
Genomic Signatures of Environmental Adaptation in Castanopsis hainanensis (Fagaceae)


Abstract
:1. Introduction
2. Results
2.1. Sequencing Quality
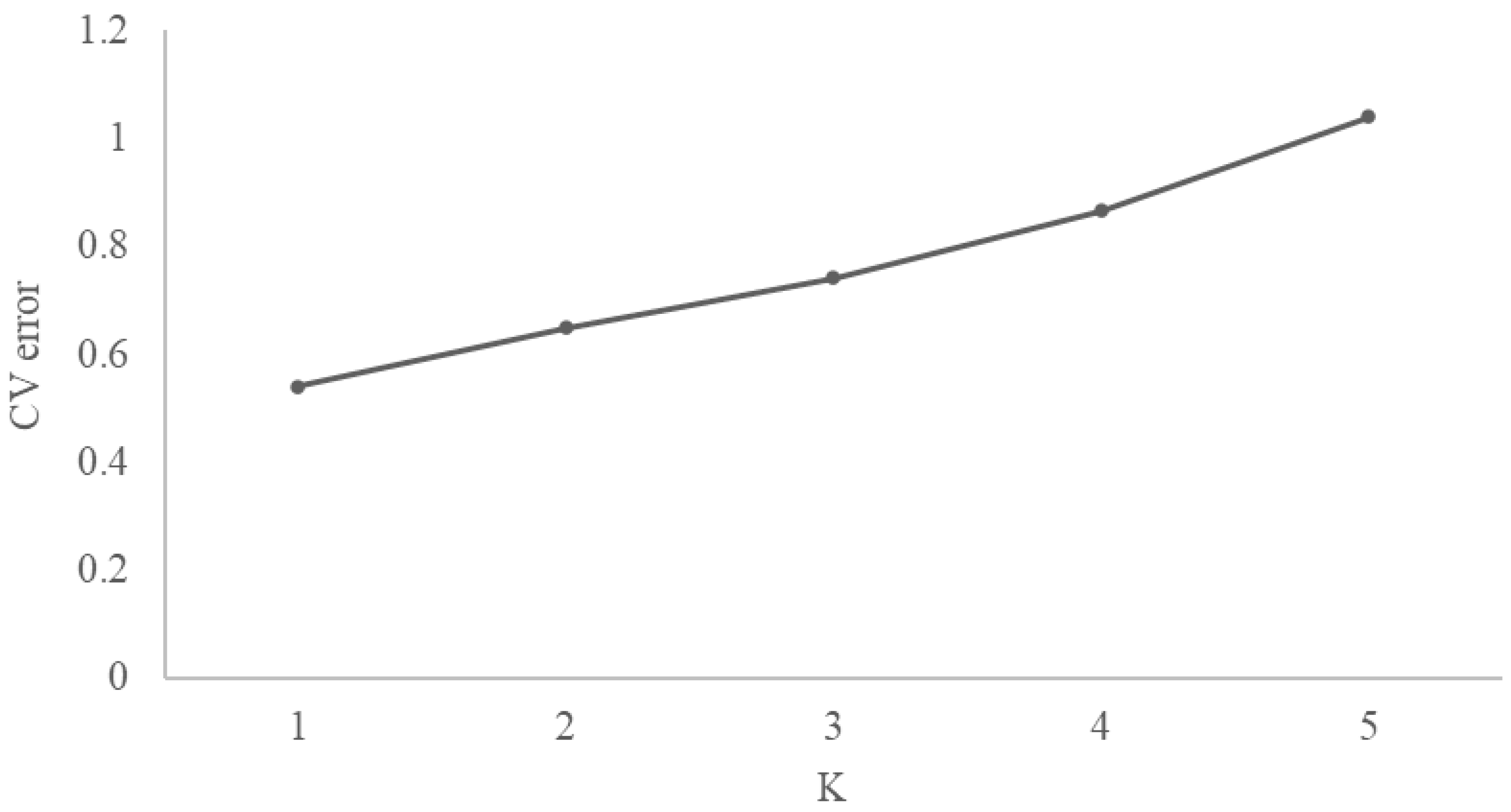
2.2. Population Structure
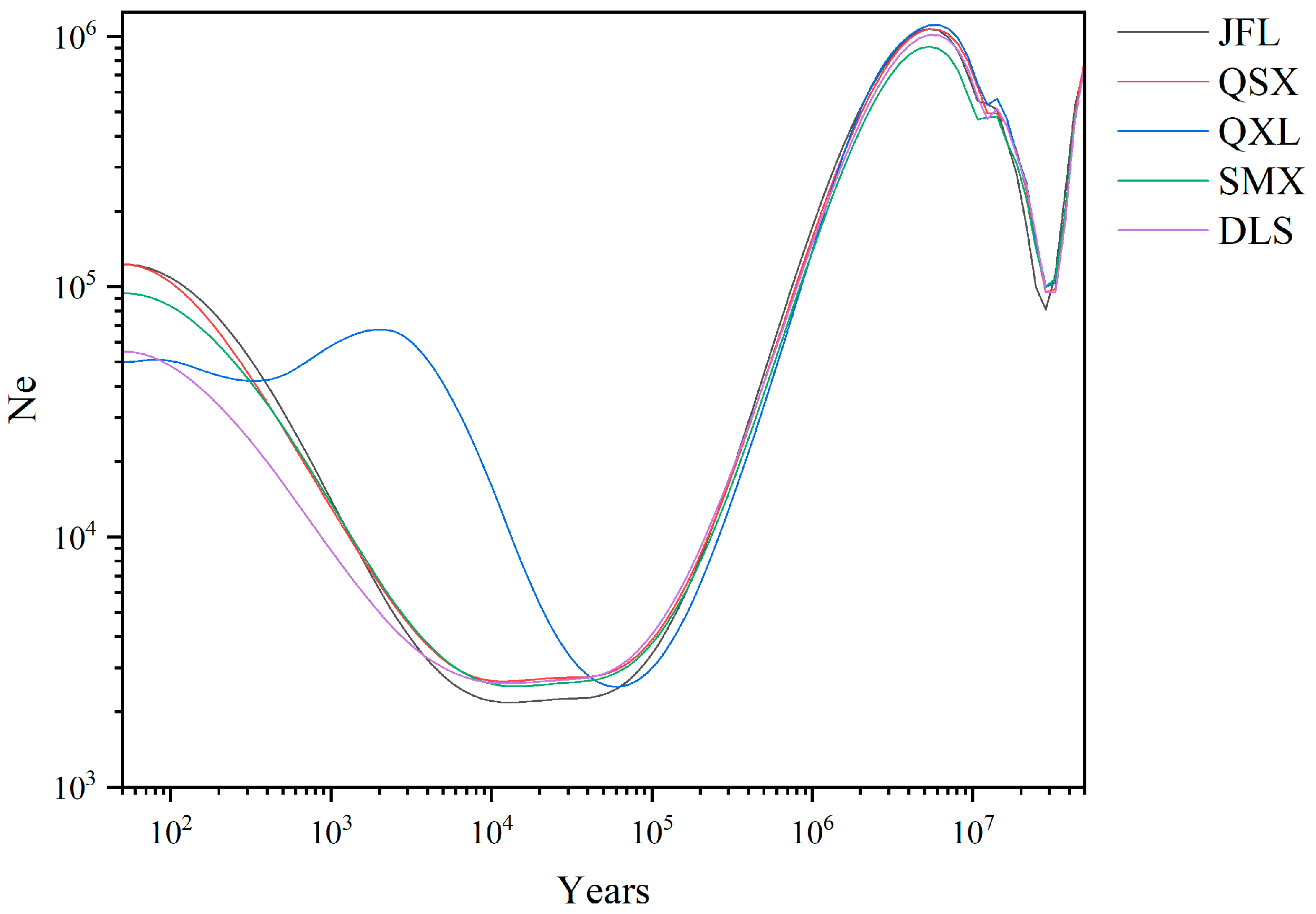
2.3. Demographic History
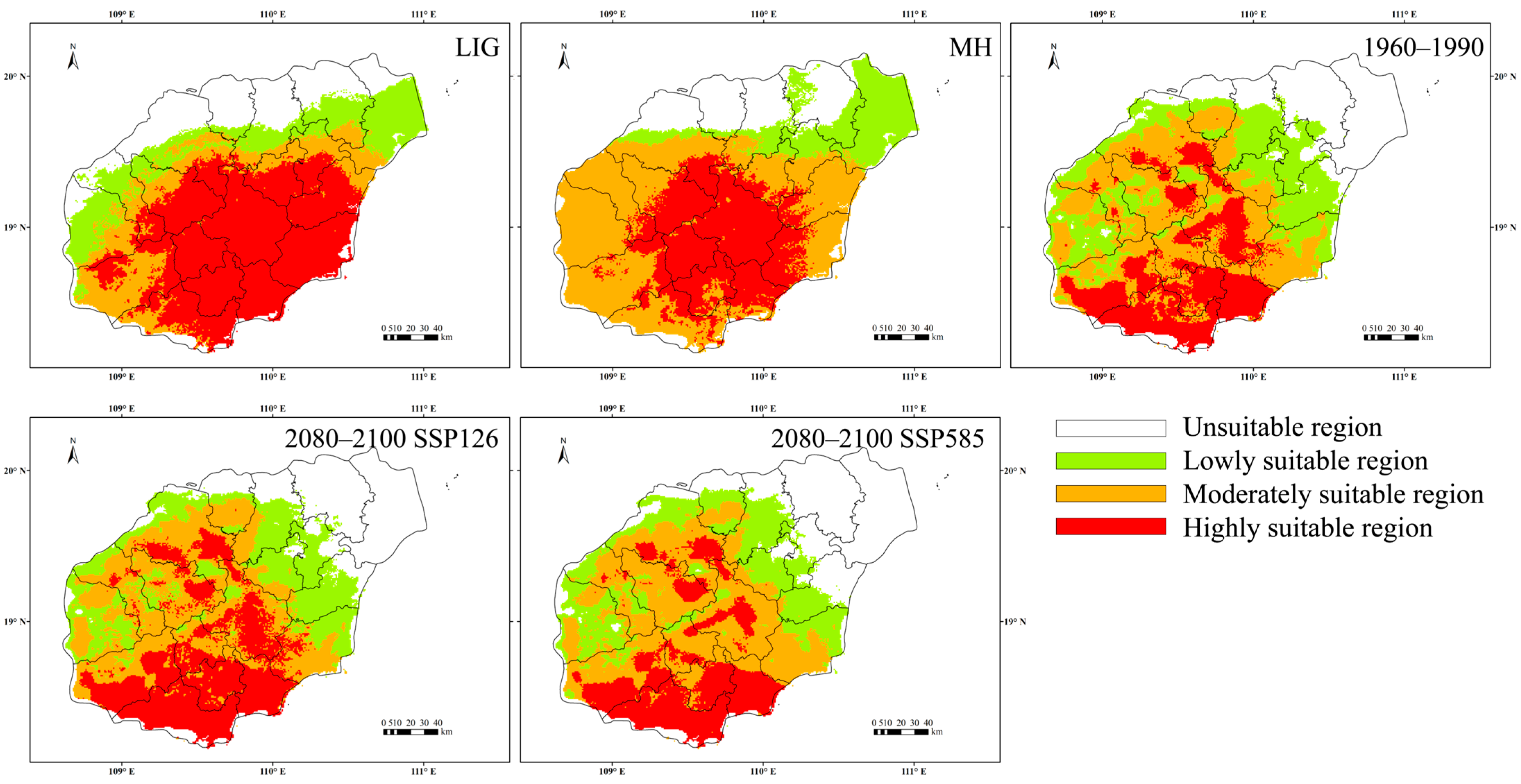
2.4. Changes in Potential Habitats
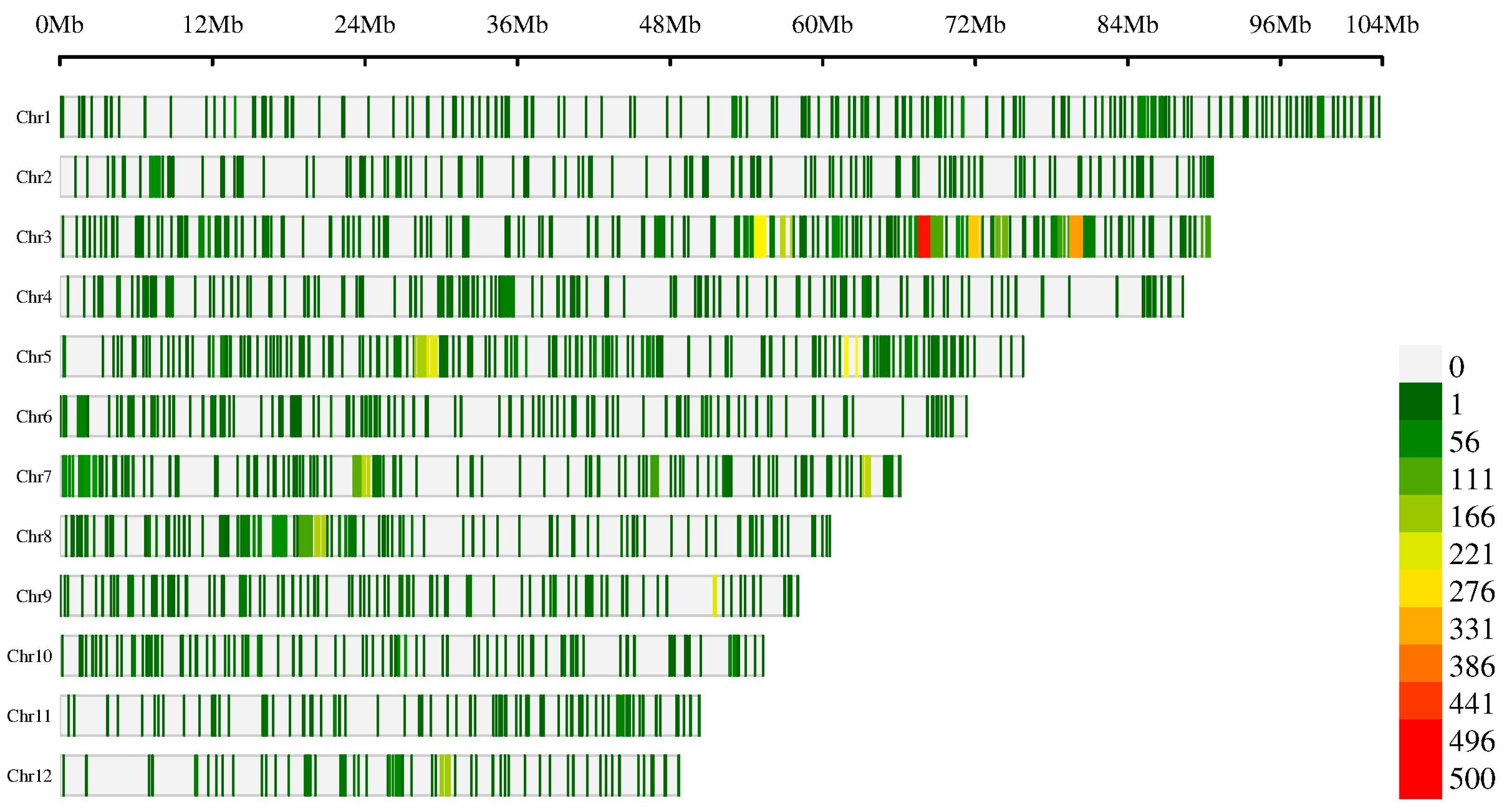
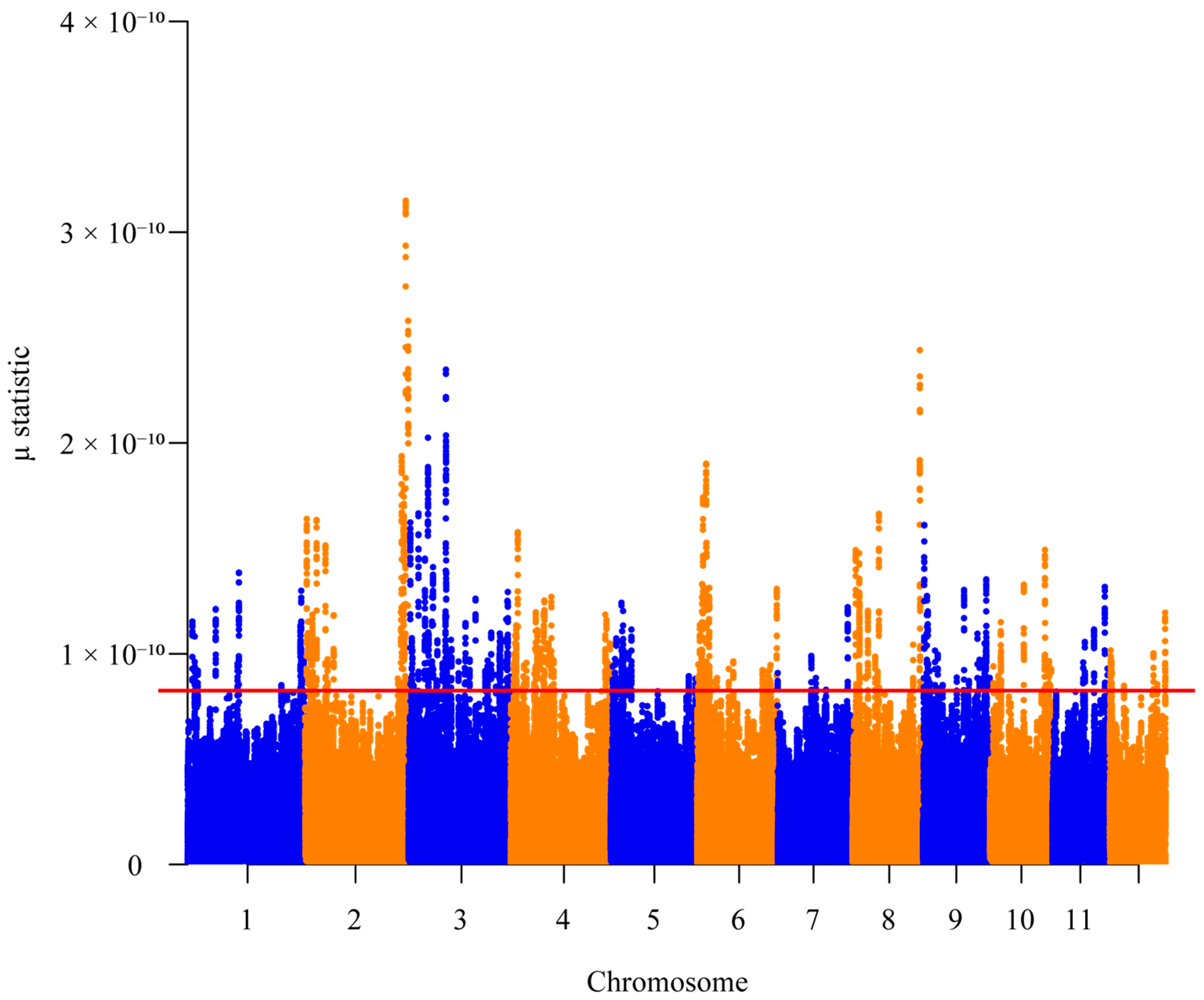
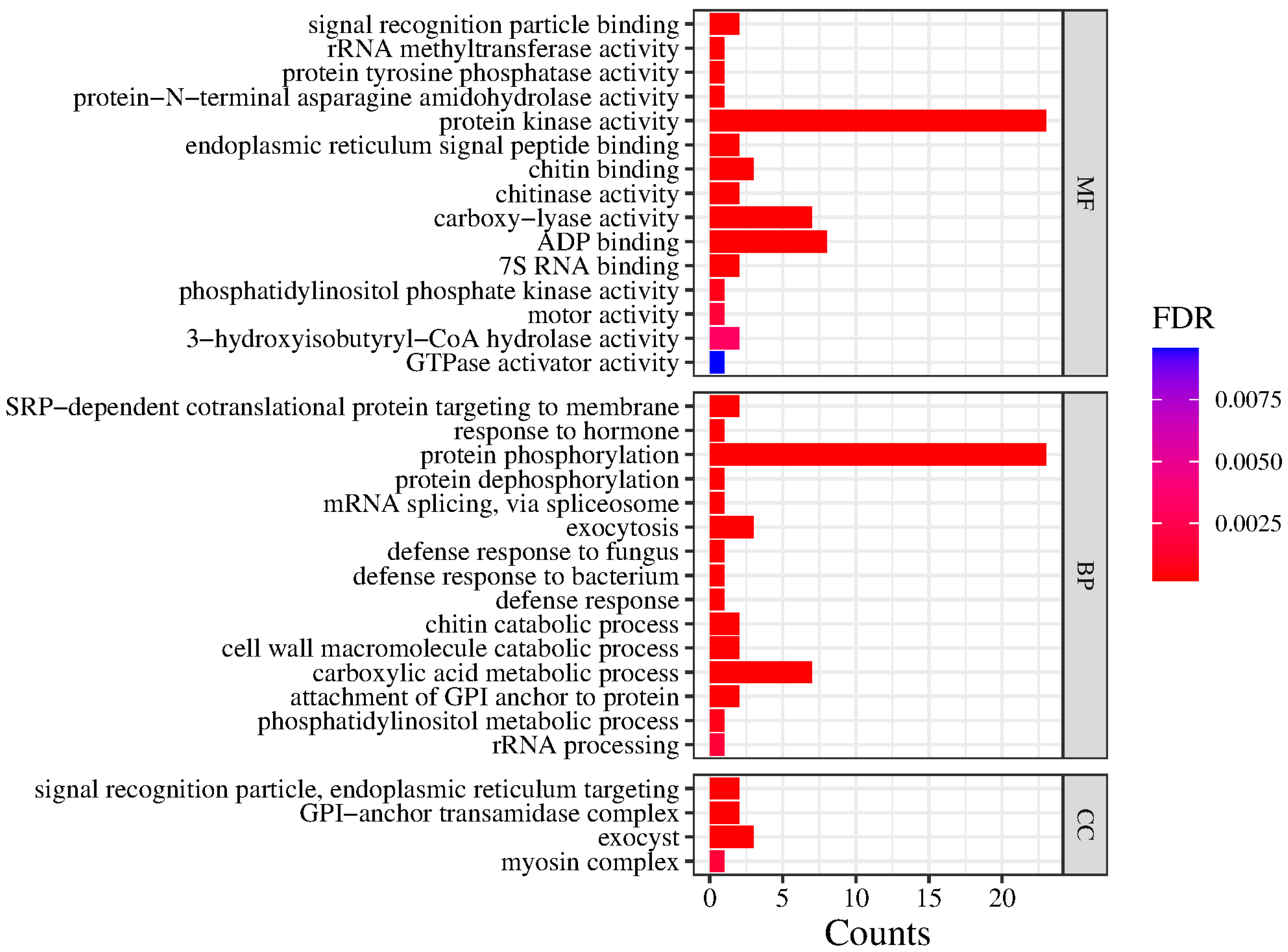
2.5. Selective Sweeps
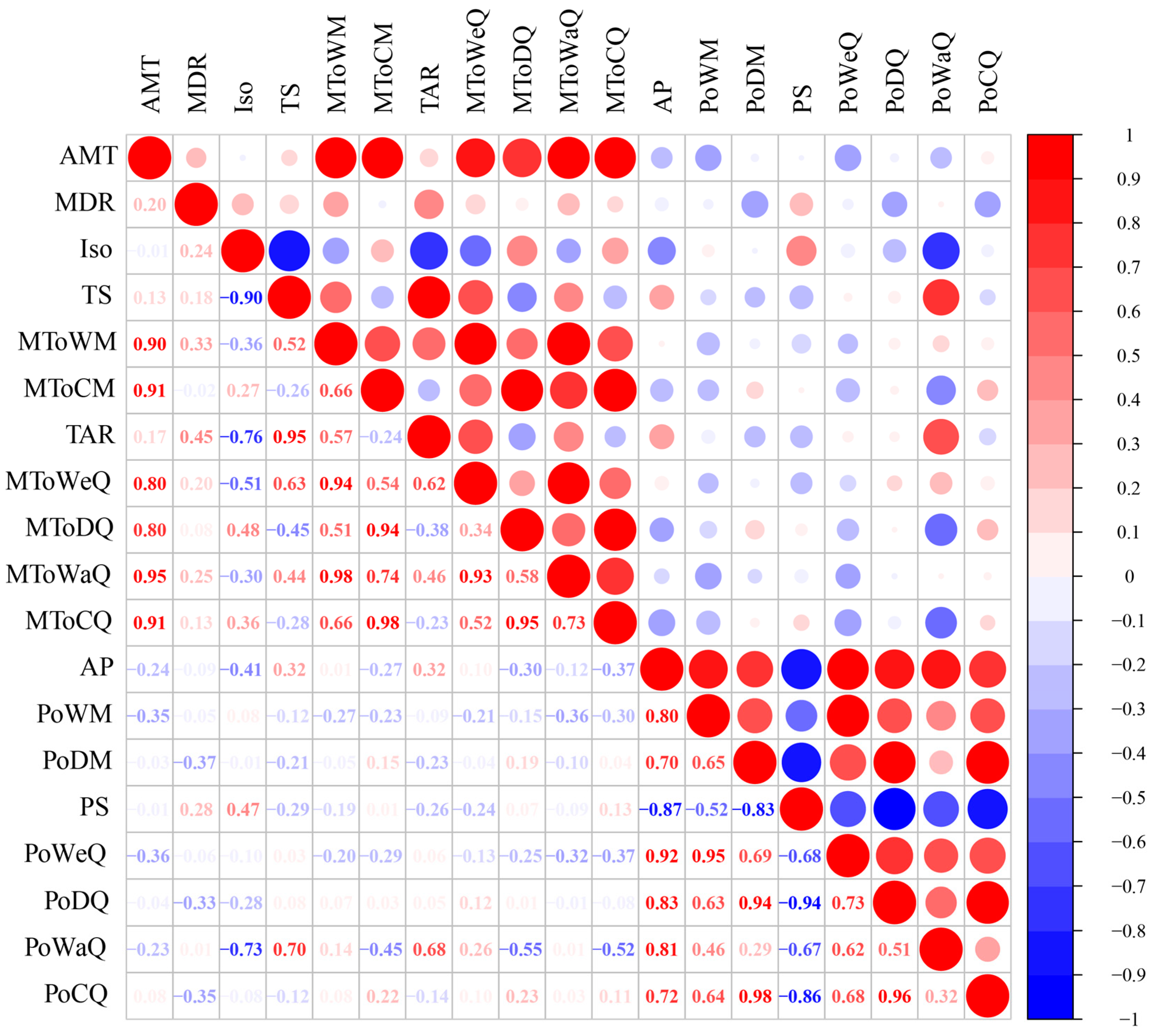
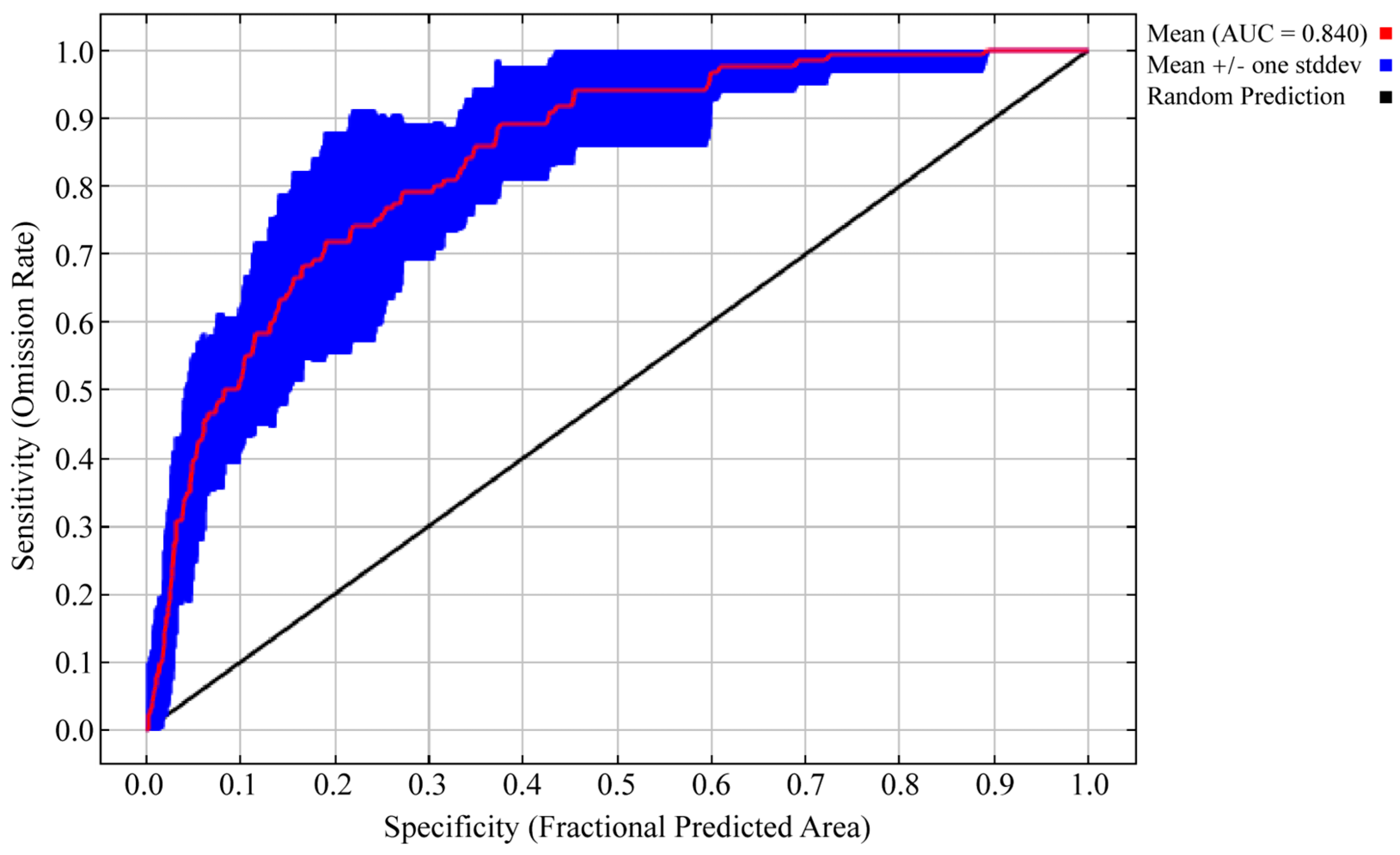
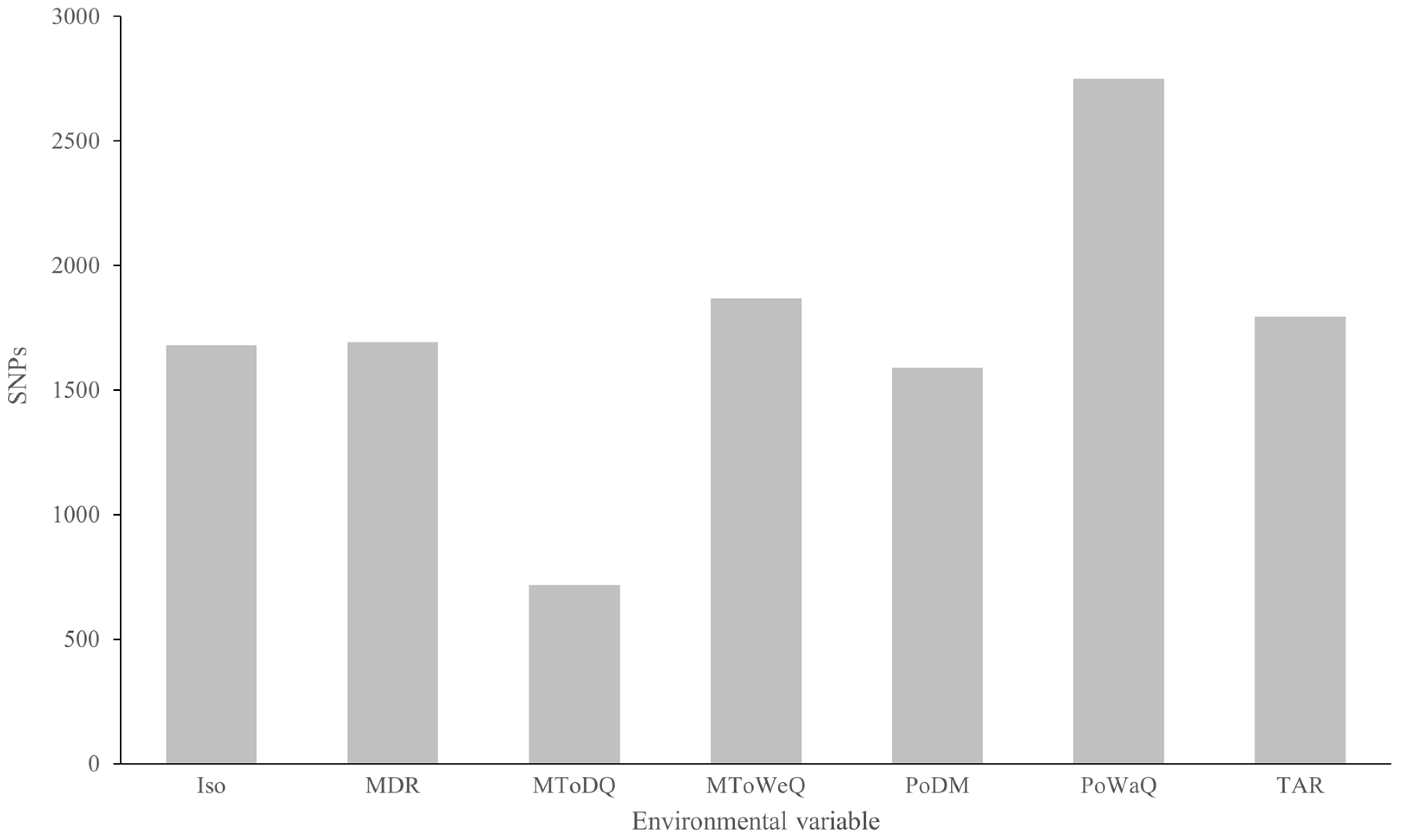
2.6. Genetic–Environment Association Analysis
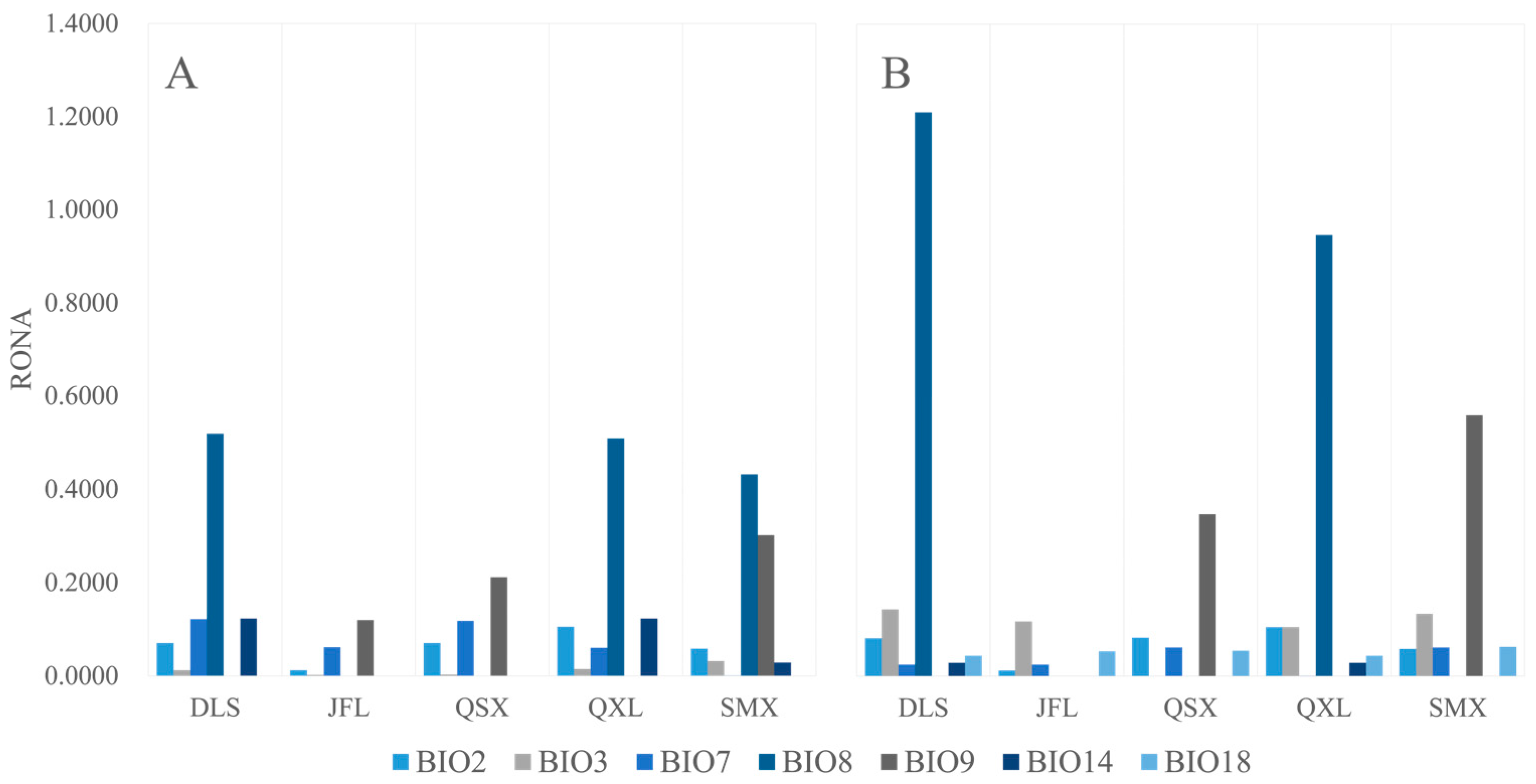
2.7. Risk of Non-Adaptedness (RONA)
3. Discussion
4. Materials and Methods
4.1. Sampling, Library Preparation, and Sequencing
4.2. SNP Calling
4.3. Population Structure Analyses
4.4. Demographic History Analyses
4.5. Species Distribution Model
4.6. Detection of Selective Sweeps
4.7. Genetic–Environment Association (GEA) Analysis
4.8. Risk of Non-Adaptedness (RONA) Under Future Climatic Scenarios
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the receiver operating characteristic (ROC) curve |
| BIO | Bioclimatic variables |
| CV | Cross-validation |
| CVH | Chinese Virtual Herbarium |
| FDR | False Discovery Rate |
| GEA | Genotype–environment association |
| GO | Gene Ontology |
| LD | Linkage disequilibrium |
| LFMM | Latent Factor Mixed Model |
| LIG | Last interglacial |
| MAF | Minor allele frequency |
| MH | Mid-Holocene |
| ML | Maximum likelihood |
| Mya | Million years ago |
| NCBI | National Center for Biotechnology Information |
| Ne | Effective population size |
| PCA | Principal component analysis |
| RONA | Risk of non-adaptedness |
| SDM | Species distribution model |
| SNP | Single-nucleotide polymorphism |
| SSP | Shared Socioeconomic Pathway |
| VCF | Variant Call Format |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Period | Unsuitable Region (km2) | Low-Suitability Region (km2) | Moderately Suitable Region (km2) | Highly Suitable Region (km2) |
|---|---|---|---|---|
| LGM | 3845.14 | 4411.81 | 11,892.36 | 8888.89 |
| MH | 4929.17 | 5463.89 | 5375.7 | 13,269.44 |
| Present | 5746.53 | 7545.83 | 9564.58 | 6420.14 |
| 2080–2100 (SSP126) | 5974.3 | 6909.03 | 8843.05 | 7550.69 |
| 2080–2100 (SSP585) | 7030.56 | 6754.86 | 9775.69 | 5715.97 |
| Variable | bio2 | bio3 | bio7 | bio8 | bio9 | bio14 | bio18 |
|---|---|---|---|---|---|---|---|
| SNPs | 1342 | 1326 | 1393 | 1418 | 630 | 1186 | 1936 |
| DLS | 0.0699 | 0.0120 | 0.1217 | 0.5188 | 0.0000 | 0.1223 | 0.0578 |
| JFL | 0.0117 | 0.0018 | 0.0614 | 0.0000 | 0.1192 | 0.0000 | 0.0530 |
| QSX | 0.0699 | 0.0024 | 0.1177 | 0.0000 | 0.2115 | 0.0000 | 0.0000 |
| QXL | 0.1049 | 0.0144 | 0.0602 | 0.5084 | 0.0000 | 0.1223 | 0.0000 |
| SMX | 0.0583 | 0.0317 | 0.0000 | 0.4321 | 0.3016 | 0.0286 | 0.0616 |
