A gene triggering flower formation in Arabidopsis. Nature 1995, 377, 522–524. [Google Scholar] [CrossRef]
- Bradley, D.; Rateliffe, O.; Vincent, C.; CarPenter, R.; Coen, E. Inflorescence commitment and architecture in Arabidopsis. Science 1997, 275, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Ho, Y.W.; Tao, C.N.; Chang, J.W. The unique LcFT1 gene sequence linked to early-flowering in the tropical litchi ‘Khom’. Acta Hortic. 2023, 1362, 389–396. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Target Gene | Forward Primers (5′ to 3′) | Reverse Primers (5′ to 3′) |
|---|---|---|---|
| C. azalea and C. japonica | GAPDH FT AP1 FUL SEP3 SVP CO EFL2 COL4 | CTGTCGATGTCTCAGTGGTTGAC ATGACCTTAGGACCTTCTACACTC CAGTCTCAATGGGAGCAGCA TCAAAGTGTCGCCAGTGGAA TTTCCGTTCTCTGCGATGCT CATGAATGAAATTGCCACCCT ATCGGAAGCTGATGGGTTCT CTGGGCTTGGAAATGGAGGT GTTGTTCCAGACCACAACGC | TGATCTCATCATAGGAAGCCTTCTT CATAACAAACCACCTCTTGCC AGCATCCAAGGTCACAAGCA GATGAAGGATGCCACCACCA GCTGTTTAGGGGGCCAAGAT AACATTAGCCACTCCCACTC ACAACCACGAACCAGACTCG GGTCAACCACCCTCCTGATG CTCGCTTCTCTGTCGATCCC |
| Arabidopsis | ACTIN LFY AP1 FUL SOC1 CO SVP | GGTAACATTGTGCTCAGTGGTGG CAGCAGCAGAGACGGAGAAA AGCAGTGGGATCAGCAGAAC AAACGGGTCAGCAAGAAGGA GCTCTCAGTGCTTTGTGATGC GGCTCCTCAGGGACTCACTA AACTCTCCGTTCTCTGCGAC | AACGACCTTAATCTTCATGCTGC CCTTGGTGGGGCATTTTTCG CGGGTTCAAGAGTCAGTTCGA TCCCCCAACTCTCTCCACAA CGGTTTGGTGCTGACTCGAT CCTGCTGCGTTATGGGAAGA GGGCGTGATCACTGTTCTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Li, J.; Yin, H.; Li, X.; Liu, W.; Fan, Z. Function of FT in Flowering Induction in Two Camellia Species. Plants 2024, 13, 784. https://doi.org/10.3390/plants13060784
Wang X, Li J, Yin H, Li X, Liu W, Fan Z. Function of FT in Flowering Induction in Two Camellia Species. Plants. 2024; 13(6):784. https://doi.org/10.3390/plants13060784
Chicago/Turabian StyleWang, Xiong, Jiyuan Li, Hengfu Yin, Xinlei Li, Weixin Liu, and Zhengqi Fan. 2024. "Function of FT in Flowering Induction in Two Camellia Species" Plants 13, no. 6: 784. https://doi.org/10.3390/plants13060784
APA StyleWang, X., Li, J., Yin, H., Li, X., Liu, W., & Fan, Z. (2024). Function of FT in Flowering Induction in Two Camellia Species. Plants, 13(6), 784. https://doi.org/10.3390/plants13060784