
Comparative Transcriptomic Analyses Reveal Key Pathways in Response to Cold Stress at the Germination Stage of Quinoa (Chenopodium quinoa Willd.) Seeds

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
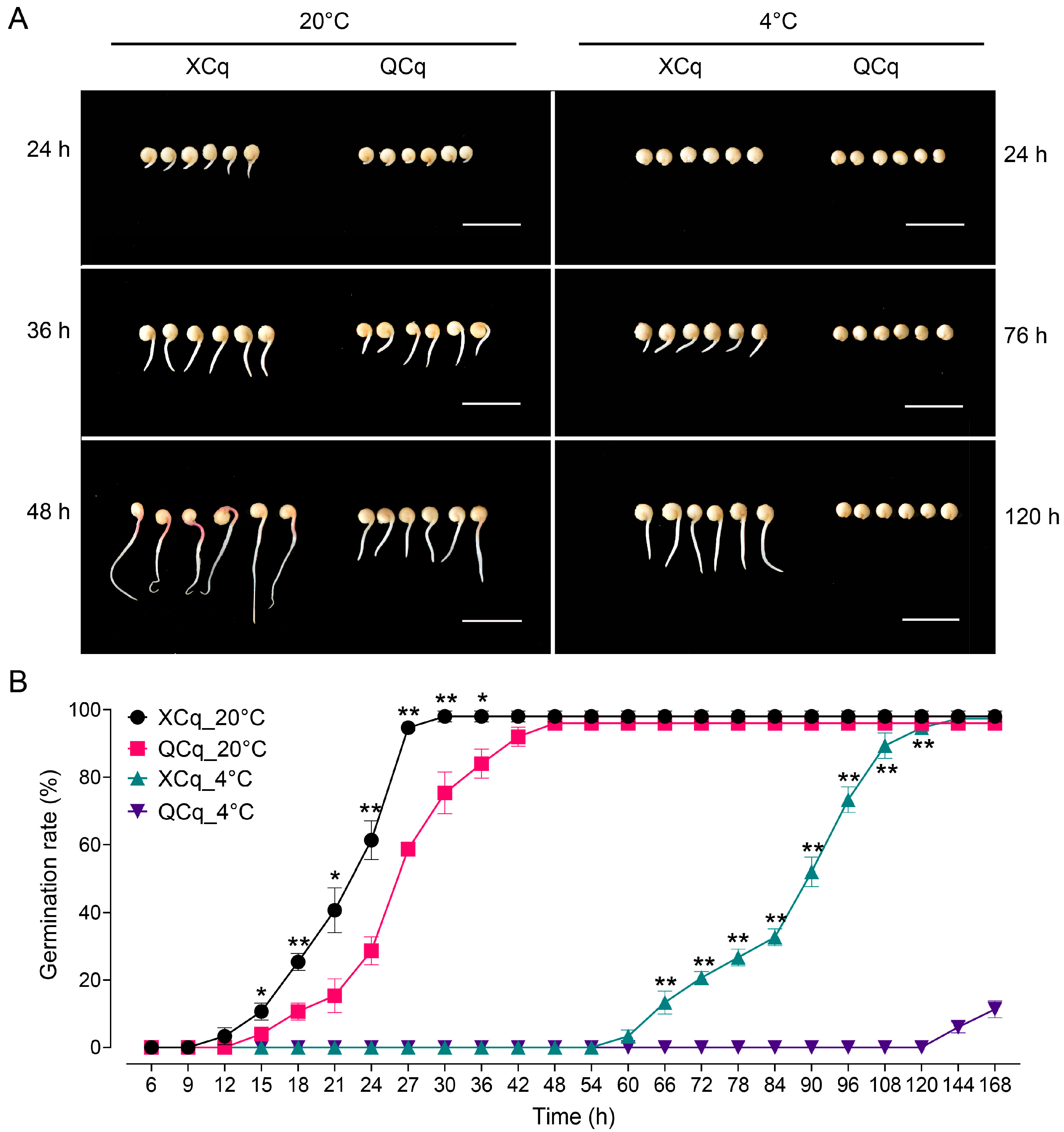
2.1. Different Germination Rates Are Observed in XCq and QCq
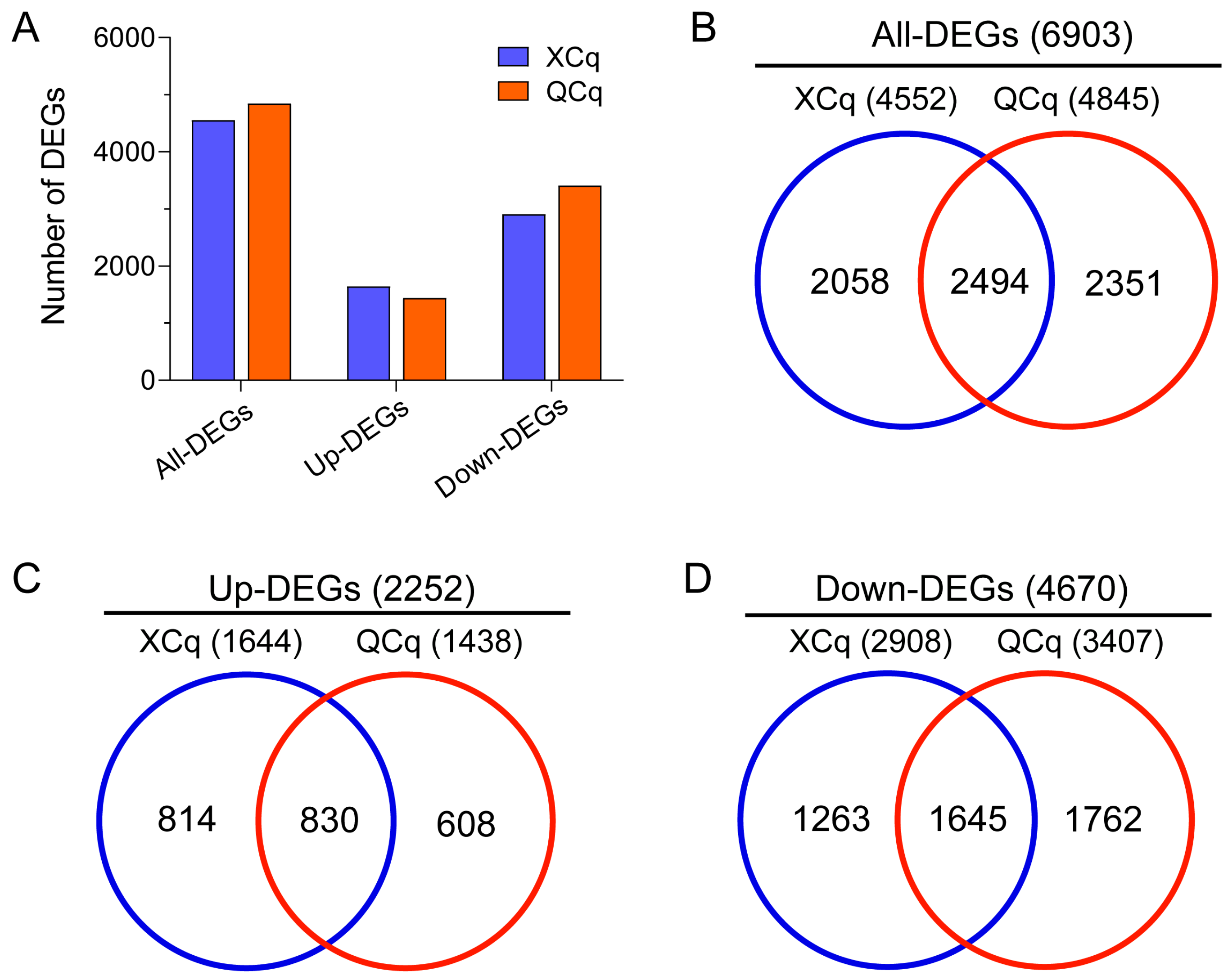
