
Characterization and Expression Analysis of the ALOG Gene Family in Rice (Oryza sativa L.)
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Plant Materials and RNA Sample Collection
2.2. Identification and Phylogenetic Analysis of the ALOG Genes in Rice
2.3. Analysis of the Characteristics of the ALOG Family
2.4. Synteny Analysis
2.5. Analysis of the Cis-Regulatory Elements
2.6. RNA Extraction, Library Construction and Sequencing
2.7. Quantitative Reverse Transcription-PCR Analysis
3. Results
3.1. Genome-Wide Identification and Phylogenetic Analysis of the ALOG Genes in Rice
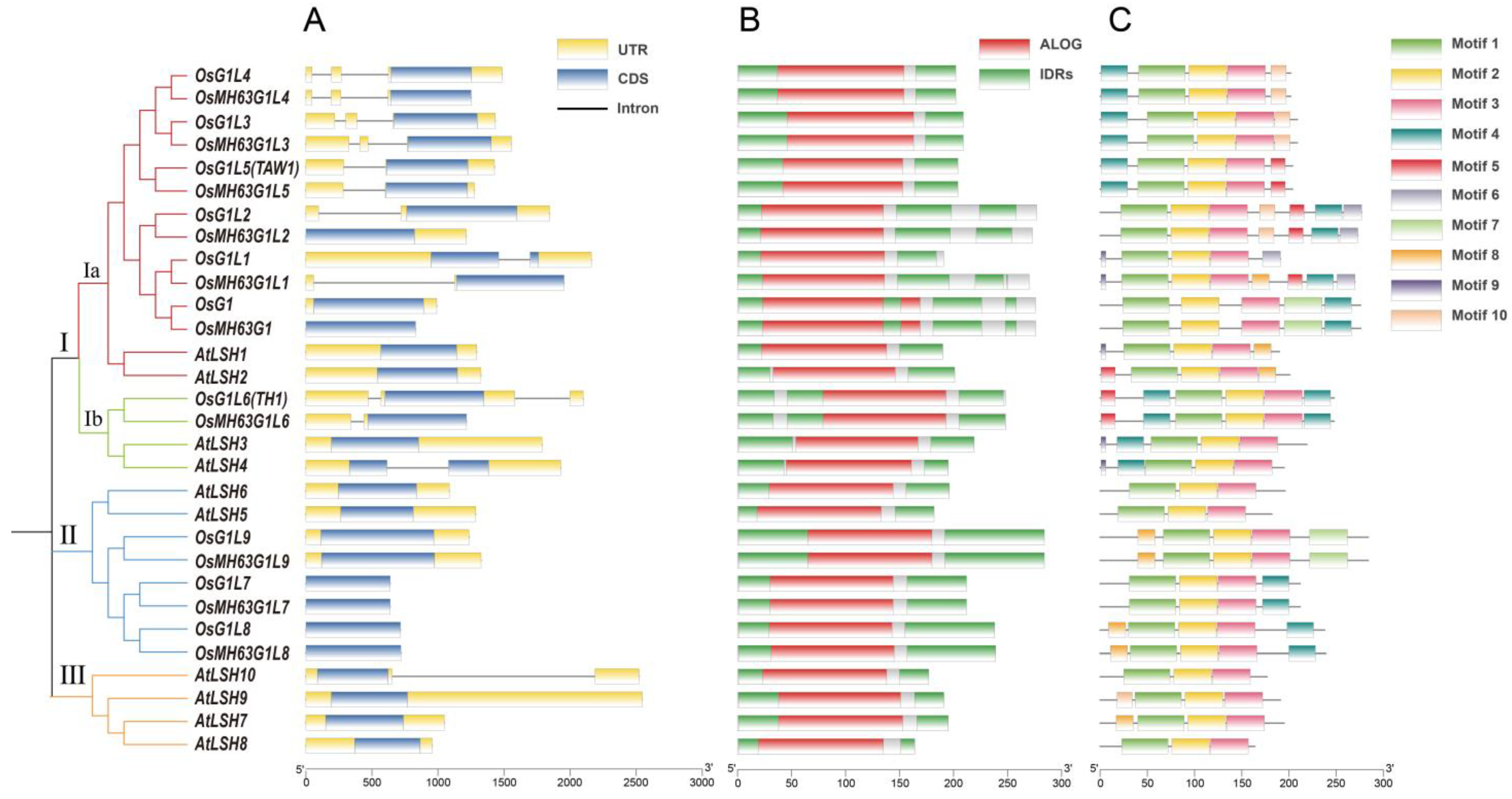
3.2. Gene Structures, Protein Domains, and Motif Compositions of the Rice ALOG Gene Family
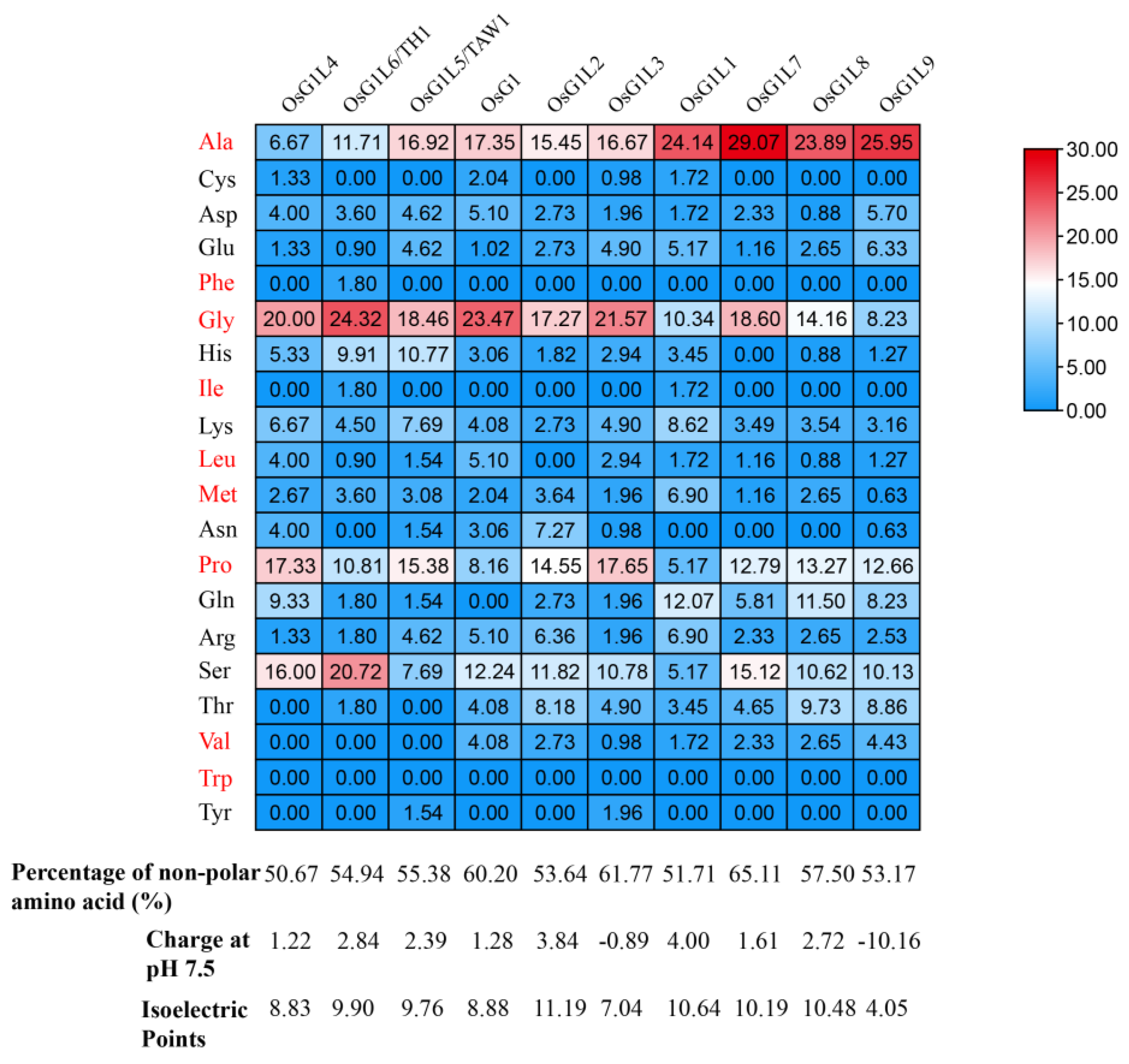
3.3. Enrichment Analysis of Residues in IDRs
3.4. Synteny Analysis of OsALOG Genes
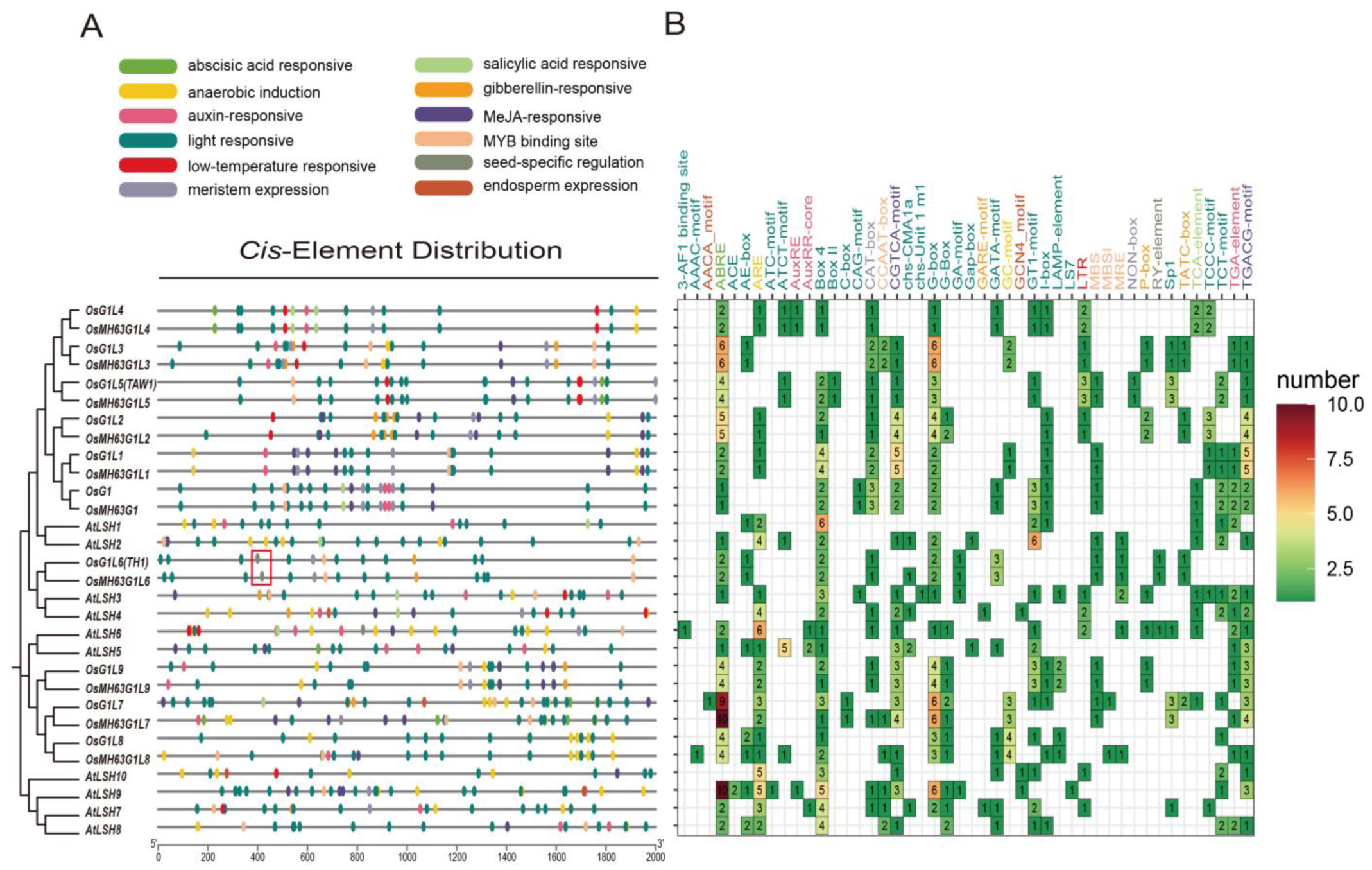
3.5. Cis-Regulatory Elements of OsALOG Genes Analysis
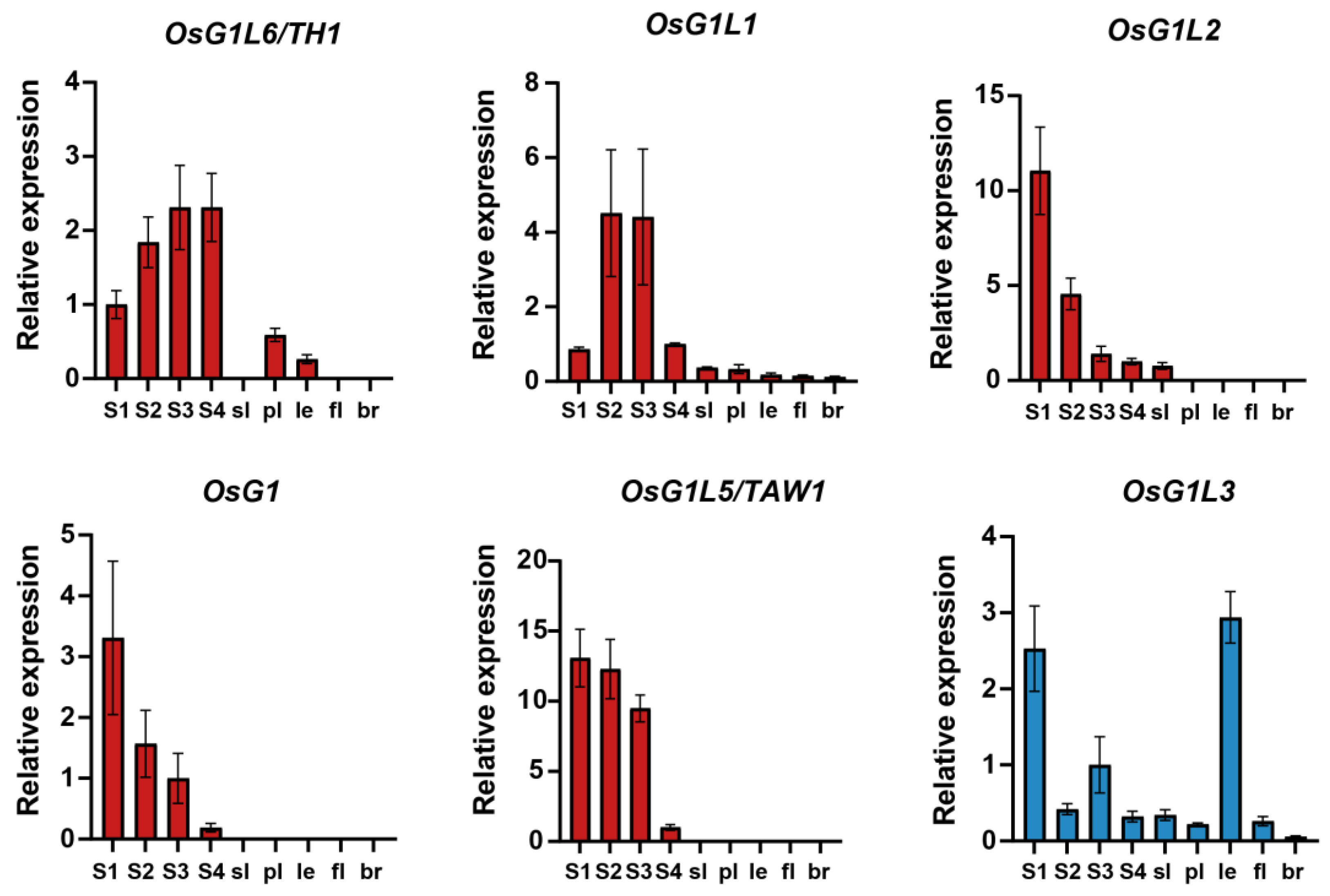
3.6. Expression Pattern of OsALOG Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yoshida, A.; Suzaki, T.; Tanaka, W.; Hirano, H.Y. The homeotic gene long sterile lemma (G1) specifies sterile lemma identity in the rice spikelet. Proc. Natl. Acad. Sci. USA 2009, 106, 20103–20108. [Google Scholar] [CrossRef] [PubMed]
- Rieu, P.; Beretta, V.M.; Caselli, F.; Thévénon, E.; Lucas, J.; Rizk, M.; Gregis, V. The ALOG domain defines a family of plant-specific transcription factors acting during Arabidopsis flower development. Proc. Natl. Acad. Sci. USA 2024, 121, e2310464121. [Google Scholar] [CrossRef]
