
A Bread Wheat Line with the Substituted Wild Emmer Chromosome 4A Results in Fragment Deletions of Chromosome 4B and Weak Plants



Abstract
1. Introduction
2. Results
2.1. Some Offspring of (CS x CASL4AL) Grows Weakly
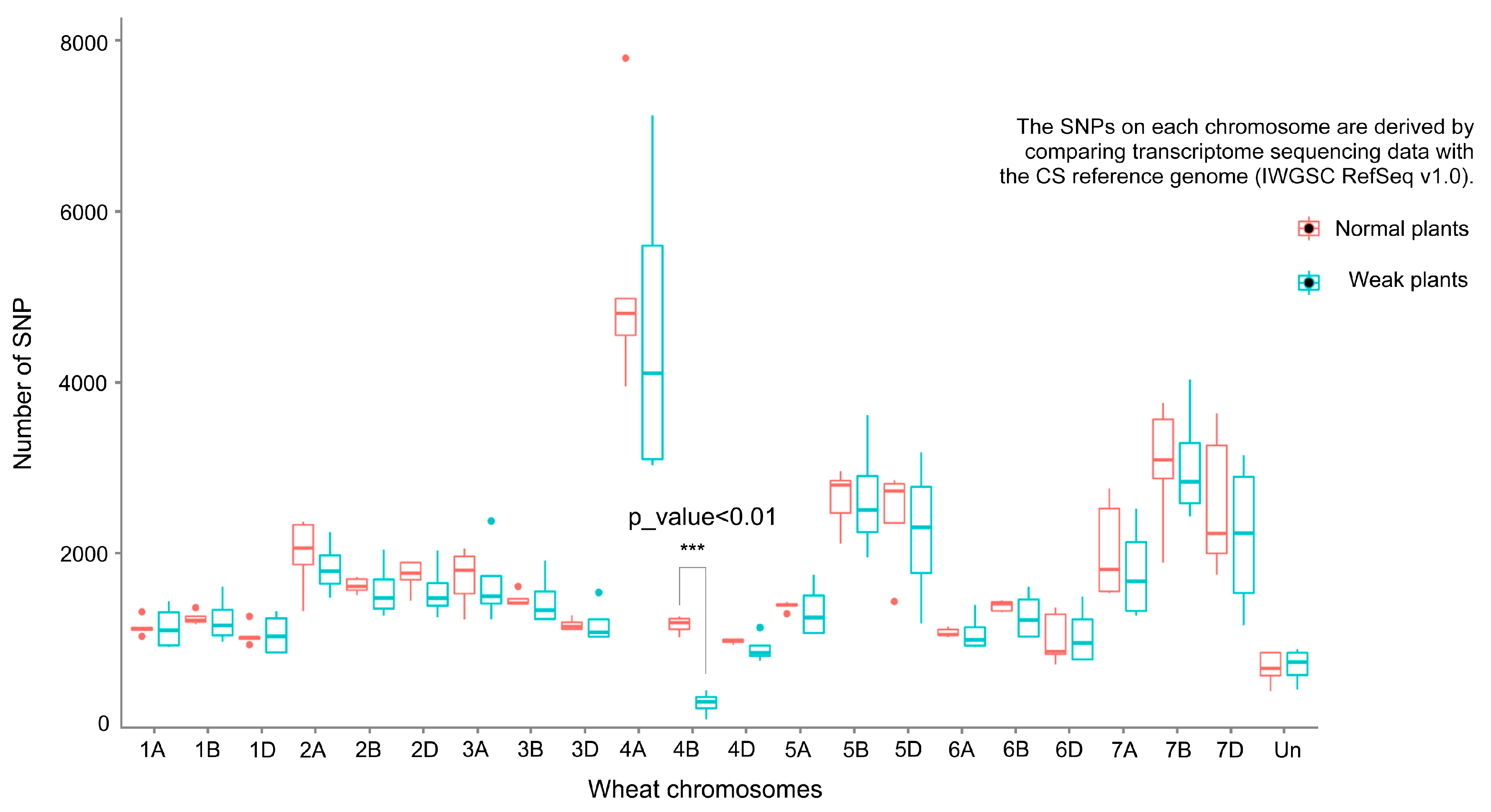
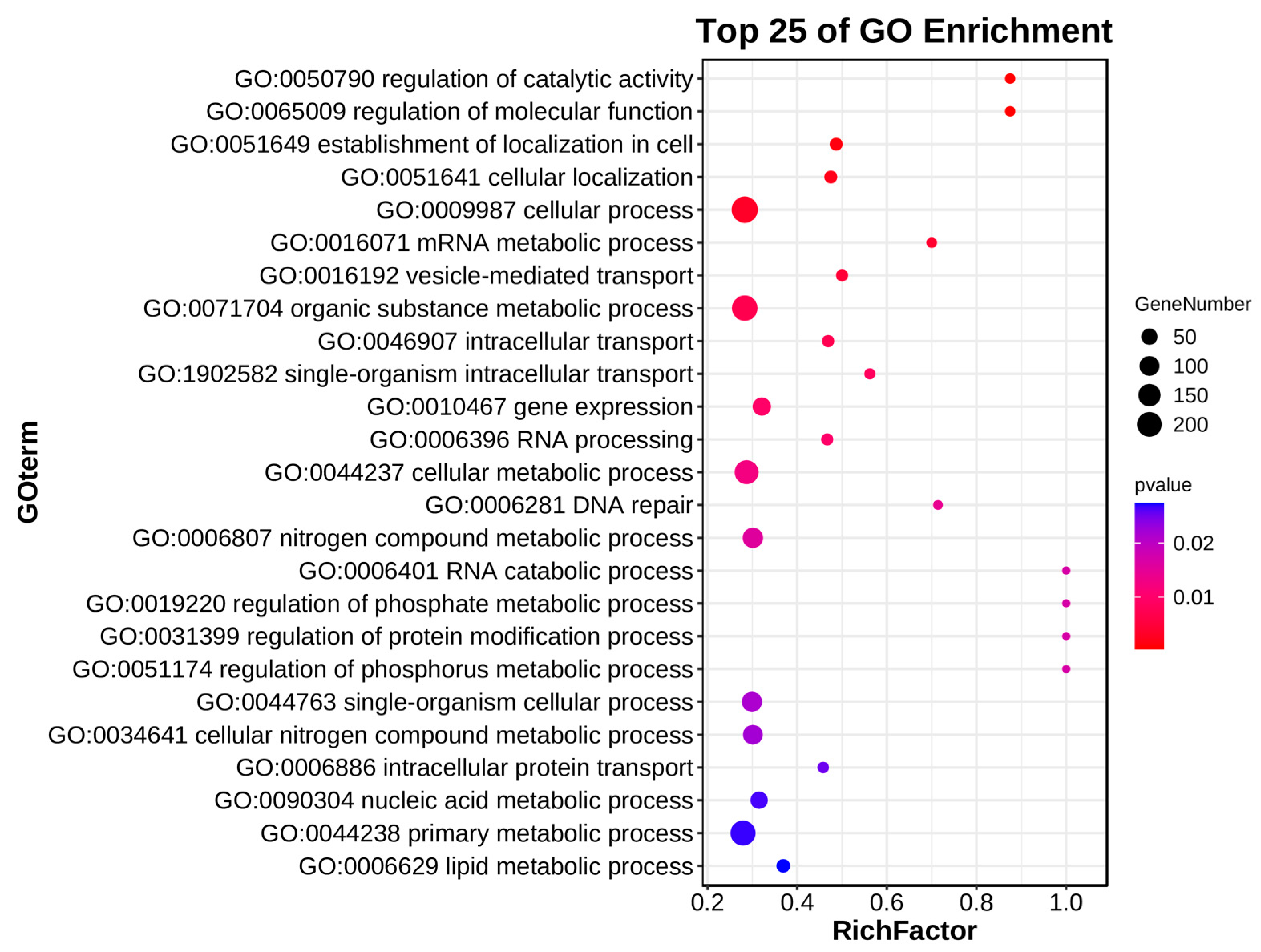
2.2. RNA-seq Analysis on Each Chromosome
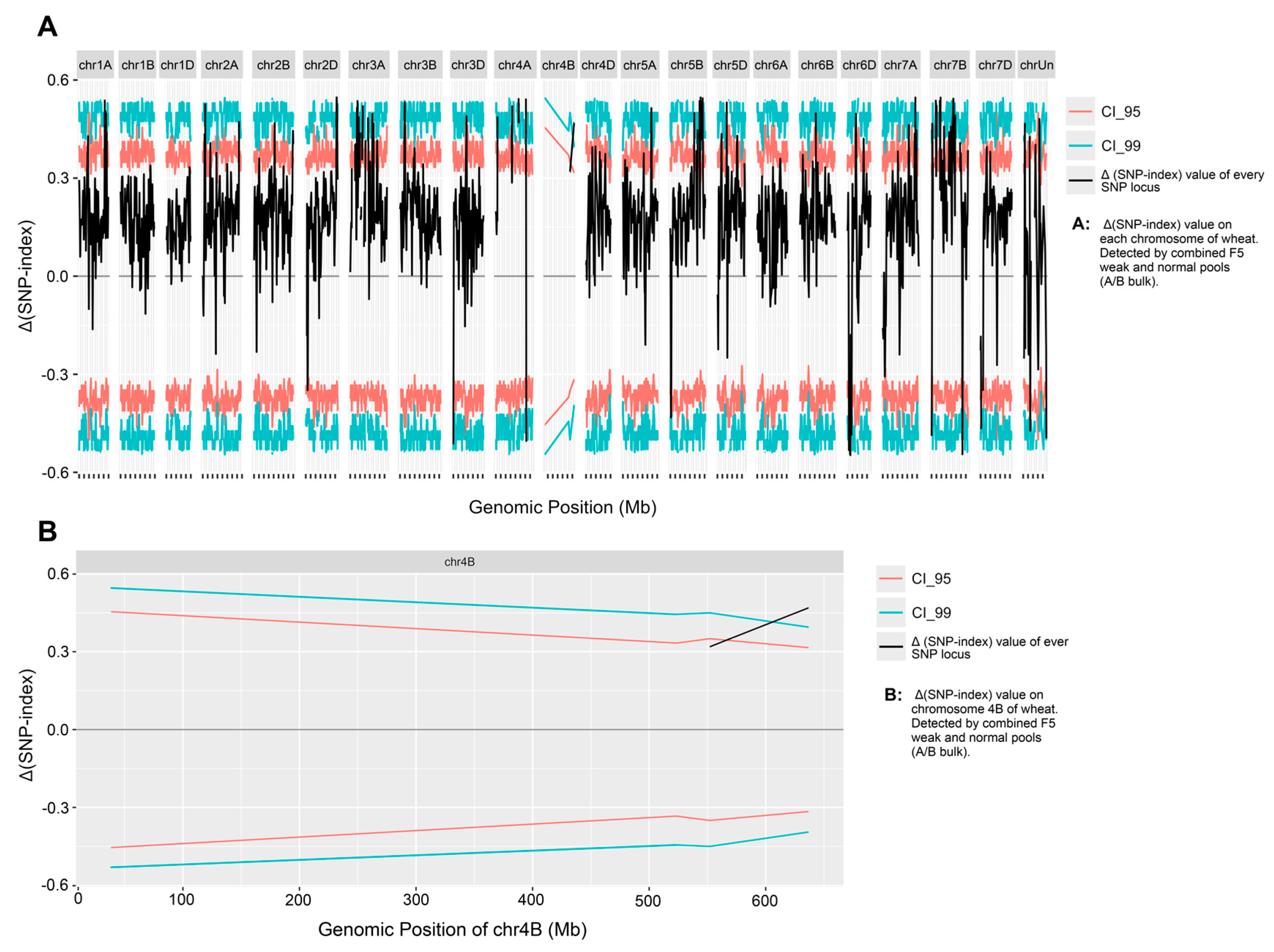
2.3. Identification of Chromosome Fragment Deletions Using SSR Maker Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Field Trials
4.2. Agronomic Trait Measurement
- At heading stage:
- At maturity:
4.3. RNA-seq Analysis
4.3.1. Sample Preparation and RNA Isolation
4.3.2. cDNA Library Preparation and Sequencing
4.3.3. Alignment of RNA-seq Reads and SNP Analysis
4.3.4. Identification and Annotation of Differentially Expressed Genes
4.4. SSR Marker Analysis
4.4.1. SSR Marker Development
4.4.2. Chromosome Structure Analysis
4.5. Statistical Analysis and Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brouns, F.; van Rooy, G.; Shewry, P.; Rustgi, S.; Jonkers, D. Adverse reactions to wheat or wheat components. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Zörb, C.; Ludewig, U.; Hawkesford, M.J. Perspective on wheat yield and quality with reduced nitrogen supply. Trends Plant Sci. 2018, 23, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Shewry, P.R.; Hey, S.J. The contribution of wheat to human diet and health. Food Energy Secur. 2015, 4, 178–202. [Google Scholar] [CrossRef]
- Consortium, I.W.G.S.; Mayer, K.F.; Rogers, J.; Doležel, J.; Pozniak, C.; Eversole, K.; Feuillet, C.; Gill, B.; Friebe, B.; Lukaszewski, A.J. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar]
- Dubcovsky, J.; Dvorak, J. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 2007, 316, 1862–1866. [Google Scholar] [CrossRef]
- Akhunov, E.D.; Akhunova, A.R.; Anderson, O.D.; Anderson, J.A.; Blake, N.; Clegg, M.T.; Coleman-Derr, D.; Conley, E.J.; Crossman, C.C.; Deal, K.R. Nucleotide diversity maps reveal variation in diversity among wheat genomes and chromosomes. BMC Genom. 2010, 11, 702. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Qiu, Y.; Lin, Z.; Wang, K.; Ye, X. Research progress on wheat improvement by using desirable genes from its relative species. Crops 2021, 37, 1–14. [Google Scholar]