2.2. A Great Number of Differentially Expressed Genes Are Identified in XCq and QCq
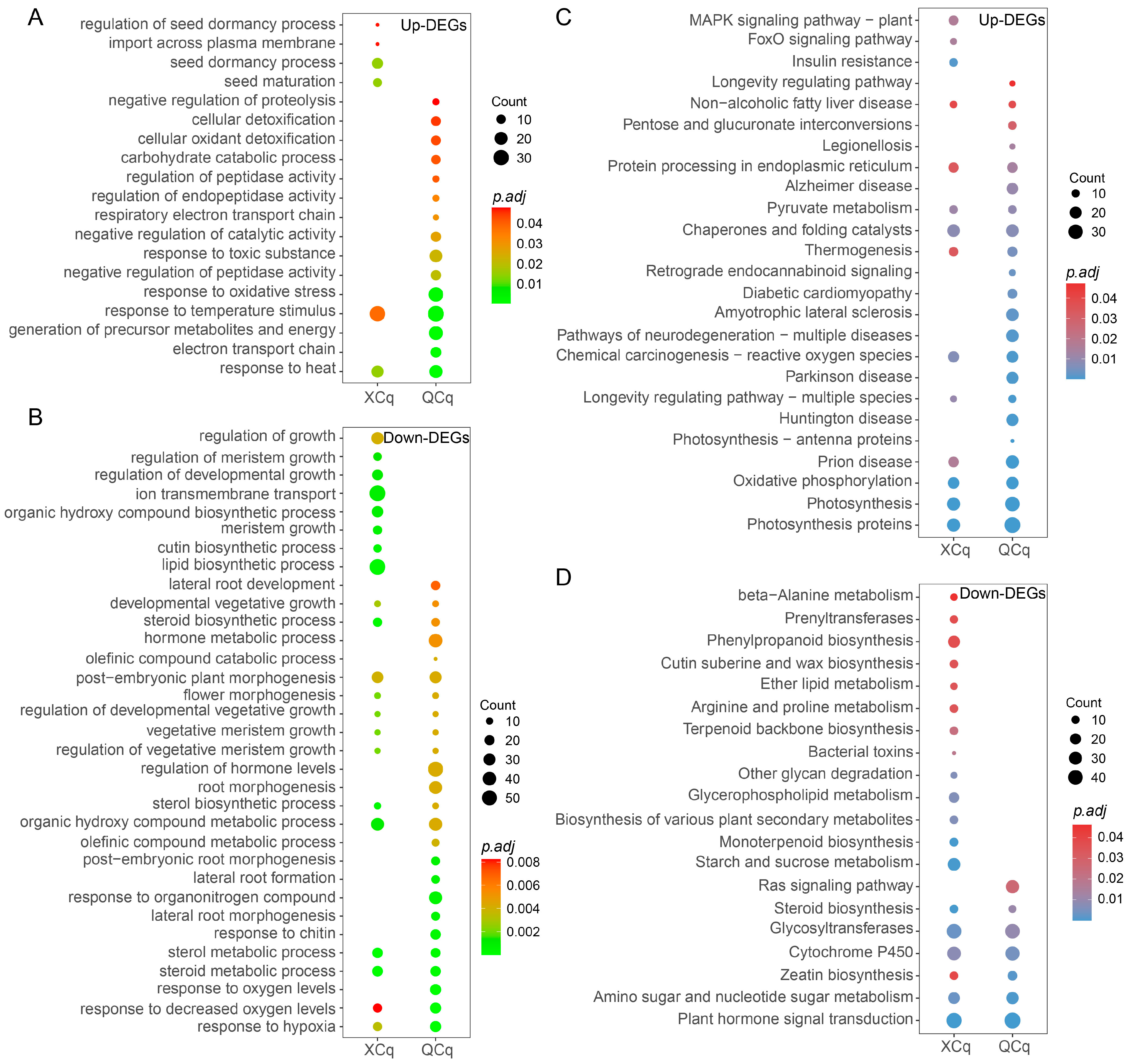
2.3. DEGs in XCq and QCq Are Enriched in