

Integrating Genome-Wide Association Study, Transcriptome and Metabolome Reveal Novel QTL and Candidate Genes That Control Protein Content in Soybean
 ,
,
Abstract
:1. Introduction
2. Results
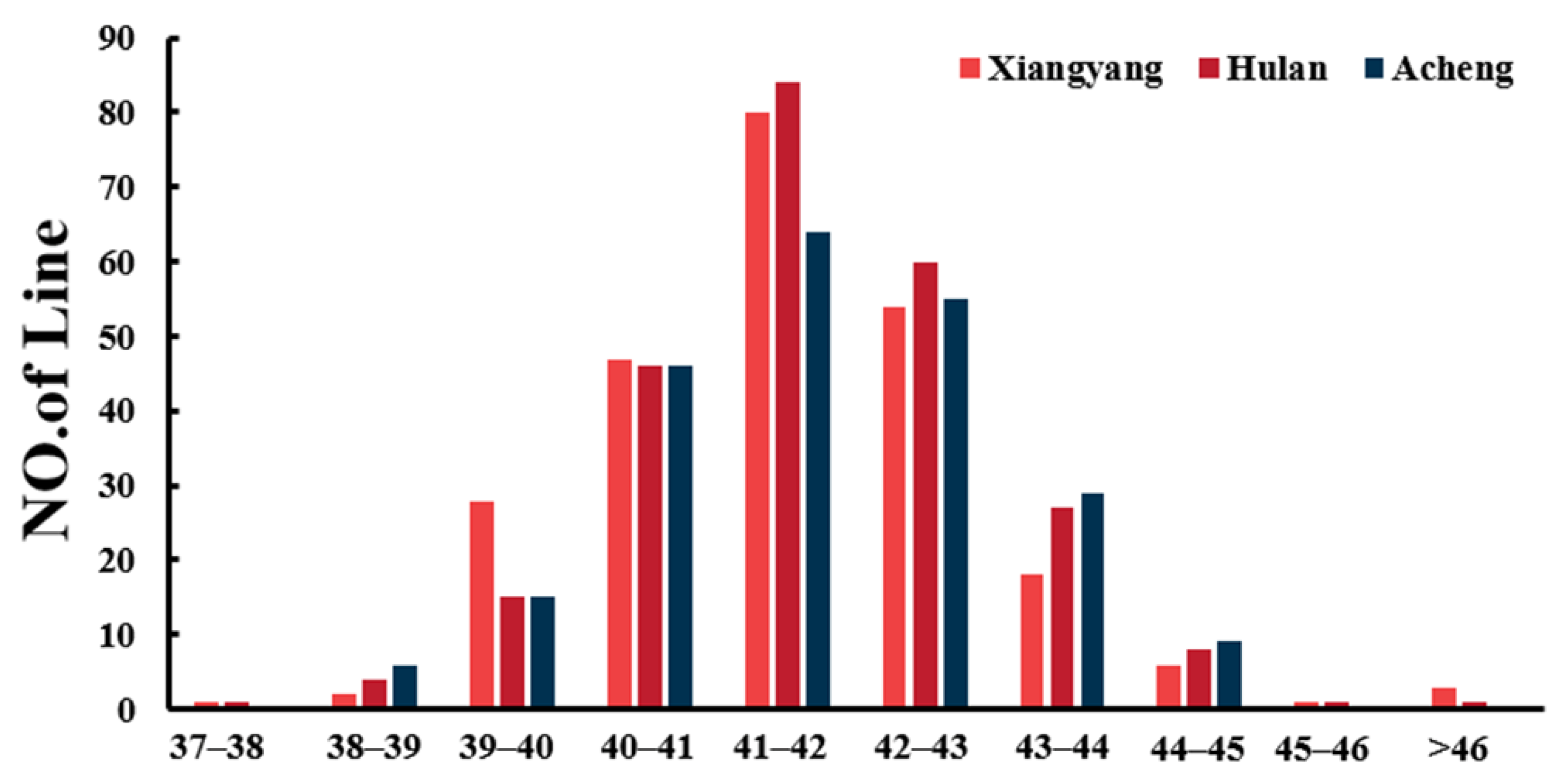
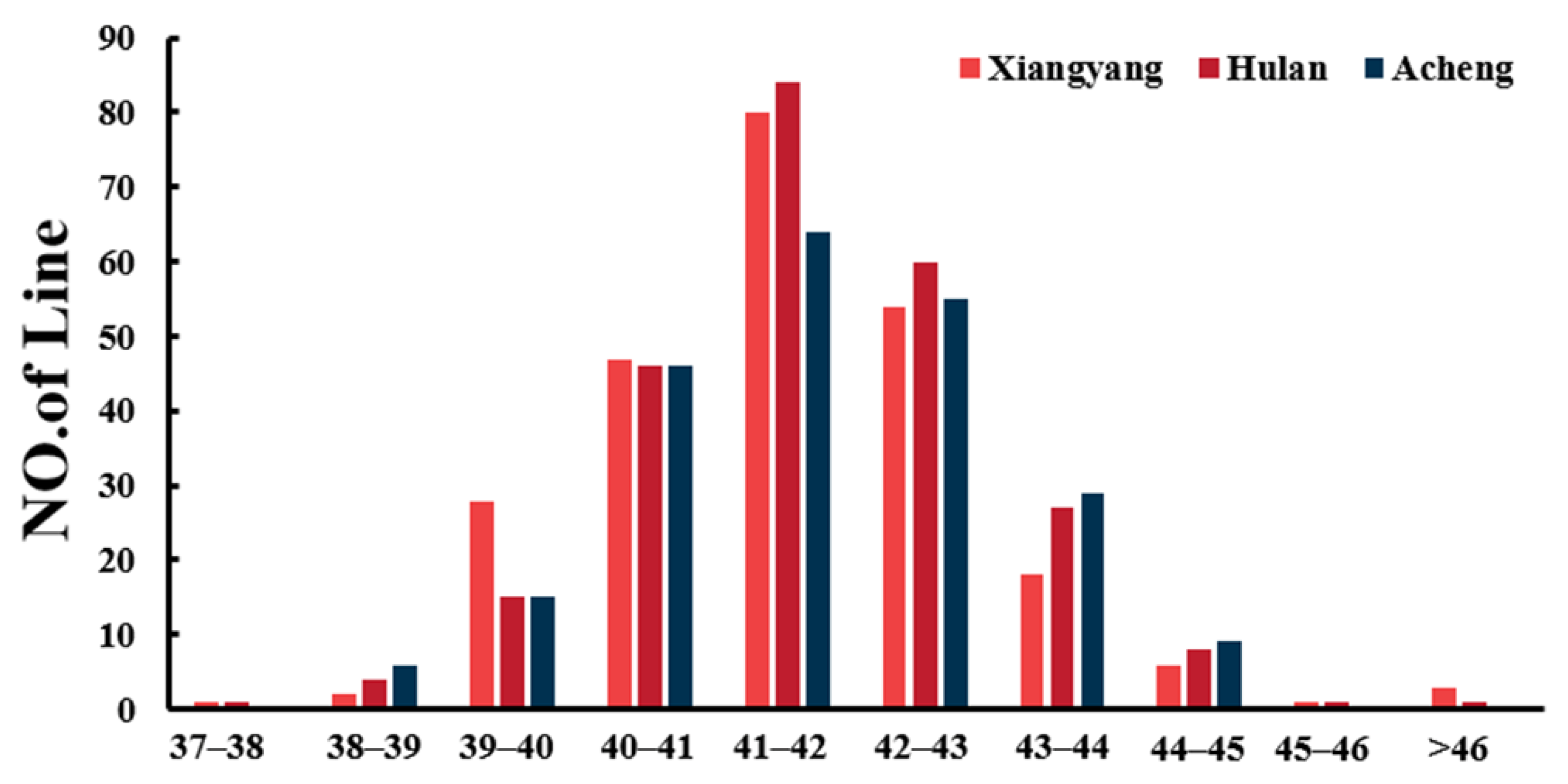
2.1. Statistical and Variation Analysis of Protein Content
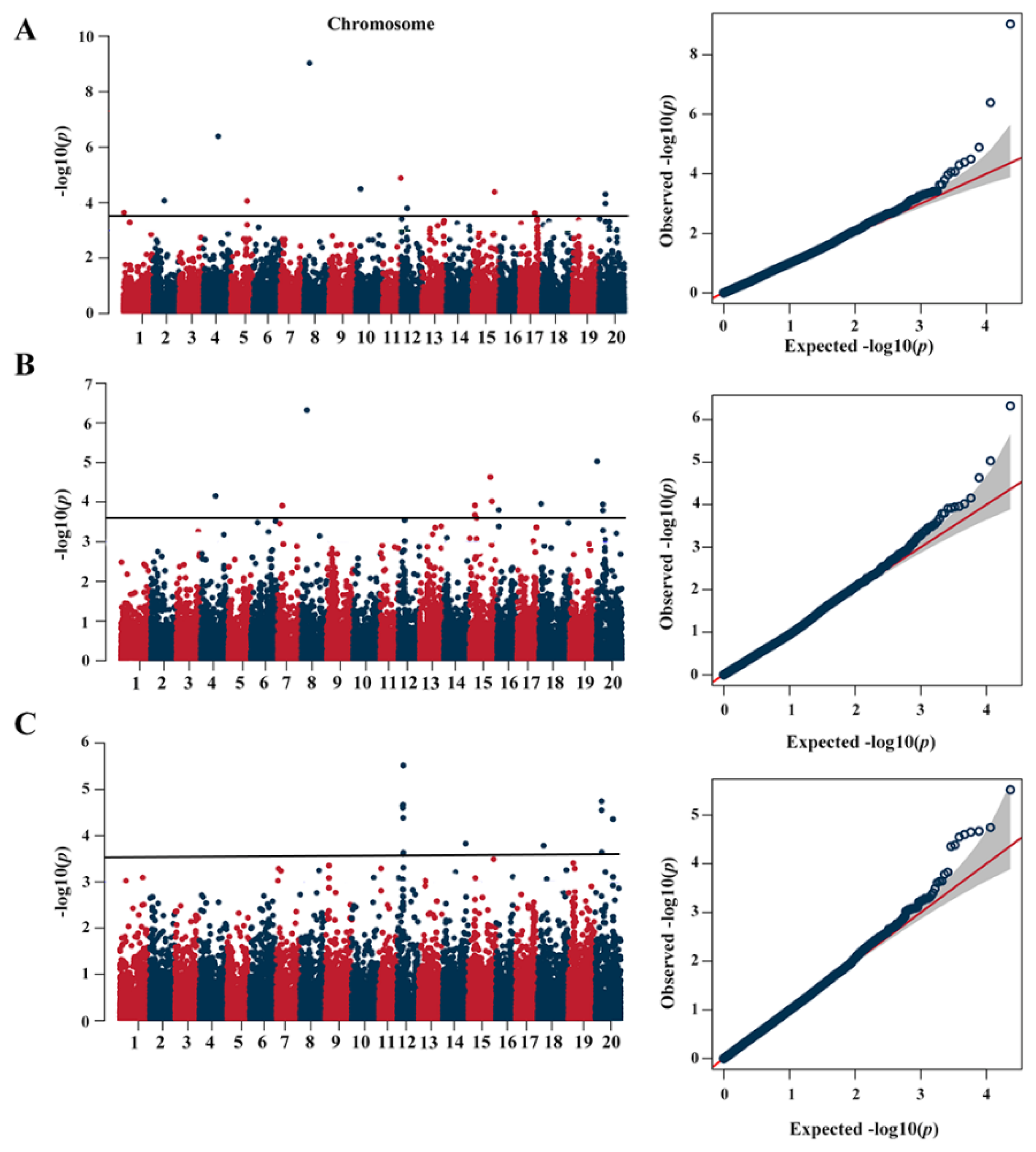
2.2. Population Structure and GWAS Analysis
2.3. Quantitative Trait Nucleotide (QTN) Associated with Protein Content by GWAS
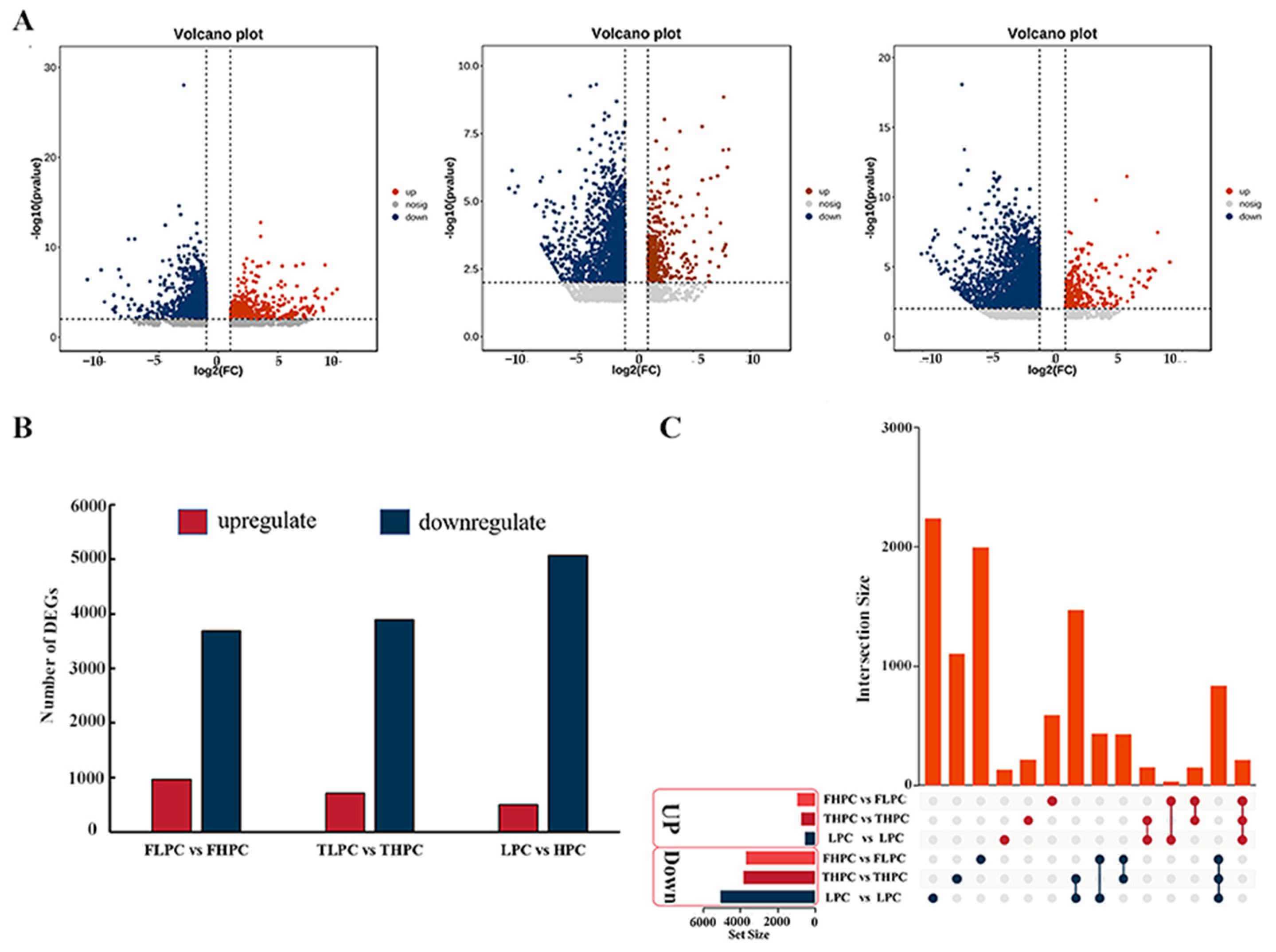
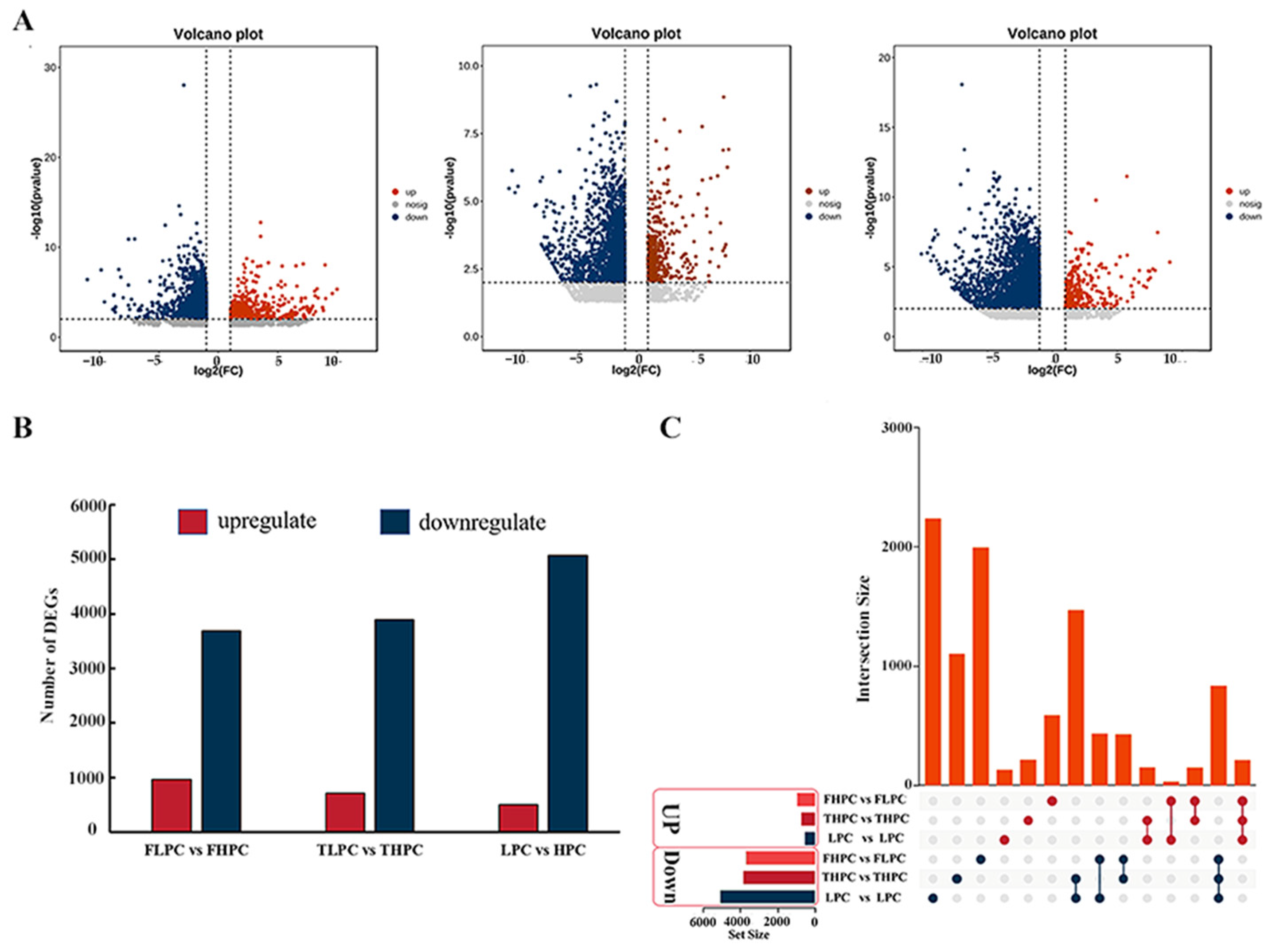
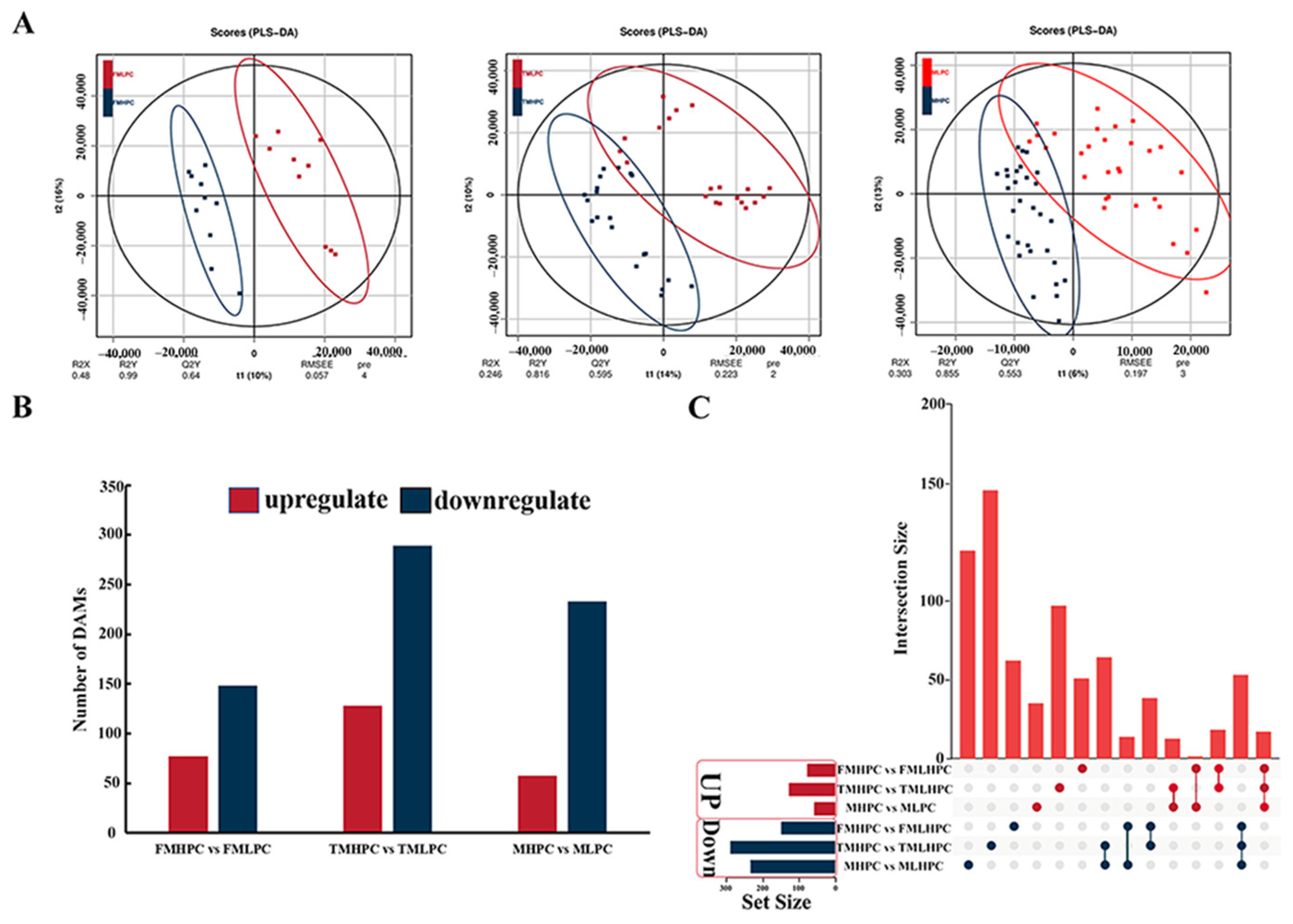
2.4. Transcriptomic Analysis of HPC and LPC Soybean Seeds
2.5. Combining GWAS and RNA Seq Results to Determine Candidate Genes for PC
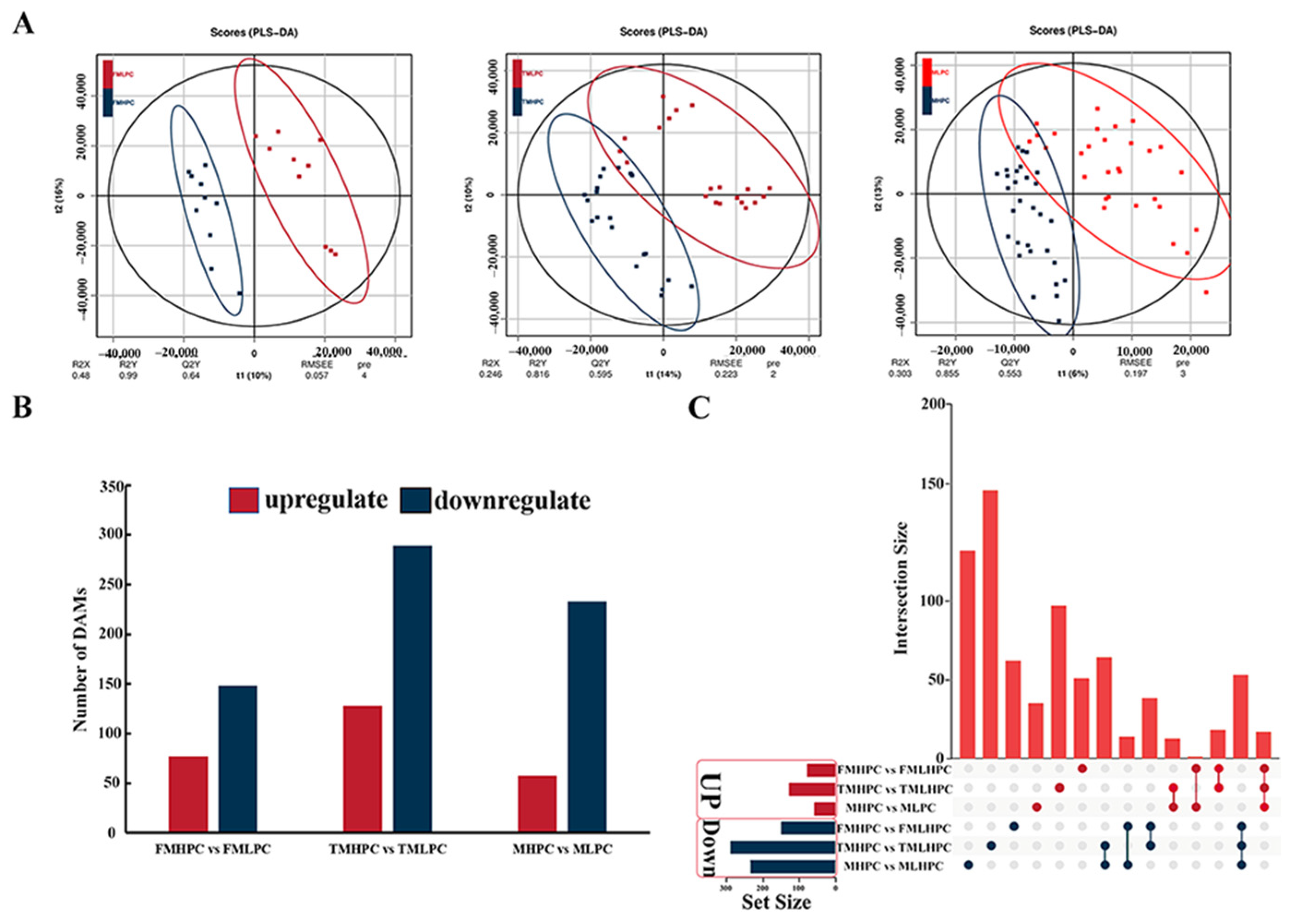
2.6. Metabolic Profiling Analysis of MHPC and MLPC Soybean Seeds
2.7. Differential Accumulation of Metabolite with HPC and LPC
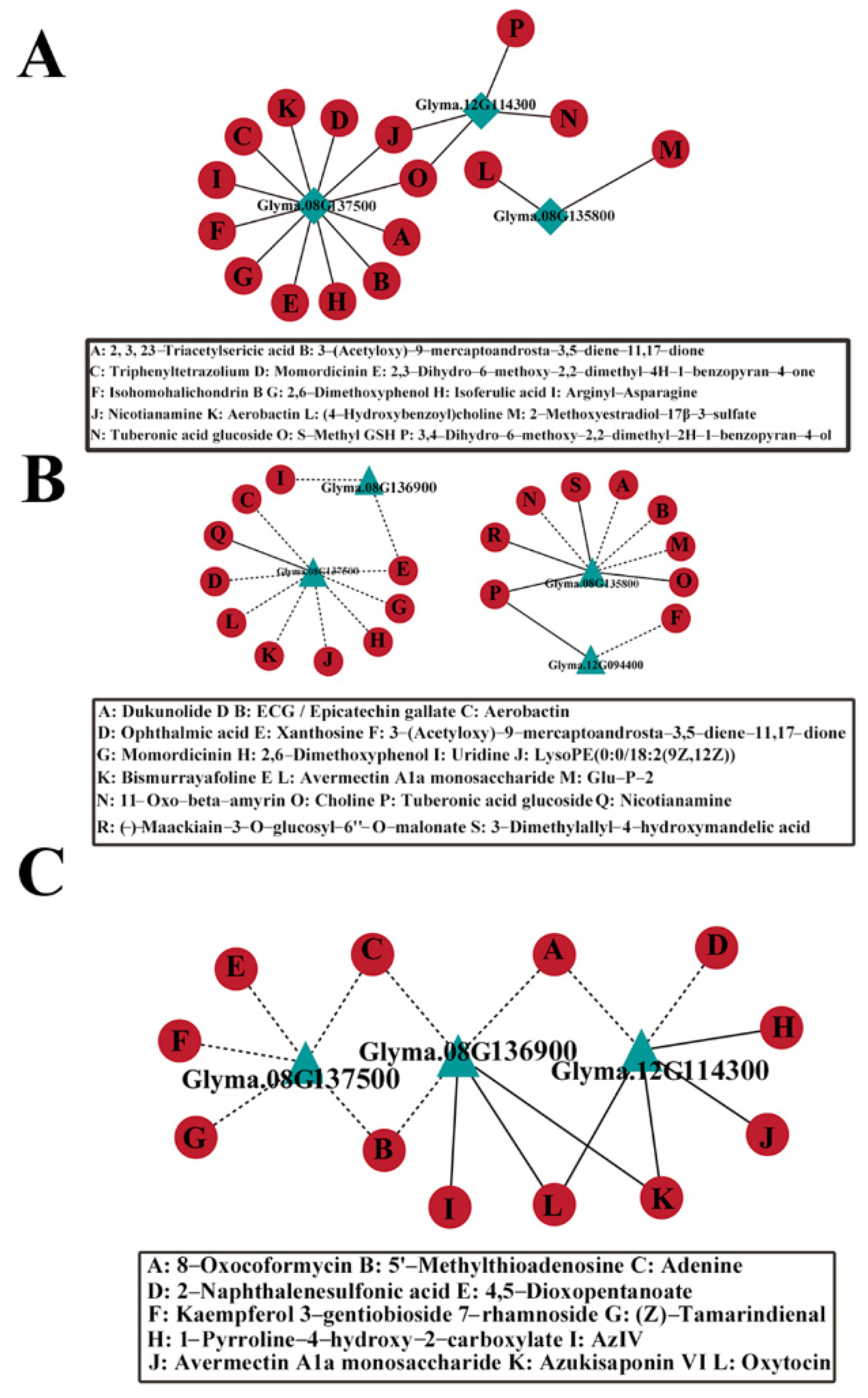
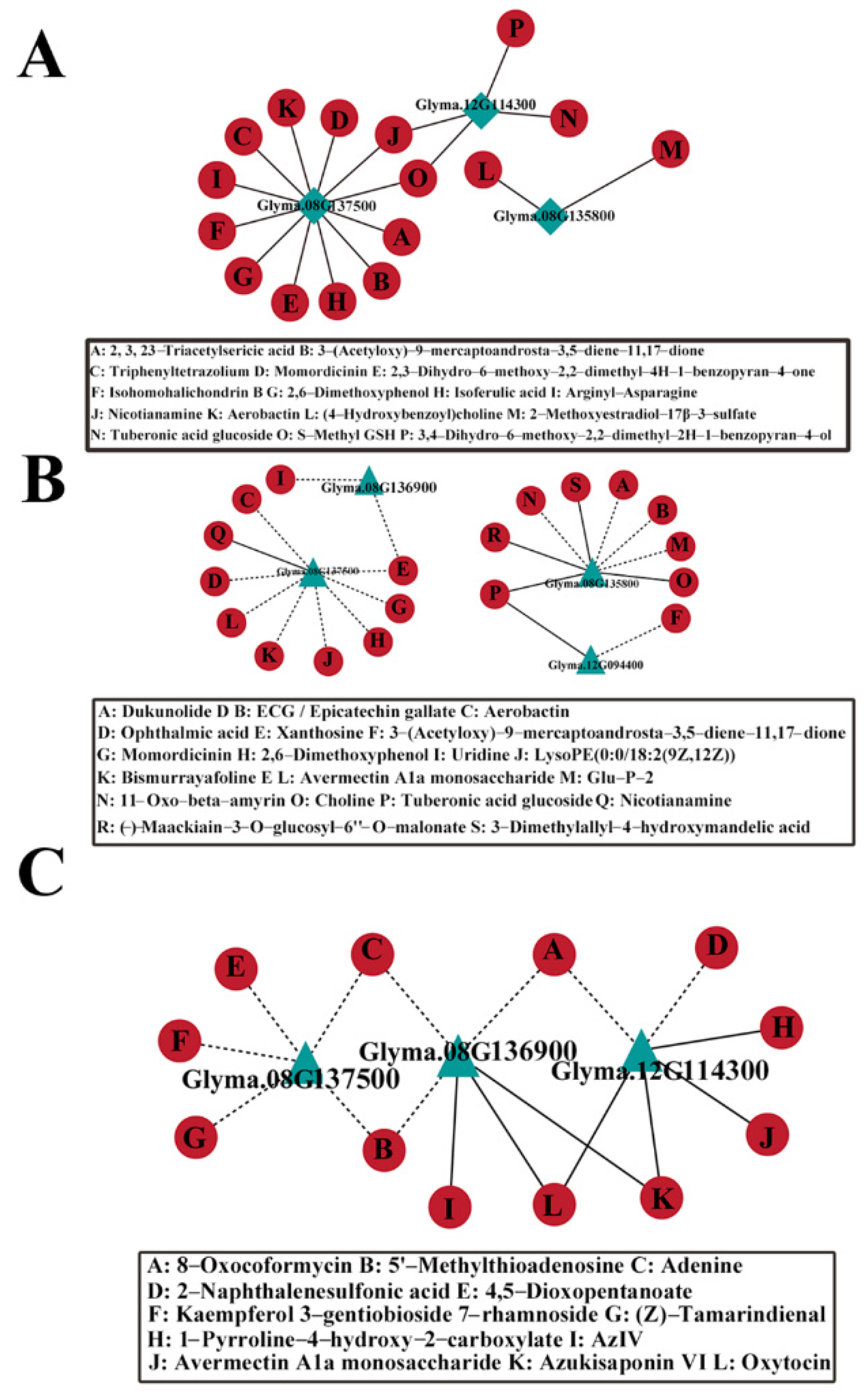
2.8. Co-Expression Analysis of Candidate Genes and DAM Metabolites
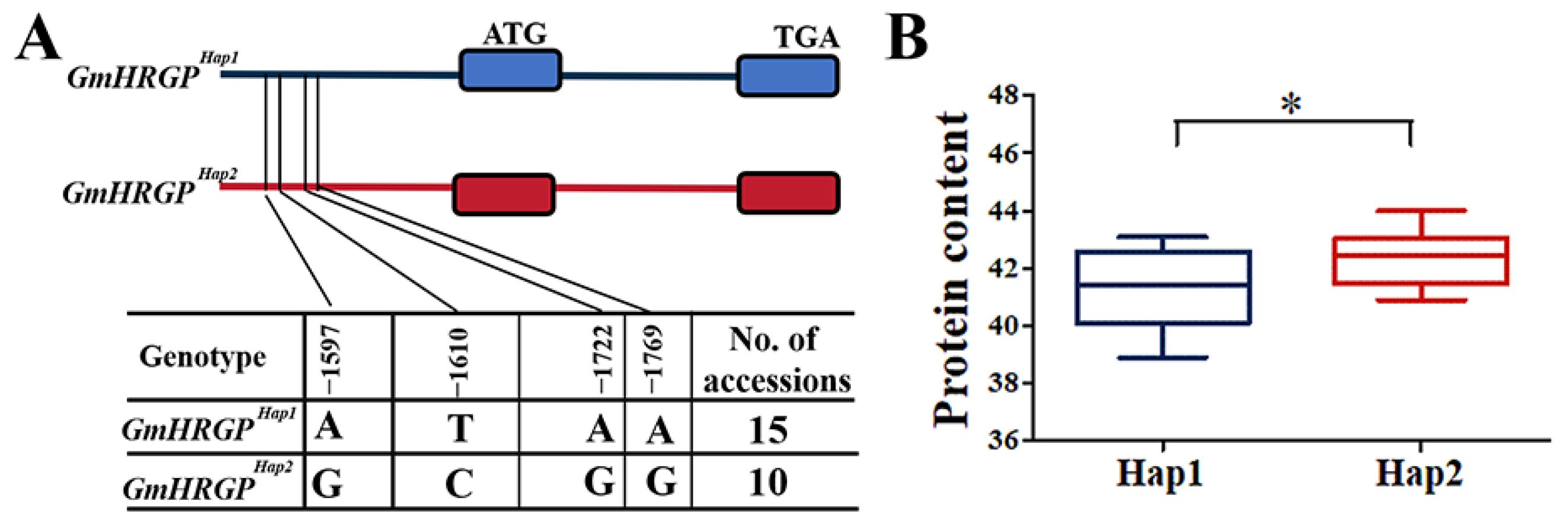
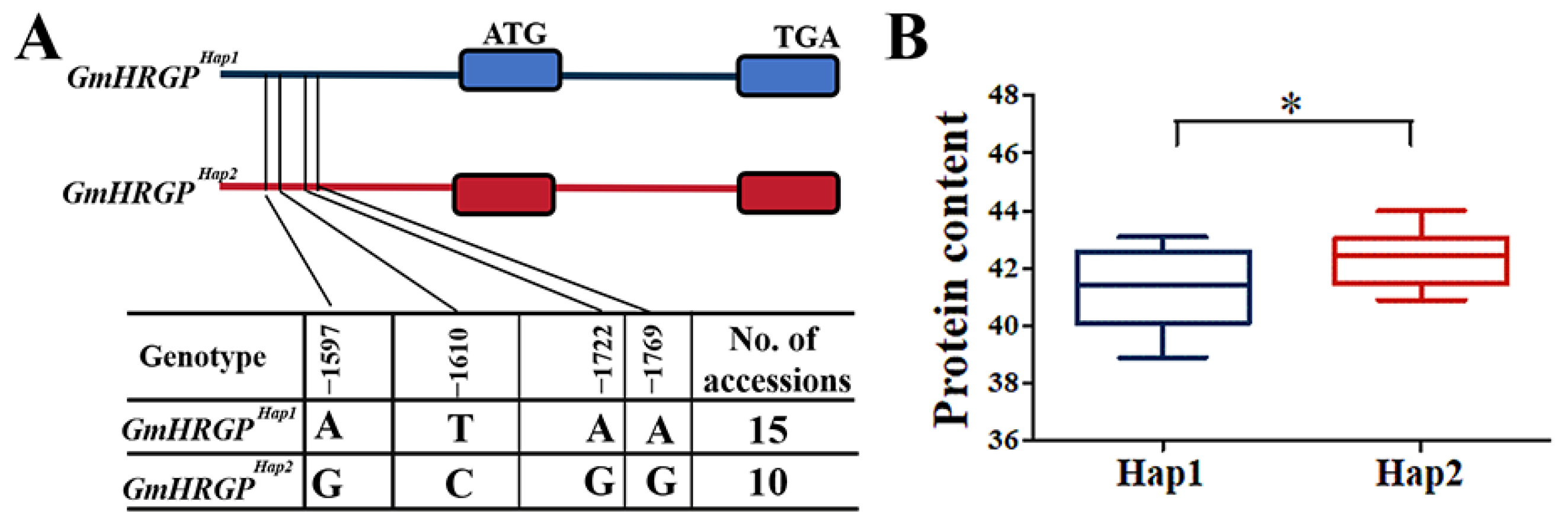
2.9. Gene-Based Association and Haplotype Analysis of Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Isolation and SNP Genotyping Data Collection
4.3. Genome-Wide Association Analysis
4.4. Metabolome Analysis
4.5. Transcriptome Analysis
4.6. Prediction of Candidate Genes
4.7. Co-Expression Analysis
4.8. Quantitative Real-Time PCR
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Natarajan, S.; Luthria, D.; Bae, H.; Lakshman, D.; Mitra, A. Transgenic soybeans and soybean protein analysis: An overview. J. Agric. Food Chem. 2013, 61, 11736–11743. [Google Scholar] [CrossRef]
- Chaudhary, J.; Patil, G.; Sonah, H.; Deshmukh, R.; Vuong, T.; Valliyodan, B. Expanding omics resources for improvement of soybean seed composition traits. Front. Plant Sci. 2015, 6, 1021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Lu, Y.; Bhusal, S.J.; Song, Q.; Cregan, P.B.; Yen, Y.; Brown, M.; Jiang, G. Genome-wide scan for seed composition provides insights into soybean quality improvement and the impacts of domestication and breeding. Mol. Plant 2018, 11, 460–472. [Google Scholar] [CrossRef]