- Ren, T.; Chen, F.; Yan, B.; Zhang, H.; Ren, Z. Genetic diversity of wheat–rye 1BL. 1RS translocation lines derived from different wheat and rye sources. Euphytica 2012, 183, 133–146. [Google Scholar] [CrossRef]
- Mago, R.; Miah, H.; Lawrence, G.; Wellings, C.; Spielmeyer, W.; Bariana, H.; McIntosh, R.; Pryor, A.; Ellis, J. High-resolution mapping and mutation analysis separate the rust resistance genes Sr31, Lr26 and Yr9 on the short arm of rye chromosome 1. Theor. Appl. Genet. 2005, 112, 41–50. [Google Scholar] [CrossRef]
- Ren, T.; Yang, Z.; Yan, B.; Zhang, H.; Fu, S.; Ren, Z. Development and characterization of a new 1BL. 1RS translocation line with resistance to stripe rust and powdery mildew of wheat. Euphytica 2009, 169, 207–213. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, S.; Liu, W.; Song, L.; Zhang, J.; Han, H.; Yang, X.; Lin, Y.; Li, X.; Li, L. Molecular cytogenetic analysis of the introgression between Agropyron cristatum P genome and wheat genome. Int. J. Mol. Sci. 2021, 22, 11208. [Google Scholar] [CrossRef]
- Han, H.; Liu, W.; Zhang, J.; Zhou, S.; Yang, X.; Li, X.; Li, L. Identification of P genome chromosomes in Agropyron cristatum and wheat-A. cristatum derivative lines by FISH. Sci. Rep. 2019, 9, 9712. [Google Scholar]
- Said, M.; Gaál, E.; Farkas, A.; Molnár, I.; Bartoš, J.; Doležel, J.; Cabrera, A.; Endo, T.R. Gametocidal genes: From a discovery to the application in wheat breeding. Front. Plant Sci. 2024, 15, 1396553. [Google Scholar]
- Endo, T. The gametocidal chromosome as a tool for chromosome manipulation in wheat. Chromosome Res. 2007, 15, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.R.; Gill, B.S. The Deletion Stocks of Common Wheat. J. Hered. 1996, 87, 295–307. [Google Scholar] [CrossRef]
- Han, H.; Ma, X.; Wang, Z.; Qi, K.; Yang, W.; Liu, W.; Zhang, J.; Zhou, S.; Lu, Y.; Yang, X. Chromosome 5P of Agropyron cristatum induces chromosomal translocation by disturbing homologous chromosome pairing in a common wheat background. Crop. J. 2023, 11, 228–237. [Google Scholar]
- Cui, Y.; Xing, P.; Qi, X.; Bao, Y.; Wang, H.; Wang, R.R.; Li, X. Characterization of chromosome constitution in three wheat-Thinopyrum intermedium amphiploids revealed frequent rearrangement of alien and wheat chromosomes. BMC Plant Biol. 2021, 21, 129. [Google Scholar]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar]