Different Biological Processes and Pathways
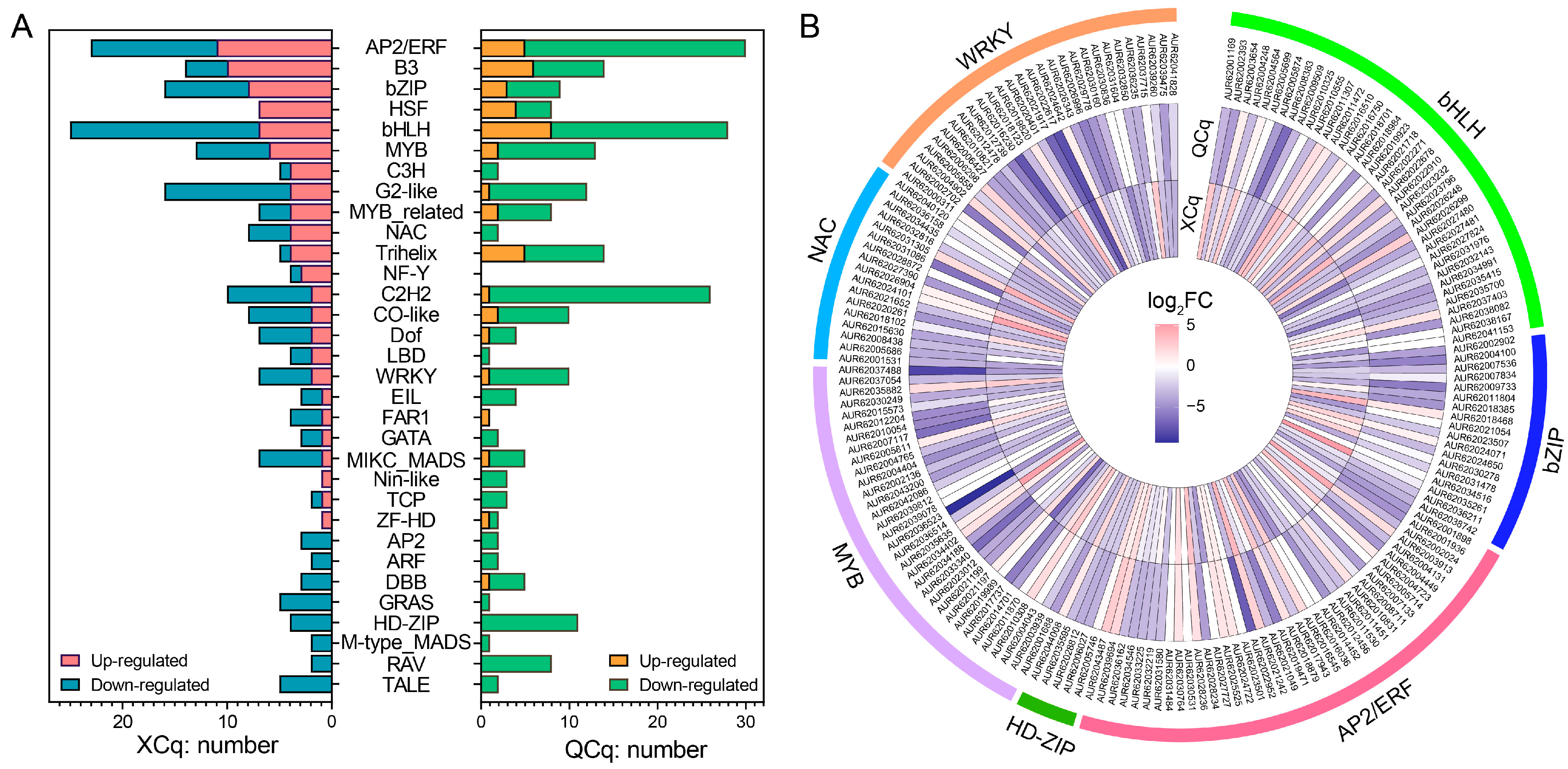
2.4. Transcription Factor Encoding Genes Are Differentially Expressed in XCq and QCq