- Wang, J.; Lin, L.; Yong, G.; Wang, Y.; Zhang, L.; Jin, L.; Guan, R.; Liu, Z.; Wang, L.; Chang, R. A Dominant locus, qBSC-1, controls beta subunit content of seed storage protein in soybean (Glycine max (L.) Merri.). J. Integr. Agr. 2014, 13, 1854–1864. [Google Scholar] [CrossRef]
- Boehm, J.D.; Nguyen, V.; Tashiro, R.M.; Anderson, D.; Shi, C.; Wu, X.; Woodrow, L.; Yu, K.; Cui, Y.; Li, Z. Genetic mapping and validation of the loci controlling 7S α’ and 11S A-type storage protein subunits in soybean [Glycine max (L.) Merr.]. Theor. Appl. Genet. 2018, 131, 659–671. [Google Scholar] [CrossRef]
- Wang, J.; Mao, L.; Zeng, Z.; Yu., X.; Lian., J.; Feng, J.; Yang, W.; An, J.; Wu, H.; Zhang, M.; et al. Genetic mapping high protein content QTL from soybean ‘Nanxiadou 25’ and candidate gene analysis. BMC Plant Biol. 2021, 21, 388. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, K.; Liu, S.; Sun, X.; Li, X.; Song, J.; Qi, Z.; Wang, Y.; Fang, Y.; Wang, J.; et al. Quantitative trait locus analysis of protein and oil content in response to planting density in soybean (Glycine max [L.] Merri.) seeds based on SNP linkage mapping. Front. Genet. 2020, 25, 563. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wang, F.; Zhao, Q.; Shi, A.; Zhao, T.; Song, Q.; Ravelombola, W.; An, H.; Yan, L.; Yang, C.; et al. Identification of candidate genes and genomic selection for seed protein in soybean breeding pipeline. Front. Plant Sci. 2022, 13, 882732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, G.; Zhang, W.; Wang, Q.; Xu, W.; Liu, X.; Cui, X.; Chen, X.; Chen, H. Identification of loci governing soybean seed protein content via genome-wide association study and selective signature analyses. Front. Plant Sci. 2022, 13, 1045953. [Google Scholar] [CrossRef]
- Jin, H.; Yang, X.; Zhao, H.; Song, X.; Tsvetkov, Y.D.; Wu, Y.; Gao, Q.; Zhang, R.; Zhang, J. Genetic analysis of protein content and oil content in soybean by genome-wide association study. Front. Plant Sci. 2023, 14, 1182771. [Google Scholar] [CrossRef]
