
Unraveling the Evolutionary Tales of Yunnanopilia longistaminea (Opiliaceae): Insights from Genetic Diversity, Climate Adaptation, and Conservation Strategies

Abstract
:1. Introduction
2. Materials and Methods
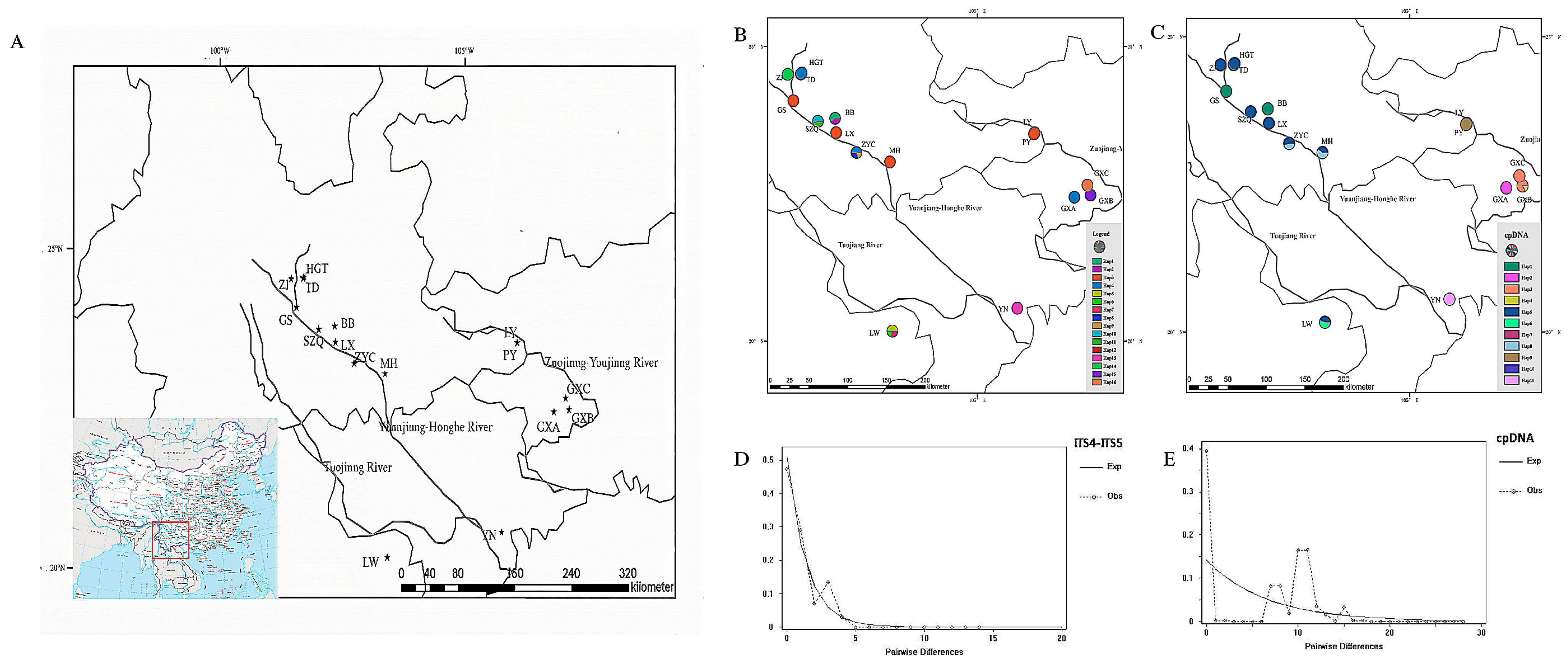
2.1. Population Sampling
2.2. Molecular Procedures
2.3. Data Analysis
2.4. Ecological Niche Modeling
3. Results
3.1. Genetic Diversity and Structure
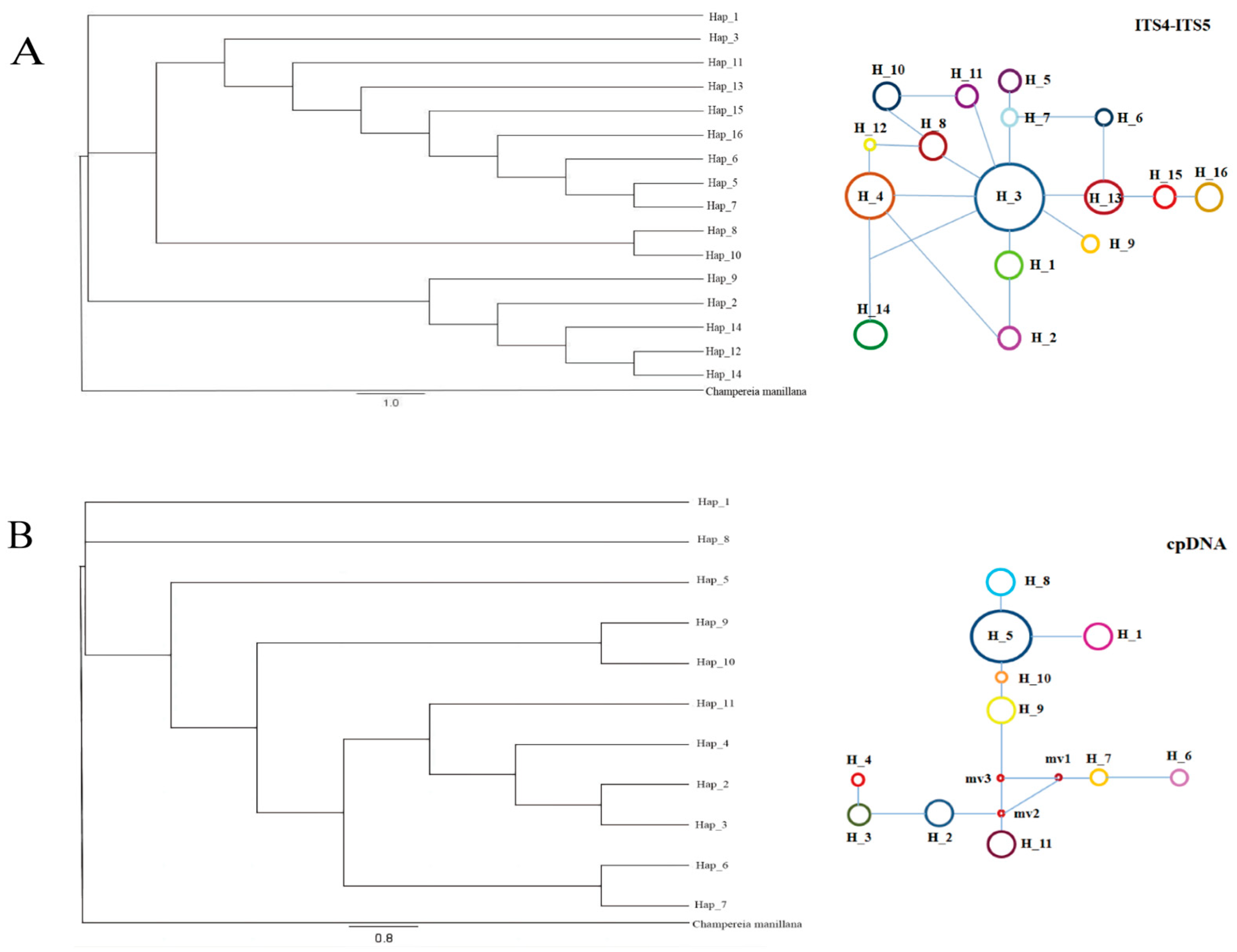
3.2. Evolutionary Network Analysis
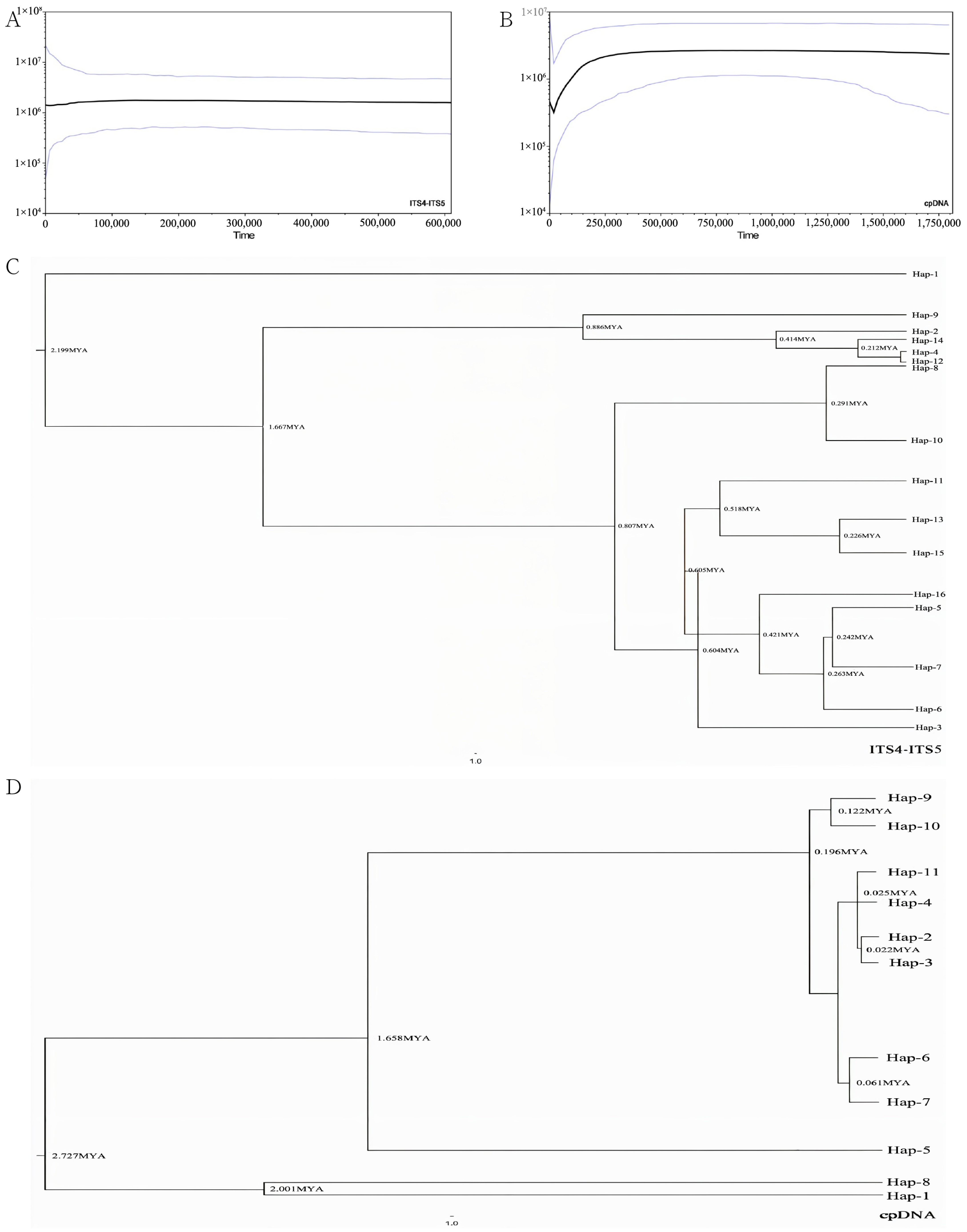
3.3. Phylogeography and Divergence Time
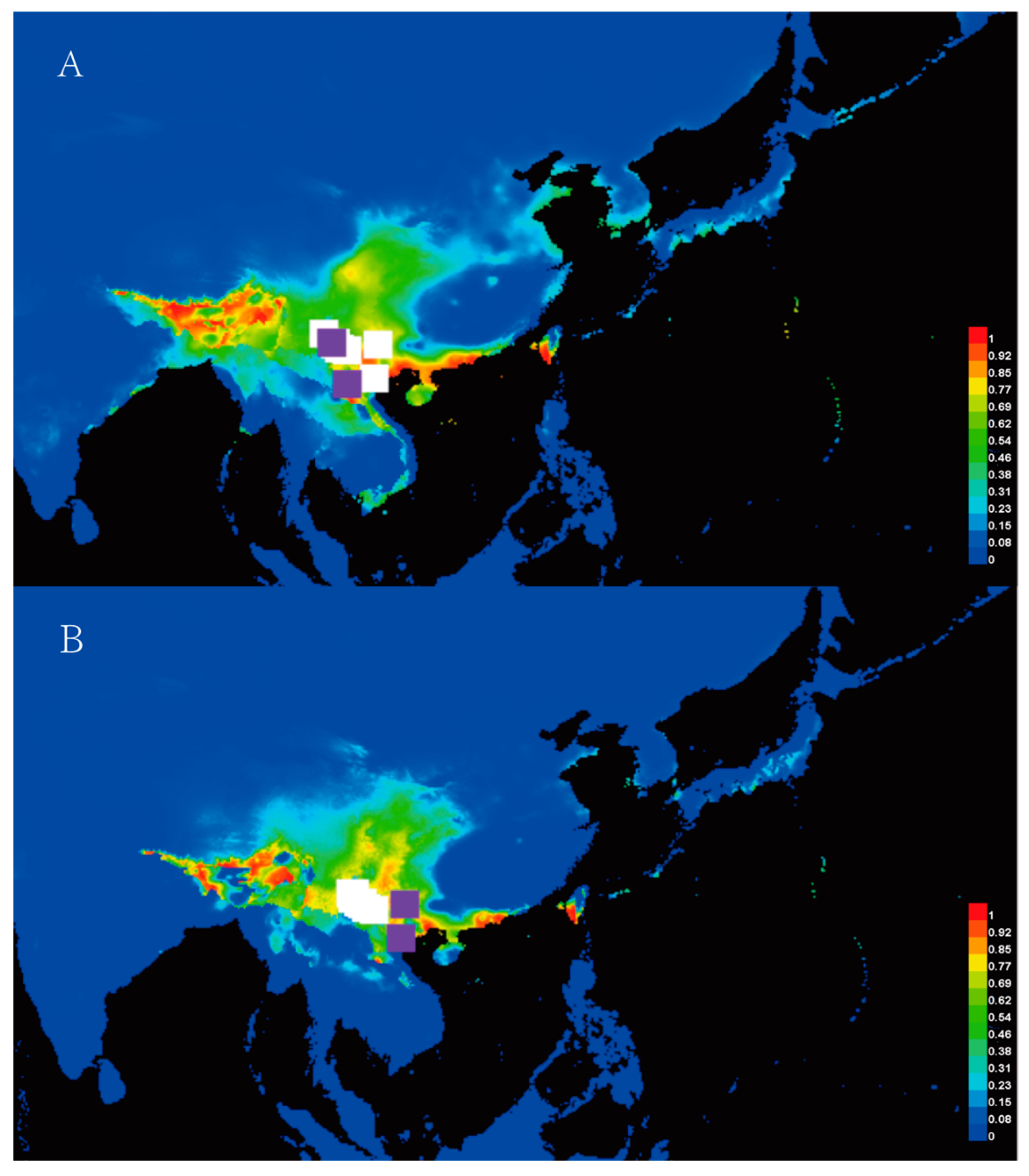
3.4. Ecological Niche Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xiao, L.Q.; Ge, X.J.; Gong, X.; Hao, G.; Zheng, S.X. ISSR variation in the endemic and endangered plant Cycas guizhouensis (Cycadaceae). Ann. Bot. 2004, 94, 133–138. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, J.; Feng, X.; Gong, X. The distribution, diversity, and conservation status of Cycas in China. Ecol. Evol. 2017, 7, 3212–3224. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Vázquez, L.; Feng, L.; Liu, Z.; Zhao, G. Climatic and soil factors shape the demographical history and genetic diversity of a deciduous oak (Quercus liaotungensis) in Northern China. Front. Plant Sci. 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Boston, MA, USA, 2000. [Google Scholar]
- Petit, R.J.; Pineau, E.; Demesure, B.; Bacilieri, R.; Ducousso, A.; Kremer, A. Chloroplast DNA footprints of postglacial recolonization by oaks. Proc. Natl. Acad. Sci. USA 1997, 94, 9996–10001. [Google Scholar] [CrossRef]
- Li, Y.; Guan, R.; Liu, Z.; Ma, Y.; Wang, L.; Li, L.; Lin, F.; Luan, W.; Chen, P.; Yan, Z. Genetic structure and diversity of cultivated soybean (Glycine max (L.) Merr.) landraces in China. Theor. Appl. Genet. 2008, 117, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Shaw, R.G. Range shifts and adaptive responses to Quaternary climate change. Science 2001, 292, 673–679. [Google Scholar] [CrossRef]