| Mean | 0.0629 | 0.0125 | 0.0722 | 0.2918 | 0.1265 | 0.0547 | 0.0345 |
| Min R2 | 0.0004 | 0.0007 | 0.0108 | 0.0020 | 0.0002 | 0.0230 | 0.0025 |
| Max R2 | 0.9212 | 0.8253 | 0.8728 | 0.7412 | 0.7146 | 0.9713 | 0.7474 |
| Average R2 | 0.2856 | 0.2379 | 0.2359 | 0.2617 | 0.2707 | 0.2139 | 0.2565 |
| Variable | bio2 | bio3 | bio7 | bio8 | bio9 | bio14 | bio18 |
|---|---|---|---|---|---|---|---|
| SNPs | 1342 | 1326 | 1393 | 1418 | 630 | 1186 | 1936 |
| DLS | 0.0807 | 0.1428 | 0.0242 | 1.2102 | 0.0000 | 0.0286 | 0.0434 |
| JFL | 0.0117 | 0.1167 | 0.0242 | 0.0000 | 0.0000 | 0.0000 | 0.0530 |
| QSX | 0.0819 | 0.0000 | 0.0614 | 0.0000 | 0.3469 | 0.0000 | 0.0543 |
| QXL | 0.1049 | 0.1048 | 0.0000 | 0.9461 | 0.0000 | 0.0285 | 0.0434 |
| SMX | 0.0583 | 0.1333 | 0.0614 | 0.0000 | 0.5592 | 0.0000 | 0.0625 |
| Mean | 0.0675 | 0.0995 | 0.0343 | 0.4313 | 0.1812 | 0.0114 | 0.0513 |
| Min R2 | 0.0004 | 0.0007 | 0.0108 | 0.0020 | 0.0002 | 0.0230 | 0.0025 |
| Max R2 | 0.9212 | 0.8253 | 0.8728 | 0.7412 | 0.7146 | 0.9713 | 0.7474 |
| Average R2 | 0.2856 | 0.2379 | 0.2359 | 0.2617 | 0.2707 | 0.2139 | 0.2565 |
References
- Dauphin, B.; Rellstab, C.; Schmid, M.; Zoller, S.; Karger, D.N.; Brodbeck, S.; Guillaume, F.; Gugerli, F. Genomic vulnerability to rapid climate warming in a tree species with a long generation time. Glob. Change Biol. 2021, 27, 1181–1195. [Google Scholar] [CrossRef]
- Aitken, S.N.; Yeaman, S.; Holliday, J.A.; Wang, T.; Curtis-McLane, S. Adaptation, migration or extirpation: Climate change outcomes for tree populations. Evol. Appl. 2008, 1, 95–111. [Google Scholar] [CrossRef]
- Leimu, R.; Fischer, M. A meta-analysis of local adaptation in plants. PLoS ONE 2008, 3, e4010. [Google Scholar] [CrossRef]
- Sork, V.L. Genomic Studies of Local Adaptation in Natural Plant Populations. J. Hered. 2017, 109, 3–15. [Google Scholar] [CrossRef]
- Hoffmann, A.A.; Sgro, C.M. Climate change and evolutionary adaptation. Nature 2011, 470, 479–485. [Google Scholar] [CrossRef]
- Dick, C.W. Phylogeography and Population Structure of Tropical Trees. Trop. Plant Biol. 2010, 3, 1–3. [Google Scholar] [CrossRef]
- Jops, K.; Dalling, J.W.; O’Dwyer, J.P. Life history is a key driver of temporal fluctuations in tropical tree abundances. Proc. Natl. Acad. Sci. USA 2025, 122, e1872619174. [Google Scholar] [CrossRef]
- Mayol, M.; Riba, M.; Cavers, S.; Grivet, D.; Vincenot, L.; Cattonaro, F.; Vendramin, G.G.; Gonzalez-Martinez, S.C. A multiscale approach to detect selection in nonmodel tree species: Widespread adaptation despite population decline in Taxus baccata L. Evol. Appl. 2020, 13, 143–160. [Google Scholar] [CrossRef]
- Capblancq, T.; Fitzpatrick, M.C.; Bay, R.A.; Exposito-Alonso, M.; Keller, S.R. Genomic Prediction of (Mal) Adaptation Across Current and Future Climatic Landscapes. Annu. Rev. Ecol. Evol. Syst. 2020, 51, 245–269. [Google Scholar] [CrossRef]