- Park, H.; Seo, J.; Kang, B.; Kim, J.; Heo, S.; Choi, M.; Ko, J.; Kim, C. QTLs and candidate genes for seed protein content in two recombinant inbred line populations of soybean. Plants 2023, 12, 3589. [Google Scholar] [CrossRef]
- Diers, B.W.; Keim, P.; Fehr, W.R.; Shoemaker, R.C. RFLP analysis of soybean seed protein and oil content. Theor. Appl. Genet. 1992, 83, 608–612. [Google Scholar] [CrossRef]
- Kim, M.; Schultz, S.; Nelson, R.L.; Diers, B.W. Identification and fine mapping of a soybean seed protein QTL from PI 407788A on chromosome 15. Crop Sci. 2016, 56, 219–225. [Google Scholar] [CrossRef]
- Warrington, C.V.; Abdel-Haleem, H.; Hyten, D.L.; Cregan, P.B.; Orf, J.H.; Killam, A.S.; Bajjalieh, N.; Li, Z.; Boerma, H. R QTL for seed protein and amino acids in the Benning x Danbaekkong soybean population. Theor. Appl. Genet. 2015, 128, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Verdier, J.; Thompson, R.D. Transcriptional regulation of storage protein synthesis during dicotyledon seed filling. Plant Cell Physiol. 2008, 49, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Severin, A.J.; Woody, J.L.; Bolon, Y.T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, Q.; Zhang, W.; Zhang, H.; Liu, X.; Song, Q.; Zhu, Y.; Cui, X.; Chen, X.; Chen, H. Using transcriptomic and metabolomic data to investigate the molecular mechanisms that determine protein and oil contents during seed development in soybean. Front. Plant Sci. 2022, 13, 1012394. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Q.; Liu, Y.; Dong, S.; Zhang, Y.; Zhu, Y.; Tian, Y.; Li, J.; Wang, Z.; Wang, Y.; et al. Overexpressing GmCGS2 improves total amino acid and protein content in soybean seed. Int. J. Mol. Sci. 2023, 24, 14125. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S.; et al. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef]
- Song, X.; Huang, W.; Shi, M.; Zhu, M.; Lin, H. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 2007, 39, 623–630. [Google Scholar] [CrossRef]
- Li, X.; Shi, S.; Tao, Q.; Tao, Y.; Miao, J.; Peng, X.; Li, C.; Yang, Z.; Zhou, Y.; Liang, G. OsGASR9 positively regulates grain size and yield in rice (Oryza sativa). Plant Sci. 2010, 286, 17–27. [Google Scholar] [CrossRef]