- Xie, W.; Nevo, E. Wild emmer: Genetic resources, gene mapping and potential for wheat improvement. Euphytica 2008, 164, 603–614. [Google Scholar] [CrossRef]
- Xie, W.; Ben-David, R.; Zeng, B.; Distelfeld, A.; Röder, M.S.; Dinoor, A.; Fahima, T. Identification and characterization of a novel powdery mildew resistance gene PmG3M derived from wild emmer wheat, Triticum dicoccoides. Theor. Appl. Genet. 2012, 124, 911–922. [Google Scholar]
- Chen, X.M.; Luo, Y.H.; Xia, X.C.; Xia, L.Q.; Chen, X.; Ren, Z.L.; He, Z.H.; Jia, J.Z. Chromosomal location of powdery mildew resistance gene Pm16 in wheat using SSR marker analysis. Plant Breed. 2008, 124, 225–228. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Ni, Z.; Nevo, E.; Yang, T. Molecular characterization of a novel powdery mildew resistance gene Pm30 in wheat originating from wild emmer. Euphytica 2002, 123, 21–29. [Google Scholar] [CrossRef]
- Geng, M.; Zhang, J.; Peng, F.; Liu, X.; Lv, X.; Mi, Y.; Li, Y.; Li, F.; Xie, C.; Sun, Q. Identification and mapping of MLIW30, a novel powdery mildew resistance gene derived from wild emmer wheat. Mol. Breed. 2016, 36, 130. [Google Scholar] [CrossRef]
- Peng, J.; Fahima, T.; Röder, M.; Huang, Q.; Dahan, A.; Li, Y.; Grama, A.; Nevo, E. High-density molecular map of chromosome region harboring stripe-rust resistance genes YrH52 and Yr15 derived from wild emmer wheat, Triticum dicoccoides. Genetica 2000, 109, 199–210. [Google Scholar] [CrossRef]
- Marais, G.; Pretorius, Z.; Wellings, C.; McCallum, B.; Marais, A. Leaf rust and stripe rust resistance genes transferred to common wheat from Triticum dicoccoides. Euphytica 2005, 143, 115–123. [Google Scholar] [CrossRef]
- Uauy, C.; Brevis, J.C.; Chen, X.; Khan, I.; Jackson, L.; Chicaiza, O.; Distelfeld, A.; Fahima, T.; Dubcovsky, J. High-temperature adult-plant (HTAP) stripe rust resistance gene Yr36 from Triticum turgidum ssp. dicoccoides is closely linked to the grain protein content locus Gpc-B1. Theor. Appl. Genet. 2005, 112, 97–105. [Google Scholar]