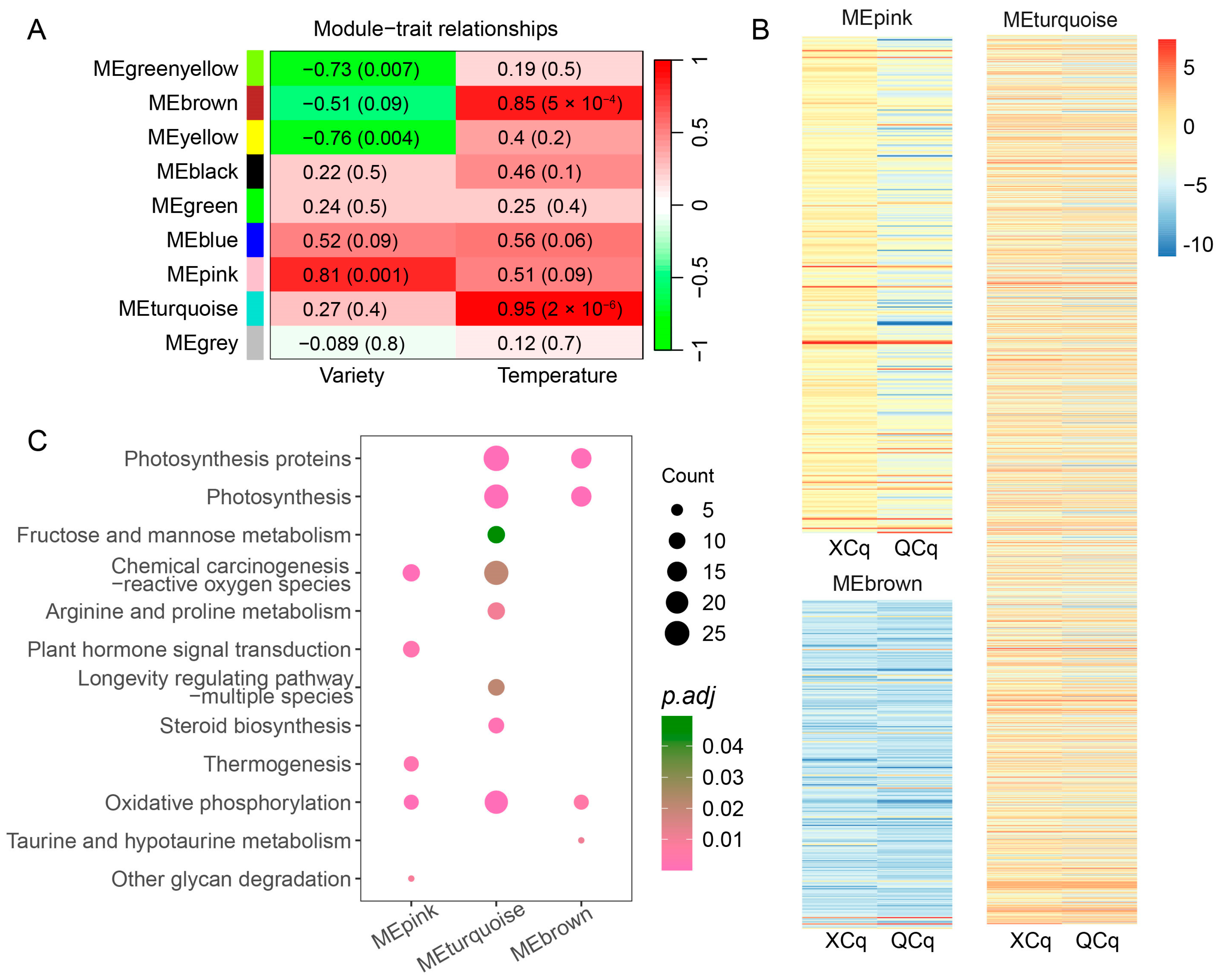
2.5. Expression of DEGs Related to Cold Stress Are Closely Correlated with the Quinoa Variety and Temperature
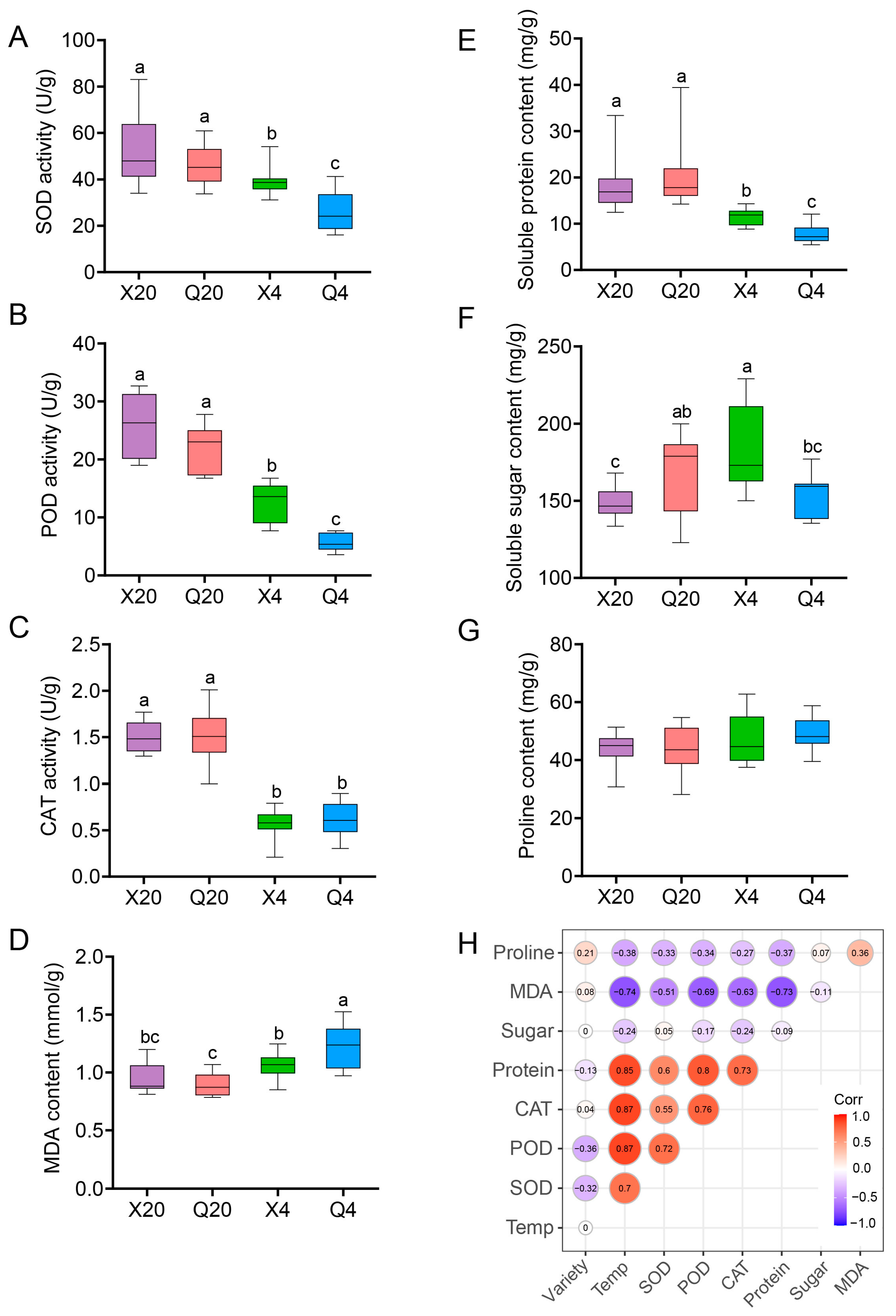