- Geng, X.; Dong, N.; Wang, Y.; Li, G.; Wang, L.; Guo, X.; Li, J.; Wen, Z.; Wei, W. RNA-seq transcriptome analysis of the immature seeds of two Brassica napus lines with extremely different thousand-seed weight to identify the candidate genes related to seed weight. PLoS ONE 2018, 13, e0191297. [Google Scholar] [CrossRef]
- Chang, Y.; Shen, E.; Wen, L.; Yu, J.; Zhu, D.; Zhao, Q. Seed-specific expression of the Arabidopsis AtMAP18 gene increases both lysine and total protein content in maize. PLoS ONE 2015, 10, e0142952. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhang, D.; Zhao, X.; Cao, X.; Dong, L.; Liu, J.; Chen, K.; Zhang, H.; Gao, C.; et al. Analysis of the functions of TaGW2 homoeologs in wheat grain weight and protein content traits. Plant J. 2018, 94, 857–866. [Google Scholar] [CrossRef]
- Lu, W.; Wen, Z.; Li, H.; Yuan, D.; Li, J.; Zhang, H.; Huang, Z.; Cui, S.; Du, W. Identification of the quantitative trait loci (QTL) underlying water soluble protein content in soybean. Theor. Appl. Genet. 2013, 126, 425–433. [Google Scholar] [CrossRef]
- Stombaugh, S.K.; Orf, J.H.; Jung, H.G.; Chase, K.; Somers, D.A. Quantitative trait loci associated with cell wall polysaccharides in soybean seed. Crop Sci. 2004, 44, 2010–2106. [Google Scholar] [CrossRef]
- Jun, T.H.; Van, K.; Kim, M.Y.; Lee, S.H.; Walker, D.R. Association analysis using SSR markers to find QTL for seed protein content in soybean. Euphytica 2008, 162, 179–191. [Google Scholar] [CrossRef]
- Teuku, T.; Satoshi, W.; Naoki, Y.; Kyuya, H. Analysis of quantitative trait loci for protein and lipid contents in soybean seeds using recombinant inbred lines. Breed. Sci. 2003, 53, 133–140. [Google Scholar]
- Chen, Q.; Zhang, Z.; Liu, C.; Xin, D.; Shan, D.; Qiu, H.; Shan, C. QTL analysis of major agronomic traits in soybean. Agric. Sci. China 2007, 4, 399–405. [Google Scholar] [CrossRef]
- Mao, T.; Jiang, Z.; Han, Y.; Teng, W.; Zhao, X.; Li, W. Identification of quantitative trait loci underlying seed protein and oil contents of soybean across multi-genetic backgrounds and environments. Plant Breeding 2013, 132, 630–641. [Google Scholar] [CrossRef]