- Iyer, L.M.; Aravind, L. ALOG domains, provenance of plant homeotic and developmental regulators from the DNA-binding domain of a novel class of DIRS1-type retroposons. Biol. Direct. 2012, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Naramoto, S.; Jones, V.A.S.; Trozzi, N.; Sato, M.; Toyooka, K.; Shimamura, M.; Kyozuka, J. A conserved regulatory mechanism mediates the convergent evolution of plant shoot lateral organs. PLoS Biol. 2019, 17, e3000560. [Google Scholar] [CrossRef]
- Hong, L.; Qian, Q.; Zhu, K.; Tang, D.; Huang, Z.; Gao, L.; Li, M.; Gu, M.; Cheng, Z. ELE restrains empty glumes from developing into lemmas. J. Genet. Genomics 2010, 37, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, H.; Su, Y.; Li, W.; Shi, C. G1/ELE functions in the development of rice lemmas in addition to determining identities of empty glumes. Front. Plant Sci. 2016, 7, 1006. [Google Scholar] [CrossRef]
- Luo, X.; Wei, Y.; Zheng, Y.; Wei, L.; Wu, F.; Cai, Q.; Xie, H.; Zhang, J. Analysis of co-expression and gene regulatory networks associated with sterile lemma development in rice. BMC Plant Biol. 2023, 23, 11. [Google Scholar] [CrossRef]
- Li, X.; Sun, L.; Tan, L.; Liu, F.; Zhu, Z.; Fu, Y.; Sun, X.; Sun, X.; Xie, D.; Sun, C. TH1, a DUF640 domain-like gene controls lemma and palea development in rice. Plant Mol. Biol. 2012, 78, 351–359. [Google Scholar] [CrossRef]