- Munns, R.; James, R.A.; Xu, B.; Athman, A.; Conn, S.J.; Jordans, C.; Byrt, C.S.; Hare, R.A.; Tyerman, S.D.; Tester, M. Wheat grain yield on saline soils is improved by an ancestral Na+ transporter gene. Nat. Biotechnol. 2012, 30, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, J.; Shi, H.; Zhang, Y.; You, Y.; Fan, J.; Chen, K.; Liu, S.; Nevo, E.; Fu, J. TdCBL6, a calcineurin B-like gene from wild emmer wheat (Triticum dicoccoides), is involved in response to salt and low-K+ stresses. Mol. Breed. 2015, 35, 50. [Google Scholar] [CrossRef]
- Kuzuoglu-Ozturk, D.; Cebeci Yalcinkaya, O.; Akpinar, B.A.; Mitou, G.; Korkmaz, G.; Gozuacik, D.; Budak, H. Autophagy-related gene, TdAtg8, in wild emmer wheat plays a role in drought and osmotic stress response. Planta 2012, 236, 1081–1092. [Google Scholar] [CrossRef]
- Lucas, S.; Dogan, E.; Budak, H. TMPIT1 from wild emmer wheat: First characterisation of a stress-inducible integral membrane protein. Gene 2011, 483, 22–28. [Google Scholar] [CrossRef]
- Distelfeld, A.; Uauy, C.; Fahima, T.; Dubcovsky, J. Physical map of the wheat high-grain protein content gene Gpc-B1 and development of a high-throughput molecular marker. New Phytol. 2006, 169, 753–763. [Google Scholar] [PubMed]
- Ma, S.W.; Wang, M.; Wu, J.H.; Guo, W.L.; Chen, Y.M.; Li, G.W.; Wang, Y.P.; Shi, W.M.; Xia, G.M.; Fu, D.L.; et al. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant 2021, 14, 1965–1968. [Google Scholar] [PubMed]
- Wang, Z.; Li, J.; Chen, S.X.; Heng, Y.F.; Chen, Z.; Yang, J.; Zhou, K.J.; Pei, J.W.; He, H.; Deng, X.W.; et al. Poaceae-specific MS1 encodes a phospholipid-binding protein for male fertility in bread wheat. Proc. Natl. Acad. Sci. USA 2017, 114, 12614–12619. [Google Scholar]
- Li, B.; Liu, D.; Li, Q.R.; Mao, X.G.; Li, A.; Wang, J.Y.; Chang, X.P.; Jing, R.L. Overexpression of wheat gene TaMOR improves root system architecture and grain yield in Oryza sativa. J. Exp. Bot. 2016, 67, 4155–4167. [Google Scholar]
- Song, R.R.; Cheng, Y.F.; Wen, M.X.; Song, X.Y.; Wang, T.; Xia, M.S.; Sun, H.J.; Cheng, M.H.; Cui, H.M.; Yuan, C.X.; et al. Transferring a new Fusarium head blight resistance locus FhbRc1 from Roegneria ciliaris into wheat by developing alien translocation lines. Theor. Appl. Genet. 2023, 136, 36. [Google Scholar]