- Shao, Y.Z.; Zhang, X.C.; Phan, L.K.; Xiang, Q.P. Elevation Shift in Abies Mill. (Pinaceae) of Subtropical and Temperate China and Vietnam-Corroborative Evidence from Cytoplasmic DNA and Ecological Niche Modeling. Front. Plant Sci. 2017, 8, 578. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.B.; Peng, X.L.; Li, L.; Liu, J.Q.; Miehe, G.; Opgenoorth, L. Molecular phylogeography and evolutionary history of Picea likiangensis in the Qinghai–Tibetan Plateau inferred from mitochondrial and chloroplast DNA sequence variation. J. Syst. Evol. 2012, 50, 341–350. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, C.-B.; Ma, X.-G.; Liang, Q.-L.; He, X.-J. Phylogeographic analysis of a temperate-deciduous forest restricted plant (Bupleurum longiradiatum Turcz.) reveals two refuge areas in China with subsequent refugial isolation promoting speciation. Mol. Phylogenet. Evol. 2013, 68, 628–643. [Google Scholar] [CrossRef]
- Zhang, J.-Q.; Meng, S.-Y.; Wen, J.; Rao, G.-Y. DNA barcoding of Rhodiola (Crassulaceae): A case study on a group of recently diversified medicinal plants from the Qinghai-Tibetan Plateau. PLoS ONE 2015, 10, e0119921. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Abbott, R.J.; Yu, Q.S.; Lin, K.; Liu, J.Q. Pleistocene climate change and the origin of two desert plant species, Pugionium cornutum and Pugionium dolabratum (Brassicaceae), in northwest China. New Phytol. 2013, 199, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.S.; Feng, J.J.; Jiang, D.C.; Mao, K.S.; Duan, Y.W.; Miehe, G.; Opgenoorth, L. The Quaternary evolutionary history, potential distribution dynamics, and conservation implications for a Qinghai–Tibet Plateau endemic herbaceous perennial, Anisodus tanguticus (Solanaceae). Ecol. Evol. 2016, 6, 1977–1995. [Google Scholar] [CrossRef]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.-L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Takeuchi, Y.; Yamasaki, M.; Sakurai, S.; Isagi, Y. Lineage admixture during postglacial range expansion is responsible for the increased gene diversity of Kalopanax septemlobus in a recently colonised territory. Heredity 2011, 107, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Tzedakis, P.; Emerson, B.C.; Hewitt, G.M. Cryptic or mystic? Glacial tree refugia in northern Europe. Trends Ecol. Evol. 2013, 28, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Li, A. Population genomics reveals demographic history and genomic differentiation of Populus davidiana and Populus tremula. Front. Plant Sci. 2020, 11, 553736. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xie, Y.; Liu, B.; New, H.Y.; Zhou, Z.; Feng, Z.; Chen, Y. A Transcriptomic Variation Map Provides Insights into The Genetic Basis of Pinus massoniana Evolution and Association of Oleoresin Yield. BMC Plant Biol. 2020, 20, 375. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.-Y.; Guo, X.-L.; Price, M.; He, X.-J.; Zhou, S.-D. Effects of mountain uplift and climatic oscillations on phylogeography and species divergence of Chamaesium (Apiaceae). Front. Plant Sci. 2021, 12, 673200. [Google Scholar] [CrossRef] [PubMed]
- Harison, T.; Chen, W.; Leloup, P. An early Miocene transition in deformation region with the Red River Fault zone, Yunnan and its significance for Indo-Asian Tectonics. J. Geophys. Res. 1992, 97, 7559–7682. [Google Scholar]
- Leloup, P.H.; Lacassin, R.; Tapponnier, P.; Schärer, U.; Zhong, D.; Liu, X.; Zhang, L.; Ji, S.; Trinh, P.T. The Ailao Shan-Red river shear zone (Yunnan, China), tertiary transform boundary of Indochina. Tectonophysics 1995, 251, 3–84. [Google Scholar] [CrossRef]
- Tapponnier, P.; Lacassin, R.; Leloup, P.H.; Schärer, U.; Dalai, Z.; Haiwei, W.; Xiaohan, L.; Shaocheng, J.; Lianshang, Z.; Jiayou, Z. The Ailao Shan/Red River metamorphic belt: Tertiary left-lateral shear between Indochina and South China. Nature 1990, 343, 431–437. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, W.; Gong, X. Species delimitation, genetic diversity and population historical dynamics of Cycas diannanensis (Cycadaceae) occurring sympatrically in the Red River region of China. Front. Plant Sci. 2015, 6, 696. [Google Scholar] [CrossRef]
- Clark, M.; Schoenbohm, L.; Royden, L.; Whipple, K.; Burchfiel, B.; Zhang, X.; Tang, W.; Wang, E.; Chen, L. Surface uplift, tectonics, and erosion of eastern Tibet from large-scale drainage patterns. Tectonics 2004, 23, TC1006. [Google Scholar] [CrossRef]