2.6. Antioxidant Resistance Is Closely Correlated with Quinoa Variety and Cold Stress
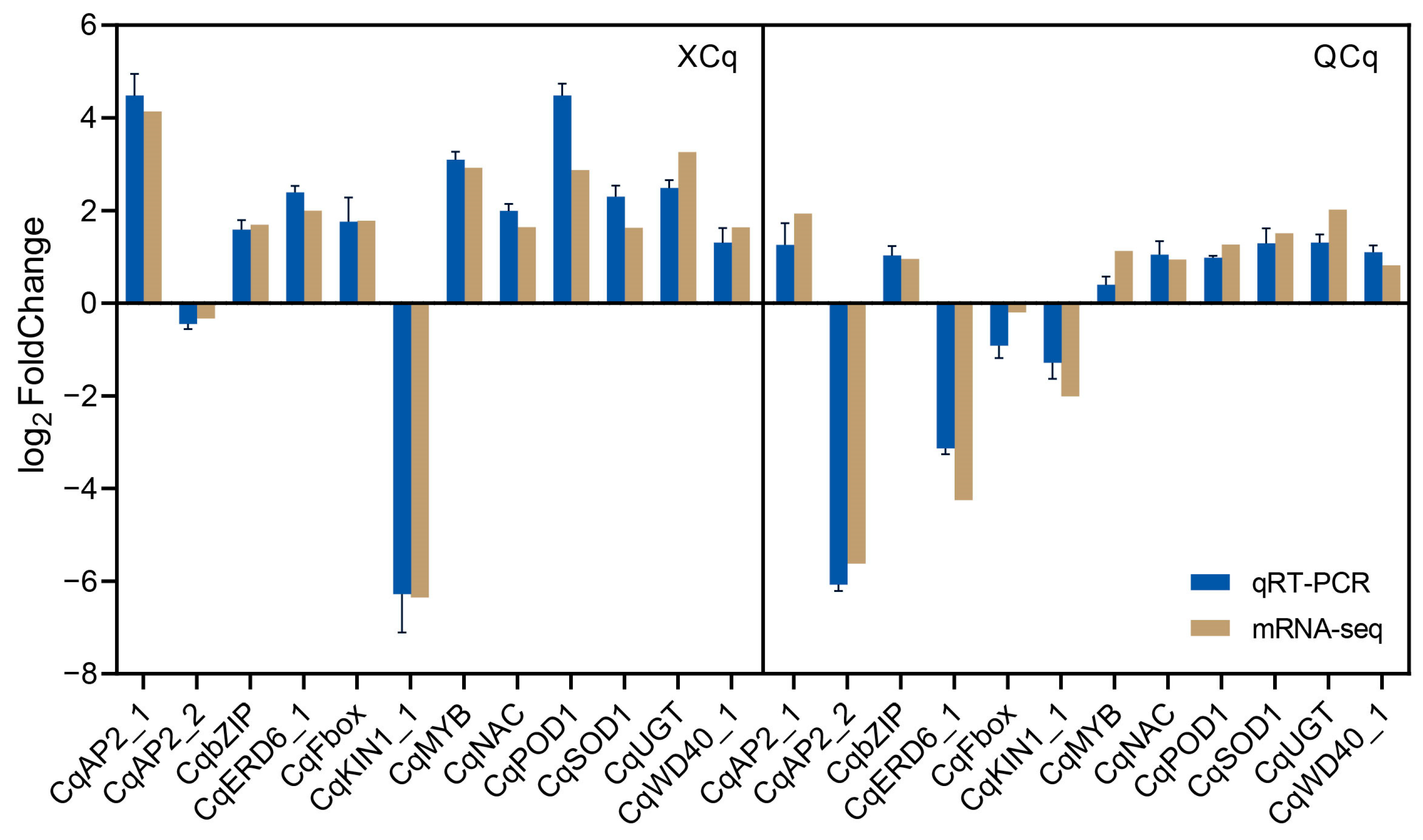
2.7. RNA-Seq Data Are Validated with qRT-PCR Assays
