
Selective Genotyping and Phenotyping for Optimization of Genomic Prediction Models for Populations with Different Diversity
 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Results
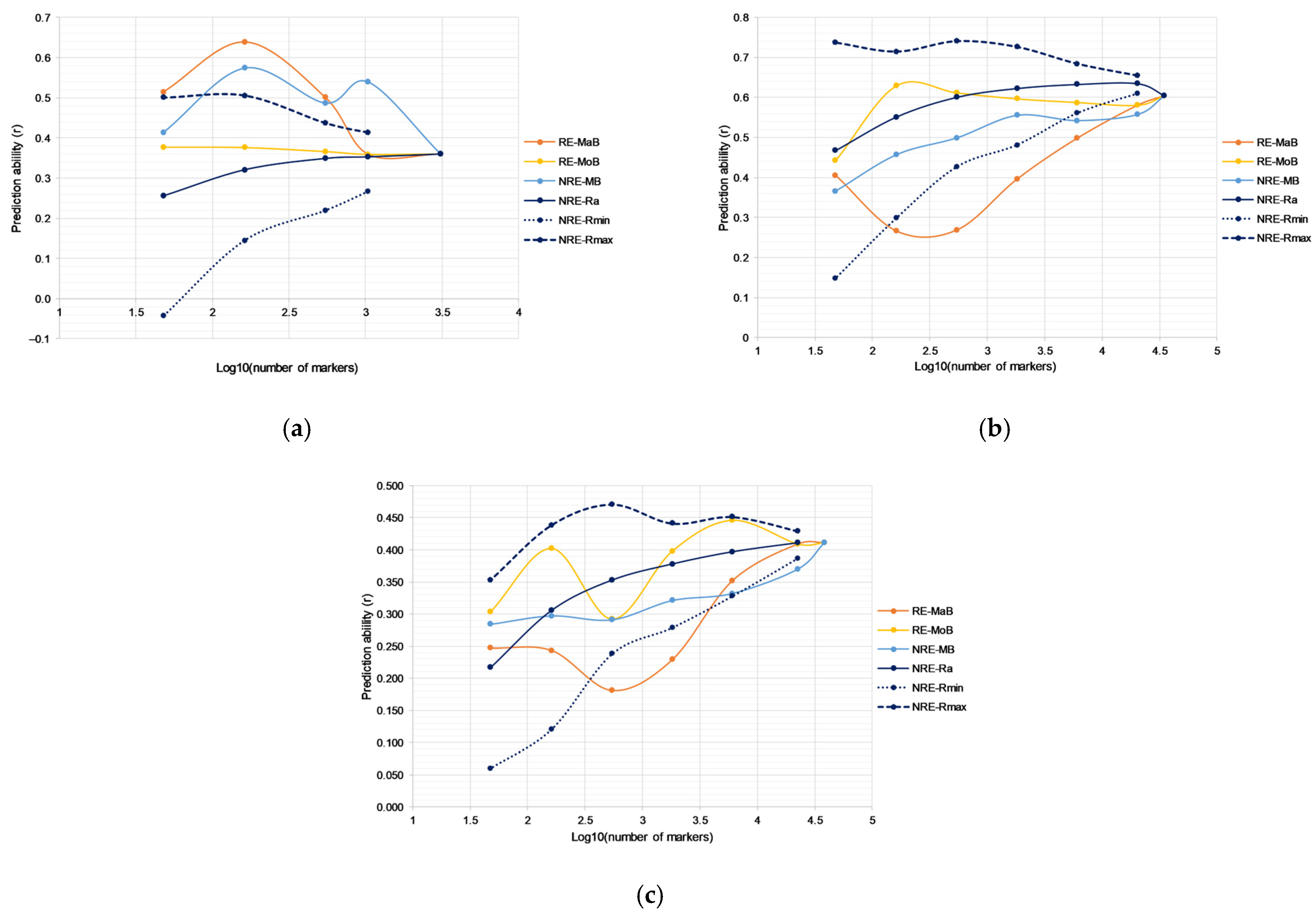
2.1. Predictive Ability across Different Methods for Selective Genotyping
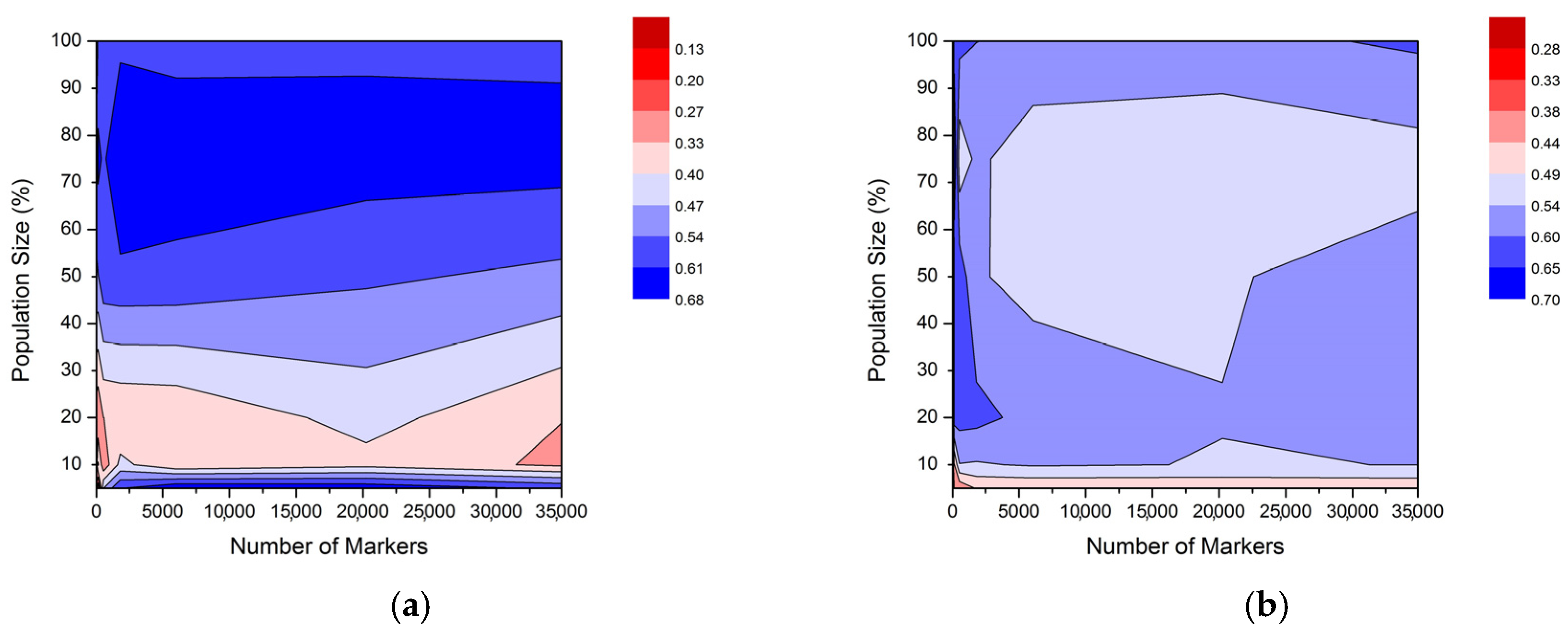
2.2. Prediction Ability across Different Methods for Selective Phenotyping
3. Discussion
4. Materials and Methods
4.1. Material
4.2. Methods
4.2.1. Genomic Prediction
- Cross-validation with 20 repetitions of a 10-fold scheme, where prediction ability (rcv) was evaluated with a validation set representing 10% of genotypes from the training set;
- External validation, where prediction ability (rev) was evaluated based on genotypes that were not included in the training set.