- Yang, G.-s.; Yang, L.; Wang, Y.-h.; Shen, S.-k. Physiological epicotyl dormancy and its alleviation in seeds of Yunnanopilia longistaminea: The first report of physiological epicotyl dormancy in China. PeerJ 2017, 5, e3435. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Y.; Gong, X. Genetic diversity, genetic structure and demographic history of Cycas simplicipinna (Cycadaceae) assessed by DNA sequences and SSR markers. BMC Plant Biol. 2014, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. DNA protocols for plants–CTAB total DNA isolation. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. [Google Scholar]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef]
- Chiang, T.; Peng, C. Phylogeography of the endemic plants of Taiwan. Proc. Conserv. Endem. Species 1998, 148–155. [Google Scholar]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Academic Press: Cambridge, MA, USA, 1990; Volume 18, pp. 315–322. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Swofford, D.L. Paup*: Phylogenetic Analysis Using Parsimony (and Other Methods) 4.0. B5; ScienceOpen, Inc.: Berlin, Germany, 2001. [Google Scholar]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Pons, O.; Petit, R. Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.; Forster, P.; Watson, J. NETWORK: A Software for Population Genetics Data Analysis, (version 4.2. 0.1); Fluxus Technology Ltd.: Clare, MI, USA, 2007. [Google Scholar]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S. Patterns of genetic variation in rare and widespread plant congeners. Am. J. Bot. 2000, 87, 783–792. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E.; Tucker, T.L.; Lang, F.A. Allozyme variability is absent in the narrow endemic Bensoniella oregona (Saxifragaceae). Conserv. Biol. 1992, 6, 131–134. [Google Scholar] [CrossRef]
- Qiu, Y.-X.; Guan, B.-C.; Fu, C.-X.; Comes, H.P. Did glacials and/or interglacials promote allopatric incipient speciation in East Asian temperate plants? Phylogeographic and coalescent analyses on refugial isolation and divergence in Dysosma versipellis. Mol. Phylogenet. Evol. 2009, 51, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Wu, H.; Wang, J.-F.; Gong, X. Genetic diversity and structure of Munronia delavayi Franch. (Meliaceae), an endemic species in the dry-hot valley of Jinsha River, south-western China. Genet. Resour. Crop Evol. 2014, 61, 1381–1395. [Google Scholar] [CrossRef]