- Ma, X.; Cheng, Z.; Wu, F.; Jin, M.; Zhang, L.; Zhou, F.; Wan, J. BEAK LIKE SPIKELET1 is required for lateral development of lemma and palea in rice. Plant Mol. Biol. Rep. 2013, 31, 98–108. [Google Scholar] [CrossRef]
- Yan, D.; Zhou, Y.; Ye, S.; Zeng, L.; Zhang, X.; He, Z. BEAK-SHAPED GRAIN 1/TRIANGULAR HULL 1, a DUF640 gene, is associated with grain shape, size and weight in rice. Sci. China Life Sci. 2013, 56, 275–283. [Google Scholar] [CrossRef]
- Sato, D.S.; Ohmori, Y.; Nagashima, H.; Toriba, T.; Hirano, H.Y. A role for TRIANGULAR HULL1 in fine-tuning spikelet morphogenesis in rice. Genes. Genet. Syst. 2014, 89, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Rao, Y.; Wu, L.; Xu, Q.; Li, Z.; Yu, H.; Zhang, Y.; Leng, Y.; Hu, J.; Zhu, L.; et al. The pleiotropic ABNORMAL FLOWER AND DWARF1 affects plant height, floral development and grain yield in rice. J. Integr. Plant Biol. 2016, 58, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Liu, L.; Fang, J.; Zhao, J.; Yuan, S.; Li, X. The rice TRIANGULAR HULL1 protein acts as a transcriptional repressor in regulating lateral development of spikelet. Sci. Rep. 2017, 7, 13712. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Wang, Y.; Huang, J.; Luo, N.; Wei, S.; Jin, J. Analysing the rice young panicle transcriptome reveals the gene regulatory network controlled by TRIANGULAR HULL1. Rice 