- Liu, H.; Harris, A.J.; Wang, Z.; Chen, H.; Li, Z.; Wei, X. The genome of the Paleogene relic tree Bretschneidera sinensis: Insights into trade-offs in gene family evolution, demographic history, and adaptive SNPs. DNA Res. 2022, 29, dsac003. [Google Scholar] [CrossRef]
- Sang, Y.; Long, Z.; Dan, X.; Feng, J.; Shi, T.; Jia, C.; Zhang, X.; Lai, Q.; Yang, G.; Zhang, H.; et al. Genomic insights into local adaptation and future climate-induced vulnerability of a keystone forest tree in East Asia. Nat. Commun. 2022, 13, 6541. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.; Potts, B.M.; Delzon, S. Genetic divergence in forest trees: Understanding the consequences of climate change. Funct. Ecol. 2014, 28, 22–36. [Google Scholar] [CrossRef]
- Rellstab, C.; Zoller, S.; Walthert, L.; Lesur, I.; Pluess, A.R.; Graf, R.; Bodenes, C.; Sperisen, C.; Kremer, A.; Gugerli, F. Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol. Ecol. 2016, 25, 5907–5924. [Google Scholar] [CrossRef]
- Feng, L.; Du, F.K. Landscape Genomics in Tree Conservation Under a Changing Environment. Front. Plant Sci. 2022, 13, 822217. [Google Scholar] [CrossRef]
- Groen, S.C.; Purugganan, M.D. Systems genetics of plant adaptation to environmental stresses. Am. J. Bot. 2016, 103, 2019–2021. [Google Scholar] [CrossRef]
- Martins, K.; Gugger, P.F.; Llanderal-Mendoza, J.; Gonzalez-Rodriguez, A.; Fitz-Gibbon, S.T.; Zhao, J.L.; Rodriguez-Correa, H.; Oyama, K.; Sork, V.L. Landscape genomics provides evidence of climate-associated genetic variation in Mexican populations of Quercus rugosa. Evol. Appl. 2018, 11, 1842–1858. [Google Scholar] [CrossRef]
- Waldvogel, A.M.; Feldmeyer, B.; Rolshausen, G.; Exposito-Alonso, M.; Rellstab, C.; Kofler, R.; Mock, T.; Schmid, K.; Schmitt, I.; Bataillon, T.; et al. Evolutionary genomics can improve prediction of species’ responses to climate change. Evol. Lett. 2020, 4, 4–18. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.W.; Li, J. Taxonomic notes on Castanopsis (Fagaceae, Castaneoideae) from China. Phytotaxa 2013, 146, 50–60. [Google Scholar]
- Chen, X.; Yu, B. A Review of the Genus of Castanopsis in Guangdong and Hainan. J. South. China Agric. Univ. 1991, 2, 87–95. [Google Scholar]
- Chen, X.; Feng, Y.; Chen, S.; Yang, K.; Wen, X.; Sun, Y. Species Delimitation and Genetic Relationship of Castanopsis hainanensis and Castanopsis wenchangensis (Fagaceae). Plants 2023, 12, 3544. [Google Scholar] [CrossRef]
- Wang, T.; Meng, H.; Wang, N.; Zheng, S.; Jiang, Y.; Lin, D.; Song, Y.; Kozlowski, G. Adaptive divergence and genetic vulnerability of relict species under climate change: A case study of Pterocarya macroptera. Ann. Bot. 2023, 132, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.J.; Qin, X.T.; Song, X.Q.; Zhang, L.N.; Ren, M.X. Genetic delimitation of Oreocharis species from Hainan Island. Phytokeys 2020, 157, 59–81. [Google Scholar]
- Oswald, J.A.; Overcast, I.; Mauck, W.M.; Andersen, M.J.; Smith, B.T. Isolation with asymmetric gene flow during the nonsynchronous divergence of dry forest birds. Mol. Ecol. 2017, 26, 1386–1400. [Google Scholar] [PubMed]