- Wang, J.F.; Gong, X.; Chiang, Y.C.; Kuroda, C. Phylogenetic patterns and disjunct distribution in Ligularia hodgsonii Hook (Asteraceae). J. Biogeogr. 2013, 40, 1741–1754. [Google Scholar] [CrossRef]
- Caicedo, A.L.; Schaal, B.A. Population structure and phylogeography of Solanum pimpinellifolium inferred from a nuclear gene. Mol. Ecol. 2004, 13, 1871–1882. [Google Scholar] [CrossRef]
- Levi, A.; Thomas, C.E.; Keinath, A.P.; Wehner, T.C. Genetic diversity among watermelon (Citrullus lanatus and Citrullus colocynthis) accessions. Genet. Resour. Crop Evol. 2001, 48, 559–566. [Google Scholar] [CrossRef]
- Havill, N.P.; Campbell, C.S.; Vining, T.F.; LePage, B.; Bayer, R.J.; Donoghue, M.J. Phylogeny and biogeography of Tsuga (Pinaceae) inferred from nuclear ribosomal ITS and chloroplast DNA sequence data. Syst. Bot. 2008, 33, 478–489. [Google Scholar] [CrossRef]
- Gong, W.; Zeng, Z.; Chen, Y.Y.; Chen, C.; Qiu, Y.X.; Fu, C.X. Glacial refugia of Ginkgo biloba and human impact on its genetic diversity: Evidence from chloroplast DNA. J. Integr. Plant Biol. 2008, 50, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. J. R. Meteorol. Soc. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Wang, Z.-W.; Chen, S.-T.; Nie, Z.-L.; Zhang, J.-W.; Zhou, Z.; Deng, T.; Sun, H. Climatic factors drive population divergence and demography: Insights based on the phylogeography of a riparian plant species endemic to the Hengduan Mountains and adjacent regions. PLoS ONE 2015, 10, e0145014. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.; Zhou, C.; Molina, I.; Fu, C.; Nakashima, J.; Li, G.; Zhang, W.; Park, J.; Tang, Y.; Jiang, Q. A class II KNOX gene, KNOX4, controls seed physical dormancy. Proc. Natl. Acad. Sci. USA 2016, 113, 6997–7002. [Google Scholar] [CrossRef]
- Javadi, F.; Tun, Y.T.; Kawase, M.; Guan, K.; Yamaguchi, H. Molecular phylogeny of the subgenus Ceratotropis (genus Vigna, Leguminosae) reveals three eco-geographical groups and Late Pliocene–Pleistocene diversification: Evidence from four plastid DNA region sequences. Ann. Bot. 2011, 108, 367–380. [Google Scholar] [CrossRef]