2019, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Sasao, M.; Yasuno, N.; Takagi, K.; Daimon, Y.; Chen, R.; Kyozuka, J. TAWAWA1, a regulator of rice inflorescence architecture, functions through the suppression of meristem phase transition. Proc. Natl. Acad. Sci. USA 2013, 110, 767–772. [Google Scholar] [CrossRef]
- Beretta, V.M.; Franchini, E.; Din, I.U.; Lacchini, E.; Broeck, L.V.; Sozzani, R.; Orozco-Arroyo, G.; Caporali, E.; Adam, H.; Jouannic, S.; et al. The ALOG family members OsG1L1 and OsG1L2 regulate inflorescence branching in rice. Plant J. 2023, 115, 351–368. [Google Scholar] [CrossRef]
- Zhao, L.; Nakazawa, M.; Takase, T.; Manabe, K.; Kobayashi, M.; Seki, M.; Matsui, M. Overexpression of LSH1, a member of an uncharacterised gene family, causes enhanced light regulation of seedling development. Plant J. 2004, 37, 694–706. [Google Scholar] [CrossRef]
- Lee, M.; Dong, X.; Song, H.; Yang, J.Y.; Kim, S.; Hur, Y. Molecular characterization of Arabidopsis thaliana LSH1 and LSH2 genes. Genes Genomics 2020, 42, 1151–1162. [Google Scholar] [CrossRef]
- Takeda, S.; Hanano, K.; Kariya, A.; Shimizu, S.; Zhao, L.; Matsui, M.; Tasaka, M.; Aida, M. CUP-SHAPED COTYLEDON1 transcription factor activates the expression of LSH4 and LSH3, two members of the ALOG gene family, in shoot organ boundary cells. Plant J. 2011, 66, 1066–1077. [Google Scholar] [CrossRef]
- Zou, J.; Li, Z.; Tang, H.; Zhang, L.; Li, J.; Li, Y.; Yao, N.; Li, Y.; Yang, D.; Zuo, Z. Arabidopsis LSH8 positively regulates ABA signaling by changing the expression pattern of ABA-responsive proteins. Int. J. Mol. Sci. 2021, 22, 10314. [Google Scholar] [CrossRef]