- Luo, J.; Dai, X.; Chen, J.; Shuang, H.; Yuan, C.; Luo, D. Study on the characteristics of genetic diversity and population structure of a rare and endangered species of Rhododendron nymphaeoides (Ericaceae) based on microsatellite markers. BMC Plant Biol. 2025, 25, 310. [Google Scholar]
- Wu, Z.; Yu, D.; Wang, Z.; Li, X.; Xu, X. Great influence of geographic isolation on the genetic differentiation of Myriophyllum spicatum under a steep environmental gradient. Sci. Rep. 2015, 5, 15618. [Google Scholar]
- Montejo-Kovacevich, G.; Meier, J.I.; Bacquet, C.N.; Warren, I.A.; Chan, Y.F.; Kucka, M.; Salazar, C.; Rueda, M.N.; Montgomery, S.H.; McMillan, W.O.; et al. Repeated genetic adaptation to altitude in two tropical butterflies. Nat. Commun. 2022, 13, 4676. [Google Scholar] [CrossRef]
- Wang, S.Q. Genetic diversity and population structure of the endangered species Paeonia decomposita endemic to China and implications for its conservation. BMC Plant Biol. 2020, 20, 510. [Google Scholar]
- Tang, L.; Long, J.; Wang, H.; Rao, C.; Long, W.; Yan, L.; Liu, Y. Conservation genomic study of Hopea hainanensis (Dipterocarpaceae), an endangered tree with extremely small populations on Hainan Island, China. Front. Plant Sci. 2024, 15, 1442807. [Google Scholar] [CrossRef]
- Huang, P.; Wang, T.; Li, M.; Lu, Z.; Su, R.; Fang, O.; Li, L.; Zhou, S.; Tan, Y.; Meng, H.; et al. RAD-seq data for Engelhardia roxburghiana provide insights into the palaeogeography of Hainan Island and its relationship to mainland China since the late Eocene. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2024, 651, 112392. [Google Scholar] [CrossRef]
- Jiang, X.; Gardner, E.M.; Meng, H.; Deng, M.; Xu, G. Land bridges in the Pleistocene contributed to flora assembly on the continental islands of South China: Insights from the evolutionary history of Quercus championii. Mol. Phylogenet Evol. 2019, 132, 36–45. [Google Scholar] [CrossRef]
- Wang, G.Q.; Scherson, R.A.; Vera, D.; Bai, Y.H.; Wen, J.; Guo, L.Y.; Hughes, A.C.; Wang, H.F. Spatial patterns and drivers of native plant diversity in Hainan, China. J. Syst. Evol. 2024, 62, 603–620. [Google Scholar] [CrossRef]
- Peng, M.S.; He, J.D.; Liu, H.X.; Zhang, Y.P. Tracing the legacy of the early Hainan Islanders—A perspective from mitochondrial DNA. BMC Evol. Biol. 2011, 11, 46. [Google Scholar] [CrossRef]
- Liu, Z.; Pagani, M.; Zinniker, D.; Deconto, R.; Huber, M.; Brinkhuis, H.; Shah, S.R.; Leckie, R.M.; Pearson, A. Global cooling during the eocene-oligocene climate transition. Science 2009, 323, 1187–1190. [Google Scholar] [CrossRef]
- Zhu, H. Biogeographical Evidences Help Revealing the Origin of Hainan Island. PLoS ONE 2016, 11, e0151941. [Google Scholar] [CrossRef]
- An, S.; Joye, S.B. Enhancement of coupled nitrification-denitrification by benthic photosynthesis in shallow estuarine sediments. Limnol. Oceanogr. 2001, 46, 62–74. [Google Scholar] [CrossRef]