- Du, Z.-Y.; Wang, Q.-F. Allopatric divergence of Stuckenia filiformis (Potamogetonaceae) on the Qinghai-Tibet Plateau and its comparative phylogeography with S. pectinata in China. Sci. Rep. 2016, 6, 20883. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.A.; Harris, N.B.; Widdowson, M.; Herman, A.B.; Guo, S.; Valdes, P.J.; Wolfe, J.A.; Kelley, S.P. Constant elevation of southern Tibet over the past 15 million years. Nature 2003, 421, 622–624. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Cai, X.; Yang, K.; Hong, Y.; Fan, F.; Wang, Q.; Zhang, B.; Hou, Q.; Leng, Y.; Qiao, G. Chromosome-level genome assembly of a rare karst-growing Rhododendron species provides insights into its evolution and environmental adaptation. J. Syst. Evol. 2024; early view. [Google Scholar]
- Jayasuriya, K.G.; Wijetunga, A.S.; Baskin, J.M.; Baskin, C.C. Recalcitrancy and a new kind of epicotyl dormancy in seeds of the understory tropical rainforest tree Humboldtia laurifolia (Fabaceae, Ceasalpinioideae). Am. J. Bot. 2010, 97, 15–26. [Google Scholar] [CrossRef] [PubMed]
- CHEN, S.C.; Zhang, L.; Zeng, J.; Shi, F.; Yang, H.; MAO, Y.R.; FU, C.X. Geographic variation of chloroplast DNA in Platycarya strobilacea (Juglandaceae). J. Syst. Evol. 2012, 50, 374–385. [Google Scholar] [CrossRef]
- Qi, X.S.; Chen, C.; Comes, H.P.; Sakaguchi, S.; Liu, Y.H.; Tanaka, N.; Sakio, H.; Qiu, Y.X. Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). New Phytol. 2012, 196, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tian, C.; Li, Y.; Li, Y. Molecular data and ecological niche modelling reveal the phylogeographic pattern of Cotinus coggygria (Anacardiaceae) in China’s warm-temperate zone. Plant Biol. 2014, 16, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Zheng, W.; Pei, K.; Ma, K. Genetic variation within and among populations of a dominant desert tree Haloxylon ammodendron (Amaranthaceae) in China. Ann. Bot. 2005, 96, 245–252. [Google Scholar] [CrossRef]
- Li, Y.; Huang, R.; Zhang, W.; Chen, Q.; Wang, Q.; Ye, J.; Xu, F. Medicinal Potential of Broussonetia papyrifera: Chemical Composition and Biological Activity Analysis. Plants 2025, 14, 523. [Google Scholar] [CrossRef]