- Han, H.M.; Yang, J.L.; Qi, K.; Zhu, H.Y.; Wu, P.Q.; Zhou, S.H.; Zhang, J.P.; Guo, B.J.; Liu, W.H.; Guo, X.M.; et al. Introgression of chromosome 5P from Agropyron cristatum enhances grain weight in a wheat background. Theor. Appl. Genet. 2024, 137, 165. [Google Scholar] [CrossRef]
- Li, G.R.; Chen, Q.H.; Jiang, W.X.; Zhang, A.H.; Yang, E.N.; Yang, Z.J. Molecular and Cytogenetic Identification of Wheat-Thinopyrum intermedium Double Substitution Line-Derived Progenies for Stripe Rust Resistance. Plants 2023, 12, 28. [Google Scholar]
- Wang, H.J.; Yu, Z.H.; Li, G.R.; Yang, Z.J. Diversified Chromosome Rearrangements Detected in a WheatDasypyrum breviaristatum Substitution Line Induced by Gamma-Ray Irradiation. Plants 2019, 8, 175. [Google Scholar] [CrossRef]
- Lu, M.J.; Lu, Y.Q.; Li, H.H.; Pan, C.L.; Guo, Y.; Zhang, J.P.; Yang, X.M.; Li, X.Q.; Liu, W.H.; Li, L.H. Transferring Desirable Genes from Agropyron cristatum 7P Chromosome into Common Wheat. PLoS ONE 2016, 11, e0159577. [Google Scholar]
- Copete-Parada, A.; Palomino, C.; Cabrera, A. Development and Characterization of Wheat-Agropyron cristatum Introgression Lines Induced by Gametocidal Genes and Wheat ph1b Mutant. Agronomy 2021, 11, 277. [Google Scholar] [CrossRef]
- Li, H.J.; Zhou, Y.; Xin, W.L.; Wei, Y.Q.; Zhang, J.L.; Guo, L.L. Wheat breeding in northern China: Achievements and technical advances. Crop J. 2019, 7, 718–729. [Google Scholar]
- Bhullar, R.; Nagarajan, R.; Bennypaul, H.; Sidhu, G.K.; Sidhu, G.; Rustgi, S.; von Wettstein, D.; Gill, K.S. Silencing of a metaphase I-specific gene results in a phenotype similar to that of the Pairing homeologous 1 (Ph1) gene mutations. Proc. Natl. Acad. Sci. USA 2014, 111, 14187–14192. [Google Scholar]
- Rey, M.D.; Calderón, M.C.; Prieto, P. The use of the ph1b mutant to induce recombination between the chromosomes of wheat and barley. Front. Plant Sci. 2015, 6, 160. [Google Scholar]
- Wan, W.T.; Xiao, J.; Li, M.L.; Tang, X.; Wen, M.X.; Cheruiyot, A.K.; Li, Y.B.; Wang, H.Y.; Wang, X. Fine mapping of wheat powdery mildew resistance gene Pm6 using 2B/2G homoeologous recombinants induced by the ph1b mutant. Theor. Appl. Genet. 2020, 133, 1265–1275. [Google Scholar] [PubMed]
- Wan, W.; Zhao, R.; Chen, T.; Wang, L.; Zhang, X.; Li, H.; Wang, X.; Bie, T. Rapid development of wheat-Dasypyrum villosum compensating translocations resistant to powdery mildew using a triple marker strategy conducted on a large ph1b-induced population. Theor. Appl. Genet. 2023, 136, 148. [Google Scholar]