4.2.2. Selective Genotyping Strategies
SNP Pruning
Approaches without and with Re-Estimation of Marker Effects
Genome-Wide Association Study (GWAS) Based Strategy
4.2.3. Selective Phenotyping Strategies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, B.; Piao, S.; Wang, X.; Lobell, D.B.; Huang, Y.; Yao, Y.; Bassu, S.; Ciais, P.; Durand, J.; et al. Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. USA 2017, 114, 9326–9331. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Bao, Y.; Vuong, T.; Meinhardt, C.; Tiffin, P.; Denny, R.; Chen, S.; Nguyen, H.T.; Orf, J.H.; Young, N.D. Potential of association mapping and genomic selection to explore PI 88788 derived soybean cyst nematode resistance. Plant Genome 2014, 7, 1–13. [Google Scholar] [CrossRef]
- Peixoto, L.A.; Moellers, T.C.; Zhang, J.; Lorenz, A.J.; Bhering, L.L.; Beavis, W.D.; Singh, A.K. Leveraging genomic prediction to scan germplasm collection for crop improvement. PLoS ONE 2017, 12, e0179191. [Google Scholar] [CrossRef]
- Ravelombola, W.S.; Qin, J.; Shi, A.; Nice, L.; Bao, Y.; Lorenz, A.; Orf, J.H.; Young, N.D.; Chen, S. Genome-wide association study and genomic selection for soybean chlorophyll content associated with soybean cyst nematode tolerance. BMC Genom. 2019, 20, 904. [Google Scholar] [CrossRef]
- Qin, J.; Shi, A.; Song, Q.; Li, S.; Wang, F.; Cao, Y.; Ravelombola, W.; Song, Q.; Yang, C.; Zhang, M. Genome Wide Association Study and Genomic Selection of Amino Acid Concentrations in Soybean Seeds. Front. Plant Sci. 2019, 10, 1445. [Google Scholar] [CrossRef]
- Riaz, A.; Raza, Q.; Kumar, A.; Dean, D.; Chiwina, K.; Phiri, T.M.; Thomas, J.; Shiet, A. GWAS and genomic selection for marker-assisted development of sucrose enriched soybean cultivars. Euphytica 2023, 219, 97. [Google Scholar] [CrossRef]
- Duhnen, A.; Gras, A.; Teyssčdre, S.; Romestant, M.; Claustre, B.; Daydé, J.; Mangin, B. Genomic selection for yield and seed protein content in soybean: A study of breeding program data and assessment of prediction accuracy. Crop Sci. 2017, 57, 1325–1337. [Google Scholar] [CrossRef]
- Matei, G.; Woyann, L.G.; Milioli, A.S.; Oliveira, I.B.; Zdziarski, A.D.; Zanella, R.; Coelho, A.S.G.; Finatto, T.; Benin, G. Genomic selection in soybean: Accuracy and time gain in relation to phenotypic selection. Mol. Breed. 2018, 38, 117. [Google Scholar] [CrossRef]
- Stewart-Brown, B.B.; Song, Q.; Vaughn, J.N.; Li, Z. Genomic Selection for Yield and Seed Composition Traits within an Applied Soybean Breeding Program. G3—Genes. Genom. Genet. 2019, 9, 2253–2265. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Song, Q.; Li, Z. Genomic selection of soybean (Glycine max) for genetic improvement of yield and seed composition in a breeding context. Plant Genome 2023, 16, e20384. [Google Scholar] [CrossRef] [PubMed]
- Jarquín, D.; Kocak, K.; Posadas, L.; Hyma, K.; Jedlicka, J.; Graef, G.; Lorenz, A. Genotyping by sequencing for genomic prediction in a soybean breeding population. BMC Genom. 2014, 15, 740. [Google Scholar] [CrossRef]
- Ma, Y.; Reif, J.C.; Jiang, Y.; Wen, Z.; Wang, D.; Liu, Z.; Guo, Y.; Wei, S.; Wang, S.; Yang, C.; et al. Potential of marker selection to increase prediction accuracy of genomic selection in soybean (Glycine max L.). Mol. Breed. 2016, 36, 113. [Google Scholar] [CrossRef] [PubMed]
- Đorđević, V.; Ćeran, M.; Miladinović, J.; Balešević-Tubić, S.; Petrović, K.; Miladinov, Z.; Marinković, J. Exploring the performance of genomic prediction models for soybean yield using different validation approaches. Mol. Breed. 2019, 39, 74. [Google Scholar] [CrossRef]
- Ravelombola, W.; Qin, J.; Shi, A.; Song, Q.; Yuan, J.; Wang, F.; Chen, P.; Yan, L.; Feng, Y.; Zhao, T.; et al. Genome-wide association study and genomic selection for yield and related traits in soybean. PLoS ONE 2021, 16, e0255761. [Google Scholar] [CrossRef] [PubMed]
- Yoosefzadeh-Najafabadi, M.; Rajcan, I.; Eskandari, M. Optimizing genomic selection in soybean: An important improvement in agricultural genomics. Heliyon 2022, 8, e11873. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Akdemir, D.; Isidro-Sánchez, J. Design of training populations for selective phenotyping in genomic prediction. Sci. Rep. 2019, 9, 1446. [Google Scholar] [CrossRef] [PubMed]