- He, Z.; Sun, Y.; Peng, L.; Luo, Z.; Qi, J.; Li, X.; Duan, X.; Ou, Z. Comparative Study of Water and Fertilizer Regimes on Water Potential, Phenolic Substances, and Photosynthetic Characteristics of Pistacia weinmannifolia. Horticulturae 2024, 10, 1302. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Populations | Location | Longitude (E°) | Latitude (N°) | Altitude (m) | N. |
|---|---|---|---|---|---|---|
| P1 | GS | Gasa town, Yunnan province | 101.5574 | 24.0793 | 1389 | 6 |
| P2 | SZQ | Qinglongchan town, Yunnan province | 102.0168 | 23.7291 | 1311 | 57 |
| P3 | BB | Baoxiu town, Yunnan province | 102.3381 | 23.7802 | 1390 | 27 |
| P4 | ZYC | Niujie town, Yunnan province | 102.3572 | 23.5371 | 1217 | 20 |
| P5 | LY | Daleng town, Yunnan province | 106.0697 | 23.5337 | 736 | 15 |
| P6 | PY | Gula town, Yunnan province | 106.0581 | 23.5169 | 826 | 15 |
| P7 | YN | Hoa Binh province, Vietnam | 105.7467 | 20.5561 | 527 | 15 |
| P8 | LW | Houphan province, Laos | 103.4102 | 20.1666 | 1100 | 15 |
| P9 | MH | Manhao town, Yunnan province | 103.3642 | 23.0406 | 783 | 15 |
| P10 | LX | Majie town, Yunnan province | 102.7385 | 23.1983 | 960 | 20 |
| P11 | ZJ | Fabiao town, Yunnan province | 101.4518 | 24.5271 | 1490 | 20 |
| P12 | TD | Tuodian town, Yunnan province | 101.6970 | 24.5320 | 1463 | 20 |
| P13 | HGT | Dutian town, Yunnan province | 101.7147 | 24.5494 | 1507 | 20 |
| P14 | GXA | Shanglong town, Guangxi province | 106.8130 | 22.4414 | 360 | 10 |
| P15 | GXB | Xiangshui town, Guangxi province | 107.117 | 22.4769 | 336 | 10 |
| P16 | GXC | Leiping town, Guangxi province | 107.058 | 22.6446 | 407 | 10 |
| Total | 295 | |||||
| Population Code | cpDNA | nrDNA | |||||
|---|---|---|---|---|---|---|---|
| Haplotyoe | Hd | Pi | Haplotyoe | Hd | Pi | ||
| 1 | GS | Hap 1(4) | 0 | 0 | Hap 3(4) | 0 | 0 |
| 2 | SZQ | Hap 5(9) | 0 | 0 | Hap 10(5) Hap11(4) | 0.556 | 0.00088 |
| 3 | BB | Hap 1(8) | 0 | 0 | Hap 1(6) Hap(4) | 0 | 0 |
| 4 | ZYC | Hap 5(10) | 0 | 0 | Hap 3(10) | 0 | 0 |
| 5 | LY | Hap 9(9) | 0.2 | 0.00016 | Hap 3(10) | 0 | 0 |
| 6 | PY | Hap 9(6) | 0 | 0 | Hap 3(6) | 0 | 0 |
| 7 | YN | Hap 11(9) | 0 | 0 | Hap13(10) | 0 | 0 |
| 8 | LW | Hap 6(5)Hap10(4) | 0 | 0 | Hap 5(5) Hap6(2) Hap7(3) | 0.688 | 0.00144 |
| 9 | MH | Hap 5(6) Hap 8(4) | 0 | 0 | Hap 3(10) | 0 | 0 |
| 10 | LX | Hap5(5) Hap8(4) | 0 | 0 | Hap 3(5) Hap8(3) Hap9(2) | 0.688 | 0.00013 |