- Brummer, E.C.; Graef, G.L.; Orf, J.H.; Wilcox, J.R.; Shoemaker, R.C. Mapping QTL for seed protein and oil content in eight soybean populations. Crop Sci. 1997, 37, 370–378. [Google Scholar] [CrossRef]
- Reinprecht, Y.; Poysa, V.W.; Yu, K.; Rajcan, I.; Ablett, G.R.; Pauls, K.P. Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 2006, 49, g06–g112. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yu, Y.; Wang, S.; Lian, Y.; Wang, T.; Wei, Y.; Gong, P.; Liu, X.; Fang, X.; Zhang, M. QTL Mapping of isoflavone, oil and protein contents in soybean (Glycine max L. Merr.). Agric. Sci. China 2010, 9, 1108–1116. [Google Scholar] [CrossRef]
- Csanadi, G.Y.; Vollmann, J.; Stift, G.; Leeley, T. Seed quality QTLs identified in a molecular map of early maturing soybean. Theor. Appl. Genet. 2001, 103, 912–919. [Google Scholar] [CrossRef]
- Sebolt, A.M.; Shoemaker, R.C.; Diers, B.W. Analysis of a quantitative trait locus allele from wild soybean that increases seed protein concentration in soybean. Crop Sci. 2000, 40, 1438–1444. [Google Scholar] [CrossRef]
- Zhou, Z.; Lin, B.; Tan, J.; Hao, P.; Hua, S.; Deng, Z. Tandem mass tag-based quantitative proteomics reveals implication of a late embryogenesis abundant protein (BnLEA57) in seed oil accumulation in Brassica napus L. Front. Plant Sci. 2022, 13, 907244. [Google Scholar] [CrossRef] [PubMed]
- Kuczak, M.; Kurczynska, E. Cell wall composition as a marker of the reprogramming of the cell fate on the example of a Daucus carota (L.) hypocotyl in which somatic embryogenesis was induced. Int. J. Mol. Sci. 2020, 21, 8126. [Google Scholar] [CrossRef] [PubMed]
- Pinski, A.; Betekhtin, A.; Kwasniewska, J.; Chajec, L.; Wolny, E.; Hasterok, R. 3,4-Dehydro-L-proline induces programmed cell death in the roots of brachypodium distachyon. Int. J. Mol. Sci. 2021, 22, 7548. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, J.; Kuang, L.; Tian, Z.; Wang, X.; Dun, X.; Tu, J.; Wang, H. Genome-wide association study and transcriptome analysis reveal key genes affecting root growth dynamics in rapeseed. Biotechnol. Biofuels 2021, 14, 178. [Google Scholar] [CrossRef]