- An, Z.; Clemens, S.C.; Shen, J.; Qiang, X.; Jin, Z.; Sun, Y.; Prell, W.L.; Luo, J.; Wang, S.; Xu, H.; et al. Glacial-interglacial Indian summer monsoon dynamics. Science 2011, 333, 719–723. [Google Scholar] [CrossRef]
- Du, F.K.; Hou, M.; Wang, W.; Mao, K.; Hampe, A. Phylogeography of Quercus aquifolioides provides novel insights into the Neogene history of a major global hotspot of plant diversity in south-west China. J. Biogeogr. 2017, 44, 294–307. [Google Scholar] [CrossRef]
- Hou, Z.; Wang, Z.; Li, A.; Zhang, J. Genetic Architecture, Demographic History and Genomic Differentiation of Populus davidiana Revealed by Whole Genome Resequencing. Evol. Appl. 2020, 13, 2582–2596. [Google Scholar]
- Chen, X.Y.; Zhou, B.F.; Shi, Y.; Liu, H.; Liang, Y.Y.; Ingvarsson, P.K.; Wang, B. Evolution of the Correlated Genomic Variation Landscape Across a Divergence Continuum in the Genus Castanopsis. Mol. Biol. Evol. 2024, 41, msae191. [Google Scholar] [CrossRef]
- Reuter, M.; Kern, A.K.; Harzhauser, M.; Kroh, A.; Piller, W.E. Global warming and South Indian monsoon rainfall—Lessons from the Mid-Miocene. Gondwana Res. 2013, 23, 1172–1177. [Google Scholar] [CrossRef]
- Xie, Z.; Chen, D.; Puxi, L.I. Discovering Climate Change during the Early 21st Century via Wasserstein Stability Analysis. Adv. Atmos. Sci. 2024, 42, 373. [Google Scholar] [CrossRef]
- Ernakovich, J.G.; Hopping, K.A.; Berdanier, A.B.; Simpson, R.T.; Kachergis, E.J.; Steltzer, H.; Wallenstein, M.D. Predicted responses of arctic and alpine ecosystems to altered seasonality under climate change. Glob. Change Biol. 2014, 20, 3256–3269. [Google Scholar] [CrossRef]
- Zhu, S.; Chen, J.; Zhao, J.; Comes, H.P.; Li, P.; Fu, C.; Xie, X.; Lu, R.; Xu, W.; Feng, Y.; et al. Genomic insights on the contribution of balancing selection and local adaptation to the long-term survival of a widespread living fossil tree, Cercidiphyllum japonicum. New Phytol. 2020, 228, 1674–1689. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Landis, J.B.; Zhang, J.; Yang, L.; Lin, N.; Zhang, H.; Guo, R.; Li, L.; Zhang, Y.; et al. Genomic insights into adaptation to heterogeneous environments for the ancient relictual Circaeaster agrestis (Circaeasteraceae, Ranunculales). New Phytol. 2020, 228, 285–301. [Google Scholar] [CrossRef]
- Aguirre-Liguori, J.A.; Ramírez-Barahona, S.; Gaut, B.S. The evolutionary genomics of species’ responses to climate change. Nat. Ecol. Evol. 2021, 5, 1350–1360. [Google Scholar] [CrossRef]
- Daewon, K.; Jiyuan, Y.; Fangwei, G.; Sungjin, P.; Jonathon, C.; Alexander, A.; Mayes, H.B.; Jeong, J.S.; Dong, B.J.; Erik, N. A temperature-sensitive FERONIA mutant allele that alters root hair growth. Plant Physiol. 2020, 185, 405–423. [Google Scholar]