- Lorenzana, R.E.; Bernardo, R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor. Appl. Genet. 2009, 120, 151–161. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Ding, X.; Bijma, P.; de Koning, D.J.; Zhang, Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE 2010, 5, e12648. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, A.J.; Chao, S.; Asoro, F.G.; Heffner, E.L.; Hayashi, T.; Iwata, H.; Smith, K.P.; Sorrells, M.E.; Jannink, J.L. Genomic Selection in Plant Breeding. Knowledge and Prospects. Adv. Agronoy 2011, 110, 77–123. [Google Scholar] [CrossRef]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Jeong, S.; Kim, J.-Y.; Kim, N. GMStool: GWAS-based marker selection tool for genomic prediction from genomic data. Sci. Rep. 2020, 10, 19653. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, M.L.; Pong-Wong, R.; Spiliopoulou, A.; Hayward, C.; Rudan, I.; Campbell, H.; Wright, A.F.; Wilson, J.F.; Agakov, F.; Navarro, P.; et al. Application of high-dimensional feature selection: Evaluation for genomic prediction in man. Sci. Rep. 2015, 5, 10312. [Google Scholar] [CrossRef] [PubMed]
- Resende, M.F.; Muñoz, P.; Resende, M.D.; Garrick, D.J.; Fernando, R.L.; Davis, J.M.; Jokela, E.J.; Martin, T.A.; Peter, G.F.; Kirst, M. Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 2012, 190, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef]
- Hoffstetter, A.; Cabrera, A.; Huang, M.; Sneller, C. Optimizing training population data and validation of genomic selection for economic traits in soft winter wheat. G3—Genes. Genom. Genet. 2016, 6, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.B.; Galli, G.; Lyra, D.H.; Granato, ĺ.S.C.; Matias, F.I.; Alves, F.C.; Fritsche-Neto, R. Increasing accuracy and reducing costs of genomic prediction by marker selection. Euphytica 2019, 215, 18. [Google Scholar] [CrossRef]
- Lado, B.; Matus, I.; Rodríguez, A.; Inostroza, L.; Poland, J.; Belzile, F.; del Pozo, A.; Quincke, M.; Castro, M.; von Zitzewitz, J. Increased genomic prediction accuracy in wheat breeding through spatial adjustment of field trial data. G3—Genes. Genom. Genet. 2013, 3, 2105–2114. [Google Scholar] [CrossRef]
- Isidro-Sánchez, J.; Jannink, J.L.; Akdemir, D.; Poland, J.; Heslot, N.; Sorrells, M.E. Training set optimization under population structure in genomic selection. Theor. Appl. Genet. 2015, 128, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Heslot, N.; Yang, H.; Sorrells, M.; Jannink, J. Genomic selection in plant breeding: A comparison of models. Crop Sci. 2012, 52, 146–160. [Google Scholar] [CrossRef]
- Jarquin, D.; Specht, J.; Lorenz, A. Prospects of Genomic Prediction in the USDA Soybean Germplasm Collection: Historical Data Creates Robust Models for Enhancing Selection of Accessions. G3—Genes. Genom. Genet. 2016, 6, 2329–2341. [Google Scholar] [CrossRef] [PubMed]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning: With Applications in R; Springer: New York, NY, USA, 2013; p. 426. [Google Scholar]
- Calus, M.P.L.; Vandenplas, J. SNPrune: An efcient algorithm to prune large SNP array and sequence datasets based on high linkage disequilibrium. Genet. Sel. Evol. 2018, 50, 34. [Google Scholar] [CrossRef] [PubMed]
- Gianola, D.; de los Campos, G.; Hill, W.G.; Manfredi, E.; Fernando, R. Additive genetic variability and the Bayesian alphabet. Genetics 2011, 189, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.A.; Hickey, J.M.; van der Werf, J.H. Different models of genetic variation and their effect on genomic evaluation. Genet. Sel. Evol. 2013, 45, 24. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2007, 8, 186. [Google Scholar] [CrossRef] [PubMed]
- Tayeh, N.; Klein, A.; Le Paslier, M.-C.; Jacquin, F.; Houtin, H.; Rond, C.; Chabert-Martinello, M.; Magnin-Robert, J.-B.; Marget, P.; Aubert, G.; et al. Genomic Prediction in Pea: Effect of Marker Density and Training Population Size and Composition on Prediction Accuracy. Front. Plant Sci. 2015, 6, 941. [Google Scholar] [CrossRef]
- Wu, X.L.; Xu, J.; Feng, G.; Wiggans, G.R.; Taylor, J.F.; He, J.; Qian, C.; Qiu, J.; Simpson, B.; Walker, J.; et al. Optimal Design of Low-Density SNP Arrays for Genomic Prediction: Algorithm and Applications. PLoS ONE 2016, 11, e0161719. [Google Scholar] [CrossRef]
- Muir, W.M. Comparison of genomic and traditional BLUP-estimated breeding value accuracy and selection response under alternative trait and genomic parameters. J. Anim. Breed. Genet. 2007, 124, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Xavier, A.; Muir, W.M.; Rainey, K.M. Assessing predictive properties of genome-wide selection in soybeans. G3—Genes. Genom. Genet. 2016, 6, 2611–2616. [Google Scholar] [CrossRef] [PubMed]