- Jiang, Y.; He, M.; Ding, M.; Rong, J. Chromosome elimination of hexaploid common wheat mediated by interaction between Chinese Spring cytoplasm and a genetic factor (s) on chromosome arm 1BL of wild emmer. Euphytica 2016, 209, 615–625. [Google Scholar] [CrossRef]
- Yang, G.; Pan, Y.; Pan, W.; Song, Q.; Zhang, R.; Tong, W.; Cui, L.; Ji, W.; Song, W.; Song, B. Combined GWAS and eGWAS reveals the genetic basis underlying drought tolerance in emmer wheat (Triticum turgidum L.). New Phytol. 2024, 242, 2115–2131. [Google Scholar]
- Nevo, E. Evolution of wild emmer wheat and crop improvement. J. Syst. Evol. 2014, 52, 673–696. [Google Scholar]
- Miraghazadeh, A.; Zhang, P.; Harding, C.; Hossain, S.; Hayden, M.; Wong, D.; Spielmeyer, W.; Chandler, P.M. The use of SNP hybridisation arrays and cytogenetics to characterise deletions of chromosome 4B in hexaploid wheat (Triticum aestivum L.). Theor. Appl. Genet. 2016, 129, 2151–2160. [Google Scholar]
- Wu, H.D.; Yu, H.P.; Zhang, Y.Y.; Yang, B.B.; Sun, W.Q.; Ren, L.Y.; Li, Y.C.; Li, Q.Q.; Liu, B.; Ding, Y.L.; et al. Unveiling RNA structure-mediated regulations of RNA stability in wheat. Nat. Commun. 2024, 15, 10042. [Google Scholar]
- Chao, H.Y.; Hu, Y.M.; Zhao, L.; Xin, S.G.; Ni, Q.Y.; Zhang, P.J.; Chen, M. Biogenesis, Functions, Interactions, and Resources of Non-Coding RNAs in Plants. Int. J. Mol. Sci. 2022, 23, 3695. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.S.; Cheng, C.; Kong, J.; Li, H.J.; Hua, J.P. Plant non-coding RNAs function in pollen development and male sterility. Front. Plant Sci. 2023, 14, 1109941. [Google Scholar] [CrossRef]
- Song, L.; Lu, Y.; Zhang, J.; Pan, C.; Yang, X.; Li, X.; Liu, W.; Li, L. Physical mapping of Agropyron cristatum chromosome 6P using deletion lines in common wheat background. Theor. Appl. Genet. 2016, 129, 1023–1034. [Google Scholar] [CrossRef]
- Huber, D.; Von Voithenberg, L.V.; Kaigala, G.V. Fluorescence in situ hybridization (FISH): History, limitations and what to expect from micro-scale FISH? Micro Nano Eng. 2018, 1, 15–24. [Google Scholar] [CrossRef]