- Yang, H.; Zhao, Y.; Chen, N.; Liu, Y.; Hu, X. A new adenylyl cyclase, putative disease resistance RPP13-like protein 3, participates in abscisic acid-mediated heat stress resistance in maize. J. Exp. Bot. 2020, 72, 283–301. [Google Scholar] [CrossRef]
- Ahmed, W.; Xia, Y.; Li, R.; Zhang, H.; Siddique, K.H.M.; Guo, P. Identification and Analysis of Small Interfering RNAs Associated with Heat Stress in Flowering Chinese Cabbage Using High-Throughput Sequencing. Front. Genet. 2021, 12, 746816. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, Z.; Zheng, X.; Shan, W.; Fan, J. How a single receptor-like kinase exerts diverse roles: Lessons from FERONIA. Mol. Hortic. 2022, 2, 25. [Google Scholar] [CrossRef]
- Kannangara, R.; Branigan, C.; Liu, Y.; Penfield, T.; Rao, V.; Mouille, G.; Hofte, H.; Pauly, M.; Riechmann, J.L.; Broun, P. The Transcription Factor WIN1/SHN1 Regulates Cutin Biosynthesis in Arabidopsis thaliana. Plant Cell 2007, 19, 1278–1294. [Google Scholar] [CrossRef]
- Jing, Y.; Yang, Z.; Yang, R.; Zhang, Y.; Qiao, W.; Zhou, Y.; Sun, J. PKL is stabilized by MMS21 to negatively regulate Arabidopsis drought tolerance through directly repressing AFL1 transcription. New Phytol. 2023, 239, 920–935. [Google Scholar] [PubMed]
- Liu, X.; Jiang, W.; Li, Y.; Nie, H.; Cui, L.; Li, R.; Tan, L.; Peng, L.; Li, C.; Luo, J.; et al. FERONIA coordinates plant growth and salt tolerance via the phosphorylation of phyB. Nat. Plants 2023, 9, 645–660. [Google Scholar] [CrossRef]
- Islam, M.M.; El-Sappah, A.; Ali, H.; Zandi, P.; Huang, Q.; Soaud, S.; Alazizi, E.M.Y.; Wafa, H.; Hossain, M.A.; Liang, Y. Pathogenesis-related proteins (PRs) countering environmental stress in plants: A review. S. Afr. J. Bot. 2023, 160, 414–427. [Google Scholar]
- Chowdhury, J.; Schober, M.S.; Shirley, N.J.; Singh, R.R.; Little, A. Down-regulation of the glucan synthase-like 6 gene (HvGsl6) in barley leads to decreased callose accumulation and increased cell wall penetration by Blumeria graminis f. sp. hordei. New Phytol. 2016, 212, 434–443. [Google Scholar]
- Duriez, P.; Vautrin, S.; Auriac, M.C.; Bazerque, J.; Boniface, M.C.; Callot, C.; Carrere, S.; Cauet, S.; Chabaud, M.; Gentou, F.; et al. A receptor-like kinase enhances sunflower resistance to Orobanche cumana. Nat. Plants 2019, 5, 1211–1215. [Google Scholar] [CrossRef]
- Kulich, I.; Pe Enková, T.; Sekere, J.; Smetana, O.E.; Fendrych, M.; Foissner, I.; Höftberger, M.; Ársky, V. Arabidopsis exocyst subcomplex containing subunit EXO70B1 is involved in autophagy-related transport to the vacuole. Traffic 2013, 14, 1155–1165. [Google Scholar]
- Xu, X.; Hayashi, N.; Wang, C.T.; Fukuoka, S.; Kawasaki, S.; Takatsuji, H.; Jiang, C.J. Rice blast resistance gene Pikahei-1(t), a member of a resistance gene cluster on chromosome 4, encodes a nucleotide-binding site and leucine-rich repeat protein. Mol. Breed. 2014, 34, 691–700. [Google Scholar]