- Kim, W.; Kang, B.; Kang, S.; Shin, S.; Chowdhury, S.; Jeong, S.; Choi, M.S.; Park, S.K.; Moon, J.K.; Ryu, J.; et al. A genome-wide association study of protein, oil, and amino acid content in wild soybean (Glycine soja). Plants 2023, 12, 1665. [Google Scholar] [CrossRef]
- Sasaki, T.; Tsuchiya, Y.; Ariyoshi, M.; Nakano, R.; Ushijima, K.; Kubo, Y.; Mori, I.C.; Higashiizumi, E.; Galis, I.; Yamamoto, Y. Two members of the aluminum-activated malate transporter family, SlALMT4 and SlALMT5, are expressed during fruit development, and the overexpression of SlALMT5 alters organic acid contents in seeds in tomato (Solanum lycopersicum). Plant Cell Physiol. 2016, 57, 2367–2379. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Liu, X.; Sun, S.; Wang, P.; Qiao, X.; Li, J.; Tang, C.; Wu, J.; Zhang, S.; Tao, S. The unique evolutionary pattern of the Hydroxyproline-rich glycoproteins superfamily in Chinese white pear (Pyrus bretschneideri). BMC Plant Biol. 2018, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, H.; Qi, H.; Zhao, J. Roles of hydroxyproline-rich glycoproteins in the pollen tube and style cell growth of tobacco (Nicotiana tabacum L.). J. Plant Physiol. 2014, 171, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Dong, H.; Chang, H.; Zhao, J.; Teng, W.; Qiu, L.; Li, W.; Han, Y. Genome wide association mapping and candidate gene analysis for hundred seed weight in soybean [Glycine max (L.) Merrill]. BMC Genom. 2019, 20, 648. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed]
- Weine, E.; McPeek, M.S.; Abney, M. Application of equal local levels to improve Q-Q plot testing bands with R package qqconf. J. Stat. Softw. 2023, 106, 10. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Smoot, M.; Ono, K.; Ideker, T.; Maere, S. PiNGO: A cytoscape plugin to find candidate genes in biological networks. Bioinformatics 2011, 27, 1030–1031. [Google Scholar] [CrossRef] [PubMed]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.; Scheible, W.R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Name | Environmental a | Chr b | Position | Effect | −Log10 (p) | MAF | Known QTLs |