- Coombes, B.; Fellers, J.P.; Grewal, S.; Rusholme-Pilcher, R.; Hubbart-Edwards, S.; Yang, C.Y.; Joynson, R.; King, I.P.; King, J.; Hall, A. Whole-genome sequencing uncovers the structural and transcriptomic landscape of hexaploid wheat/Ambylopyrum muticum introgression lines. Plant Biotechnol. J. 2023, 21, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Jiang, C.; Zhao, Y.; Yang, S.; Li, Y.; Yan, K.; Wu, S.; Luo, B.; Du, Y.; Jin, H. Wheat heat tolerance is impaired by heightened deletions in the distal end of 4AL chromosomal arm. Plant Biotechnol. J. 2021, 19, 1038–1051. [Google Scholar] [CrossRef]
- Lu, F.; Chen, M.X.; Zhao, Y.H.; Wu, S.X.; Yasir, M.; Zhang, H.; Hu, X.; Rong, J.K. Genetic Mapping and Candidate Gene Prediction of a QTL Related to Early Heading on Wild Emmer Chromosome 7BS in the Genetic Background of Common Wheat. Agronomy 2022, 12, 1089. [Google Scholar] [CrossRef]
- Feldman, M.; Millet, E.; Abbo, S. Exploitation of wild emmer wheat to increase yield and protein content in durum and common wheat. In Proceedings of the Genetic Resources Section Meeting of Eucarpia, Clermont-Ferrand, France, 15–18 March 1994. [Google Scholar]
- Chen, T.T.; Chen, X.; Zhang, S.S.; Zhu, J.W.; Tang, B.X.; Wang, A.K.; Dong, L.L.; Zhang, Z.W.; Yu, C.X.; Sun, Y.L.; et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef]
- Xue, Y.B.; Bao, Y.M.; Zhang, Z.; Zhao, W.M.; Xiao, J.F.; He, S.M.; Zhang, G.Q.; Li, Y.X.; Zhao, G.P.; Chen, R.S.; et al. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [PubMed]
- Mansfeld, B.N.; Grumet, R. QTLseqr: An R Package for Bulk Segregant Analysis with Next-Generation Sequencing. Plant Genome 2018, 11, 180006. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome Range (Mb) | CS RefSeqv1.0 | Normal Plant Samples | Weak Plant Samples |
|---|---|---|---|
| 0–203 | 1444 | 998 | 319 |
| 203–674 | 2425 | 1599 | 1382 |
| 0–674 | 3869 | 2597 | 1701 |
| Plants Number | PH | FLL | FLW | SW | SL | SLN | GPS |
|---|---|---|---|---|---|---|---|
| 491-07-05 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 491-07-05-01 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 491-07-05-02 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 491-07-05-03 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 491-07-05-04 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 491-07-05-05 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 491-07-05-06 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 491-07-05-07 | ↓ | ns. | ns. | ↓ | ns. | ↓ | ↓ |