- Zhang, X.; Wang, G.; Qu, X.; Wang, M.; Guo, H.; Zhang, L.; Li, T.; Wang, Y.; Zhang, H.; Ji, W. A truncated CC-NB-ARC gene TaRPP13L1-3D positively regulates powdery mildew resistance in wheat via the RanGAP-WPP complex-mediated nucleocytoplasmic shuttle. Planta 2022, 225, 60. [Google Scholar]
- Sreekanta, S.; Bethke, G.; Hatsugai, N.; Tsuda, K.; Thao, A.; Wang, L.; Katagiri, F.; Glazebrook, J. The receptor-like cytoplasmic kinase PCRK1 contributes to pattern-triggered immunity against Pseudomonas syringae in Arabidopsis thaliana. New Phytol. 2015, 207, 78–90. [Google Scholar] [CrossRef]
- Mingqin, C.; Shu-Zon, W.; Ryken, S.E.; O’Sullivan, J.E.; Magdalena, B. COPII Sec23 proteins form isoform-specific endoplasmic reticulum exit sites with differential effects on polarized growth. Plant Cell 2021, 34, 333–350. [Google Scholar]
- Hung, F.Y.; Feng, Y.R.; Hsin, K.T.; Shih, Y.H.; Chang, C.H.; Zhong, W.; Lai, Y.C.; Xu, Y.; Yang, S.; Sugimoto, K. Arabidopsis histone H3 lysine 9 methyltransferases KYP/SUVH5/6 are involved in leaf development by interacting with AS1-AS2 to repress KNAT1 and KNAT2. Commun. Biol. 2023, 6, 219. [Google Scholar]
- Somashekar, H.; Mimura, M.; Tsuda, K.; Nonomura, K. Rice GLUCAN SYNTHASE-LIKE5 promotes anther callose deposition to maintain meiosis initiation and progression. Plant Physiol. 2023, 191, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, M.L.; Fischer, G.A. The impact of climate change on the distribution of Castanopsis (Fagaceae) species in south China and Indo-China region. Glob. Ecol. Conserv. 2021, 26, e01388. [Google Scholar] [CrossRef]
- Brady, S.P.; Bolnick, D.I.; Angert, A.L.; Gonzalez, A.; Barrett, R.D.H.; Crispo, E.; Derry, A.M.; Eckert, C.G.; Fraser, D.J.; Fussmann, G.F.; et al. Causes of maladaptation. Evol. Appl. 2019, 12, 1229–1242. [Google Scholar] [CrossRef]
- Kottler, E.J.; Dickman, E.E.; Sexton, J.P.; Emery, N.C.; Franks, S.J. Draining the Swamping Hypothesis: Little Evidence that Gene Flow Reduces Fitness at Range Edges. Trends Ecol. Evol. 2021, 36, 533–544. [Google Scholar] [CrossRef]
- Monnahan, P.J.; Colicchio, J.; Kelly, J.K. A genomic selection component analysis characterizes migration-selection balance. Evolution 2015, 69, 1713–1727. [Google Scholar] [CrossRef]
- Eva-Maria, W.; Christine, D.; Cock, V.O.; Jerome, C. Estimates of Genetic Differentiation Measured by FST Do Not Necessarily Require Large Sample Sizes When Using Many SNP Markers. PLoS ONE 2012, 7, e42649. [Google Scholar]
- Nazareno, A.G.; Bemmels, J.B.; Dick, C.W.; Lohmann, L.G. Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 2017, 17, 1136–1147. [Google Scholar]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, J.; Zeng, X.; Chen, R.; Feng, Y.; Chen, S.; Yang, K. Chromosome-scale genome assembly of Castanopsis tibetana provides a powerful comparative framework to study the evolution and adaptation of Fagaceae trees. Mol. Ecol. Resour. 2022, 22, 1178–1189. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Terhorst, J.; Kamm, J.A.; Song, Y.S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 2017, 49, 303–309. [Google Scholar] [CrossRef]