| 11 | ZJ | Hap5(8) | 0 | 0 | Hap 14(9) | 0 | 0 |
| 12 | TD | Hap5(10) | 0 | 0 | Hap3(2) Hap4(4) Hap8(3) | 0.533 | 0.00084 |
| 13 | HGT | Hap5(10) | 0 | 0 | Hap 4(10) | 0 | 0 |
| 14 | GXA | Hap2(3) | 0 | 0 | Hap 4(5) | 0 | 0 |
| 15 | GXB | Hap3(5) Hap4(1) | 0 | 0 | Hap 15(4) | 0 | 0 |
| 16 | GXC | Hap(10) | 0 | 0 | Hap 16(5) | 0 | 0 |
| total | 0.605 | 0.0024 | 0.526 | 0.00151 | |||
| Markers | HS | HT | GST | NST |
|---|---|---|---|---|
| cpDNA | 0.330 | 0.953 | 0.949 | 0.983 |
| nrDNA | 0.203 | 0.883 | 0.651 | 0.833 |
| Markes | Source of Variation | d.f. | Sum of Squares | Percentage of Variation (%) | FST |
|---|---|---|---|---|---|
| cpDNA | Among populations | 15 | 1701.294 | 98.5% | 0.985 |
| Within populations | 116 | 24.767 | 1.50% | 0.015 | |
| nrDNA | Among populations | 14 | 103.915 | 87.62% | 0.876 |
| Within populations | 106 | 13.622 | 12.38% | 0.124 |
| Markers | Tajima’ D | Fu and Li’ D * | Fu and Li’ F * | Fu’ Fs |
|---|---|---|---|---|
| cpDNA | 0.992 | 1.563 | 1.384 | 0.00 |
| ITS4-ITS5 | −0.570 | 1.172 | 0.682 | −1.919 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Yang, L.; Shen, S.; Wang, Y.; Wang, Y. Unraveling the Evolutionary Tales of Yunnanopilia longistaminea (Opiliaceae): Insights from Genetic Diversity, Climate Adaptation, and Conservation Strategies. Plants 2025, 14, 706. https://doi.org/10.3390/plants14050706
Yang G, Yang L, Shen S, Wang Y, Wang Y. Unraveling the Evolutionary Tales of Yunnanopilia longistaminea (Opiliaceae): Insights from Genetic Diversity, Climate Adaptation, and Conservation Strategies. Plants. 2025; 14(5):706. https://doi.org/10.3390/plants14050706
Chicago/Turabian StyleYang, Guansong, Liu Yang, Shikang Shen, Yuehua Wang, and Yuying Wang. 2025. "Unraveling the Evolutionary Tales of Yunnanopilia longistaminea (Opiliaceae): Insights from Genetic Diversity, Climate Adaptation, and Conservation Strategies" Plants 14, no. 5: 706. https://doi.org/10.3390/plants14050706
APA StyleYang, G., Yang, L., Shen, S., Wang, Y., & Wang, Y. (2025). Unraveling the Evolutionary Tales of Yunnanopilia longistaminea (Opiliaceae): Insights from Genetic Diversity, Climate Adaptation, and Conservation Strategies. Plants, 14(5), 706. https://doi.org/10.3390/plants14050706