- Beche, E.; Gillman, J.D.; Song, Q.; Nelson, R.; Beissinger, T.; Decker, J.; Shannon, G.; Scaboo, A.M. Genomic prediction using training population design in interspecific soybean populations. Mol. Breed. 2021, 41, 15. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef]
- Brøndum, R.F.; Su, G.; Janss, L.; Sahana, G.; Guldbrandtsen, B.; Boichard, D.; Lund, M.S. Quantitative trait loci markers derived from whole genome sequence data increases the reliability of genomic prediction. J. Dairy. Sci. 2015, 98, 4107–4116. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, F.; Lange, T.M.; Kircher, M.; Ramzan, F.; Schmitt, A.O.; Gultas, M. Exploring the potential of incremental feature selection to improve genomic prediction accuracy. Genet. Sel. Evol. 2023, 55, 78. [Google Scholar] [CrossRef] [PubMed]
- Jannink, J.L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E.; Hayes, B.J. Genomic selection. J. Anim. Breed. Genet. 2007, 124, 323–330. [Google Scholar] [CrossRef]
- VanRaden, P.M.; Van Tassell, C.P.; Wiggans, G.R.; Sonstegard, T.S.; Schnabel, R.D.; Taylor, J.F.; Schenkel, F.S. Invited review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy. Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef]
- Isidro-Sánchez, J.; Akdemir, D. Training Set Optimization for Sparse Phenotyping in Genomic Selection: A Conceptual Overview. Front. Plant Sci. 2021, 12, 715910. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, J.; Akdemir, D.; Isidro-Sánchez, J. A comparison of methods for training population optimization in genomic selection. Theor. Appl. Genet. 2023, 136, 30. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.-Y.; Ou, J.-H.; Liao, C.-T. Sample size determination for training set optimization in genomic prediction. Theor. Appl. Genet. 2023, 136, 57. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Song, Q.; Yan, L.; Quigley, C.; Jordan, B.D.; Fickus, E.; Schroeder, S.; Song, B.-H.; Charles An, Y.-Q.; Hyten, D.; Nelson, R.; et al. Genetic characterization of the soybean nested association mapping population. Plant Genome 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing data inference for whole genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-bysequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Hyten, D.L.; Jia, G.; Quigley, C.V.; Fickus, E.W.; Nelson, R.L.; Cregan, P.B. Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS ONE 2013, 8, e54985. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- De Beukelaer, H.; Davenport, G.F.; Fack, V. Core Hunter 3: Flexible core subset selection. BMC Bioinform. 2018, 19, 203. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Training Set Size (CV) | Training/Validation Set Size (EV) | Initial SNPs | Pruned SNPs |
|---|---|---|---|---|
| RIL | 137 | 117/20 | 3088 | 1044 |
| MDL | 227 | 227/21 | 34,889 | 20,242 |
| GPL | 1107 | 1107/301 | 38,184 | 22,470 |
| Population | Sample Size | Markers | Correlation Rate (Mean ± Std.) | ||||
|---|---|---|---|---|---|---|---|
| Training | Validation | Test | All | Selected | Training | Validation | |
| MDL | 151 | 76 | 21 | 34,889 | 306 | 0.948 ± 0.015 | 0.941 ± 0.058 |
| GPL | 886 | 221 | 301 | 38,184 | 1112 | 0.917 ± 0.043 | 0.886 ± 0.070 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ćeran, M.; Đorđević, V.; Miladinović, J.; Vasiljević, M.; Đukić, V.; Ranđelović, P.; Jaćimović, S. Selective Genotyping and Phenotyping for Optimization of Genomic Prediction Models for Populations with Different Diversity. Plants 2024, 13, 975. https://doi.org/10.3390/plants13070975
Ćeran M, Đorđević V, Miladinović J, Vasiljević M, Đukić V, Ranđelović P, Jaćimović S. Selective Genotyping and Phenotyping for Optimization of Genomic Prediction Models for Populations with Different Diversity. Plants. 2024; 13(7):975. https://doi.org/10.3390/plants13070975
Chicago/Turabian StyleĆeran, Marina, Vuk Đorđević, Jegor Miladinović, Marjana Vasiljević, Vojin Đukić, Predrag Ranđelović, and Simona Jaćimović. 2024. "Selective Genotyping and Phenotyping for Optimization of Genomic Prediction Models for Populations with Different Diversity" Plants 13, no. 7: 975. https://doi.org/10.3390/plants13070975