|---|---|---|---|---|---|---|---|
| rs8158 | E1, E2 | 8 | 10,476,797 | 1.32 | 9.03/6.32 | 0.02 | [25] |
| rs4186 | E1, E2 | 4 | 27,955,520 | 2.02/1.49 | 6.39/4.16 | 0.02 | [26] |
| rs12036 | E1 | 11 | 33,830,623 | −1.12 | 4.89 | 0.04 | |
| rs10510 | E1 | 10 | 9,242,053 | −1.493 | 4.5 | 0.04 | [27] |
| rs16341 | E1, E2 | 15 | 41,855,327 | 1.1111111 | 4.38/4.02 | 0.04 | |
| rs22328 | E1, E2, E3 | 20 | 12,157,089 | 0.68/0.63/0.72 | 4.30/3.79/4.74 | 0.12 | [28] |
| rs1724 | E1 | 2 | 20,263,396 | 0.83 | 4.07 | 0.07 | [29] |
| rs5276 | E1 | 5 | 30,572,600 | 1.16 | 4.06 | 0.05 | [30] |
| rs22326 | E1, E2, E3 | 20 | 12,137,051 | 0.64/0.64/0.69 | 3.97/3.94/4.55 | 0.13 | [31] |
| rs12338 | E1, E2, E3 | 12 | 11,269,928 | 0.41/0.38/0.45 | 3.80/3.54/5.52 | 0.39 | [25] |
| rs34 | E1 | 1 | 858,919 | −0.63 | 3.64 | 0.11 | [31] |
| rs18647 | E1 | 17 | 28,541,923 | −0.54 | 3.62 | 0.37 | [30] |
| rs22112 | E2 | 20 | 1,396,122 | 0.94 | 5.03 | 0.07 | [32] |
| rs16229 | E2 | 15 | 38,871,682 | 1.09 | 4.63 | 0.04 | [29] |
| rs19145 | E2 | 18 | 3,775,534 | −1.27 | 3.96 | 0.03 | [33] |
| rs15253 | E2 | 15 | 9,792,514 | −0.44 | 3.92 | 0.38 | |
| rs7157 | E2 | 7 | 8,398,920 | 0.49 | 3.91 | 0.25 | |
| rs16837 | E2 | 16 | 3,244,871 | −0.78 | 3.8 | 0.07 | |
| rs15251 | E2 | 15 | 9,752,108 | −0.42 | 3.68 | 0.42 | |
| rs15349 | E2 | 15 | 12,923,409 | −0.85 | 3.6 | 0.06 | |
| rs6741 | E2 | 6 | 47,134,144 | 0.73 | 3.52 | 0.08 | [34] |
| rs12331 | E3 | 12 | 10,978,300 | 0.56 | 4.67 | 0.18 | [25] |
| rs12316 | E3 | 12 | 9,835,536 | −0.43 | 4.65 | 0.43 | [25] |
| rs12325 | E3 | 12 | 10,505,242 | −0.47 | 4.6 | 0.26 | [25] |
| rs12329 | E3 | 12 | 10,956,419 | −0.51 | 4.38 | 0.19 | [25] |
| rs22678 | E3 | 20 | 33,768,863 | 0.44 | 4.36 | 0.38 | [12] |
| rs14891 | E3 | 14 | 4,366,8761 | −0.8 | 3.83 | 0.05 | [33] |
| rs19362 | E3 | 18 | 11,173,355 | −0.68 | 3.78 | 0.08 | [30] |
| rs22335 | E3 | 20 | 12,352,049 | −0.62 | 3.64 | 0.1 | [35] |
| rs12340 | E3 | 12 | 11,328,123 | 0.45 | 3.63 | 0.25 | [25] |
| rs12330 | E3 | 12 | 10,960,320 | 0.47 | 3.61 | 0.17 | [25] |