| 491-07-05-08 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 494-05-03 | ↓ | ns. | ns. | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-01 | ↓ | ↓ | ns. | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-02 | ↓ | ↓ | ns. | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-04 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 494-05-03-07 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 494-05-03-11 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-12 | ns. | ns. | ns. | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-13 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-14 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-18 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-24 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 494-05-03-25 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 494-05-03-27 | ns. | ns. | ns. | ns. | ns. | ns. | ns. |
| 494-05-03-28 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.; Lu, F.; Yang, B.; Hu, X.; Zhao, Y.; Ding, M.; Yang, L.; Rong, J. A Bread Wheat Line with the Substituted Wild Emmer Chromosome 4A Results in Fragment Deletions of Chromosome 4B and Weak Plants. Plants 2025, 14, 1134. https://doi.org/10.3390/plants14071134
Qiu Y, Lu F, Yang B, Hu X, Zhao Y, Ding M, Yang L, Rong J. A Bread Wheat Line with the Substituted Wild Emmer Chromosome 4A Results in Fragment Deletions of Chromosome 4B and Weak Plants. Plants. 2025; 14(7):1134. https://doi.org/10.3390/plants14071134
Chicago/Turabian StyleQiu, Yu, Fei Lu, Bohao Yang, Xin Hu, Yanhao Zhao, Mingquan Ding, Lei Yang, and Junkang Rong. 2025. "A Bread Wheat Line with the Substituted Wild Emmer Chromosome 4A Results in Fragment Deletions of Chromosome 4B and Weak Plants" Plants 14, no. 7: 1134. https://doi.org/10.3390/plants14071134
APA StyleQiu, Y., Lu, F., Yang, B., Hu, X., Zhao, Y., Ding, M., Yang, L., & Rong, J. (2025). A Bread Wheat Line with the Substituted Wild Emmer Chromosome 4A Results in Fragment Deletions of Chromosome 4B and Weak Plants. Plants, 14(7), 1134. https://doi.org/10.3390/plants14071134