- Vo Phan, M.S.; Keren, I.; Tran, P.T.; Lapidot, M.; Citovsky, V. Arabidopsis LSH10 transcription factor and OTLD1 histone deubiquitinase interact and transcriptionally regulate the same target genes. Commun. Biol. 2023, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Xiao, N.; Zou, Y.; Xie, Y.; Tang, L.; Zhang, Y.; Yu, Y.; Li, Y.; Xu, C. Heterotypic transcriptional condensates formed by prion-like paralogous proteins canalize flowering transition in tomato. Genome Biol. 2022, 23, 78. [Google Scholar] [CrossRef]
- Takanashi, H.; Kajiya-Kanegae, H.; Nishimura, A.; Yamada, J.; Ishimori, M.; Kobayashi, M.; Sakamoto, W. DOMINANT AWN INHIBITOR encodes the ALOG protein originating from gene duplication and inhibits awn elongation by suppressing cell proliferation and elongation in sorghum. Plant Cell Physiol. 2022, 63, 901–918. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Kovacs, D.; Tompa, P. Diverse functional manifestations of intrinsic structural disorder in molecular chaperones. Biochem. Soc. Trans. 2012, 40, 963–968. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Skeymkowitz, J.; Wolozin, B.; Bosch, L.V.D.; Tompa, P.; et al. Protein phase separation, a new phase in cell biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Sunohara, H.; Nagato, Y. Developmental course of inflorescence and spikelet in rice. Breed. Sci. 2004, 54, 147–156. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Bateman, A. Pfam, The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search, HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit, a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4, molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II, A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3, precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy, the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model, application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Paterson, A.H. MCScanX, a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0, a toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannet, N.M.; et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 2018, 175, 1842–1855. [Google Scholar] [CrossRef]
- Michelitsch, M.D.; Weissman, J.S. A census of glutamine/asparagine-rich regions, implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 2000, 97, 11910–11915. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Alberti, S. Prion-like low-complexity sequences, Key regulators of protein solubility and phase behavior. J. Biol. Chem. 2019, 294, 7128–7136. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sampling Point | Length of Inflorescence | Apical Spikelet Development Stage |
|---|---|---|---|
| S1 | Young panicle | 1~2 mm | Formation of palea, elongation of Sterile lemma and lemma. |
| S2 | Young panicle | 2~5 mm | Formation of stamen and pistil primordia. |
| S3 | Young panicle | 5~10 mm | Stamen and pistil begin to differentiate, Palea and lemma close up gradually. |
| S4 | Young panicle | 10~15 mm | Formation of pollen and ovule. |
| sl | Sterile lemma | - | Booting stage |
| pl | Palea | - | Booting stage |
| le | Lemma | - | Booting stage |
| fl | Inner floral organs | - | Booting stage |
| br | Branch | - | Booting stage |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, X.; Wang, H.; Wei, Y.; Wu, F.; Zhu, Y.; Xie, H.; Xie, H.; Zhang, J. Characterization and Expression Analysis of the ALOG Gene Family in Rice (Oryza sativa L.). Plants 2025, 14, 1208. https://doi.org/10.3390/plants14081208
Luo X, Wang H, Wei Y, Wu F, Zhu Y, Xie H, Xie H, Zhang J. Characterization and Expression Analysis of the ALOG Gene Family in Rice (Oryza sativa L.). Plants. 2025; 14(8):1208. https://doi.org/10.3390/plants14081208
Chicago/Turabian StyleLuo, Xi, Hongfei Wang, Yidong Wei, Fangxi Wu, Yongsheng Zhu, Hongguang Xie, Huaan Xie, and Jianfu Zhang. 2025. "Characterization and Expression Analysis of the ALOG Gene Family in Rice (Oryza sativa L.)" Plants 14, no. 8: 1208. https://doi.org/10.3390/plants14081208
APA StyleLuo, X., Wang, H., Wei, Y., Wu, F., Zhu, Y., Xie, H., Xie, H., & Zhang, J. (2025). Characterization and Expression Analysis of the ALOG Gene Family in Rice (Oryza sativa L.). Plants, 14(8), 1208. https://doi.org/10.3390/plants14081208