| Gene ID | Marker | Chr | Position (bp) | FHPC vs. FLPC (Log2FC) | THPC vs. TLPC (Log2FC) | HPC vs. LPC (Log2FC) | Arabidopsis Homologs | Gene Function |
|---|---|---|---|---|---|---|---|---|
| Glyma.08G137500 | rs8158 | 8 | 10,476,797 | 3.17 | 1.37 | 1.93 | AT5G65140 | Haloacid dehalogenase-like hydrolase (HAD) superfamily protein |
| Glyma.08G136900 | rs8158 | 8 | 10,476,797 | 4.66 | 2.83 | 4.27 | AT2G27080 | Late embryogenesis abundant (LEA) hydroxyproline-rich glycoprotein family |
| Glyma.08G135600 | rs8158 | 8 | 10,476,797 | −1.20 | −1.12 | −1.65 | AT5G10250 | Phototropic-responsive NPH3 family protein |
| Glyma.08G137900 | rs8158 | 8 | 10,476,797 | −1.32 | −1.58 | −1.09 | AT4G39790 | Protein of unknown function (DUF630 and DUF632) |
| Glyma.12G114100 | rs12338 | 12 | 11,269,928 | 6.26 | NA | NA | AT4G28350 | Concanavalin A-like lectin protein kinase family protein |
| Glyma.08G135800 | rs8158 | 8 | 104,76,797 | 2.89 | 1.97 | 2.46 | AT5G65000 | Nucleotide-sugar transporter family protein |
| Glyma.12G114300 | rs12338 | 12 | 11,269,928 | 2.77 | 1.22 | 1.33 | AT4G20970 | basic helix–loop–helix (bHLH) DNA-binding superfamily protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Zhu, H.; Liu, F.; Wang, J.; Zhou, C.; Yuan, M.; Zhao, X.; Li, Y.; Teng, W.; Han, Y.; et al. Integrating Genome-Wide Association Study, Transcriptome and Metabolome Reveal Novel QTL and Candidate Genes That Control Protein Content in Soybean. Plants 2024, 13, 1128. https://doi.org/10.3390/plants13081128
Zhao X, Zhu H, Liu F, Wang J, Zhou C, Yuan M, Zhao X, Li Y, Teng W, Han Y, et al. Integrating Genome-Wide Association Study, Transcriptome and Metabolome Reveal Novel QTL and Candidate Genes That Control Protein Content in Soybean. Plants. 2024; 13(8):1128. https://doi.org/10.3390/plants13081128
Chicago/Turabian StyleZhao, Xunchao, Hanhan Zhu, Fang Liu, Jie Wang, Changjun Zhou, Ming Yuan, Xue Zhao, Yongguang Li, Weili Teng, Yingpeng Han, and et al. 2024. "Integrating Genome-Wide Association Study, Transcriptome and Metabolome Reveal Novel QTL and Candidate Genes That Control Protein Content in Soybean" Plants 13, no. 8: 1128. https://doi.org/10.3390/plants13081128