- Kass, J.M.; Muscarella, R.; Galante, P.J.; Bohl, C.L.; Pinilla-Buitrago, G.E.; Boria, R.A.; Soley-Guardia, M.; Anderson, R.P. ENMeval 2.0: Redesigned for customizable and reproducible modelling of species’ niches and distributions. Methods Ecol. Evol. 2021, 12, 1602–1608. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Urbani, F.; D’Alessandro, P.; Frasca, R.; Biondi, M. Maximum entropy modeling of geographic distributions of the flea beetle species endemic in Italy (Coleoptera: Chrysomelidae: Galerucinae: Alticini). Zool. Anz. A J. Comp. Zool. 2015, 258, 99–109. [Google Scholar] [CrossRef]
- Elith, J.; Phillips, S.J.; Hastie, T.; Dudík, M.; Chee, Y.E.; Yates, C.J. A statistical explanation of MaxEnt for ecologists. Divers. Distrib. 2011, 17, 43–57. [Google Scholar] [CrossRef]
- Alachiotis, N.; Pavlidis, P. RAiSD detects positive selection based on multiple signatures of a selective sweep and SNP vectors. Commun. Biol. 2018, 1, 79. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Schlotterer, C. Gowinda: Unbiased analysis of gene set enrichment for genome-wide association studies. Bioinformatics 2012, 28, 2084–2085. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.D.; Xu, H. A general method for accurate estimation of false discovery rates in identification of differentially expressed genes. Bioinformatics 2014, 30, 2018–2025. [Google Scholar] [CrossRef]
- Caye, K.; Jumentier, B.; Lepeule, J.; Francois, O. LFMM 2: Fast and Accurate Inference of Gene-Environment Associations in Genome-Wide Studies. Mol. Biol. Evol. 2019, 36, 852–860. [Google Scholar] [CrossRef]






| Population | Sampling Location | Elevation (m) | Longitude (°) | Latitude (°) |
|---|---|---|---|---|
| DLS | Diaoluoshan Mountain, Lingshui County | 377 | 109.9160 | 18.6598 |
| JFL | Jianfengling Mountain, Ledong County | 283 | 108.8379 | 18.6957 |
| QSX | Qingsong Village, Baisha County | 396 | 109.2745 | 19.1304 |
| QXL | Qixianling Mountain, Baoting County | 264 | 109.6759 | 18.7034 |
| SMX | Shuiman Village, Wuzhishan City | 658 | 109.6629 | 18.8830 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Chen, X.; Wu, Y.; Sun, Y. Genomic Signatures of Environmental Adaptation in Castanopsis hainanensis (Fagaceae). Plants 2025, 14, 1128. https://doi.org/10.3390/plants14071128
Li S, Chen X, Wu Y, Sun Y. Genomic Signatures of Environmental Adaptation in Castanopsis hainanensis (Fagaceae). Plants. 2025; 14(7):1128. https://doi.org/10.3390/plants14071128
Chicago/Turabian StyleLi, Sha, Xing Chen, Yang Wu, and Ye Sun. 2025. "Genomic Signatures of Environmental Adaptation in Castanopsis hainanensis (Fagaceae)" Plants 14, no. 7: 1128. https://doi.org/10.3390/plants14071128
APA StyleLi, S., Chen, X., Wu, Y., & Sun, Y. (2025). Genomic Signatures of Environmental Adaptation in Castanopsis hainanensis (Fagaceae). Plants, 14(7), 1128. https://doi.org/10.3390/plants14071128