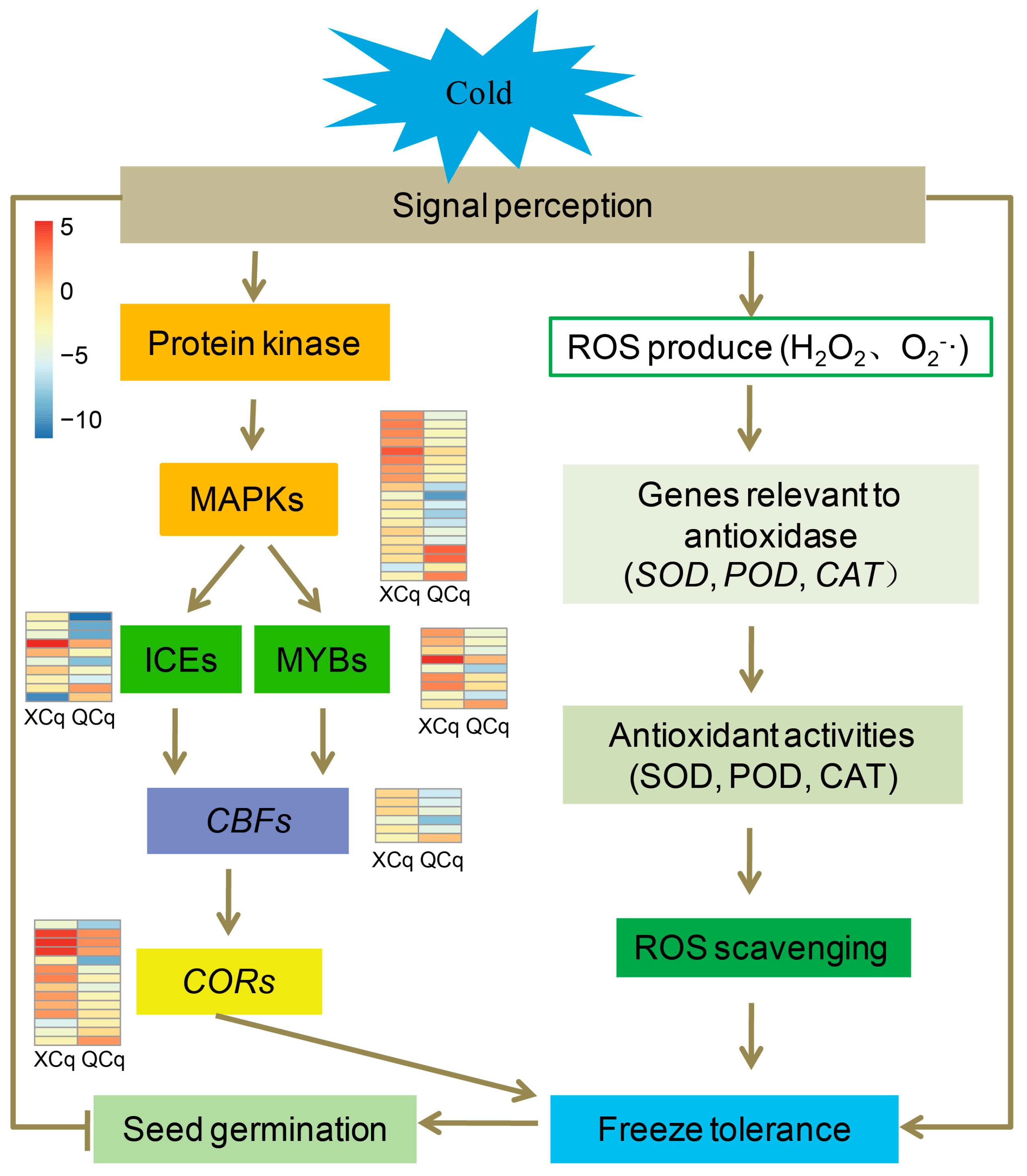
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. RNA-Seq Analysis
4.3. Identification of Co-Expression Modules
4.4. Physiological Indicator Analysis
4.5. Validation of RNA-Seq Data
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Eom, S.H.; Ahn, M.A.; Kim, E.; Lee, H.J.; Lee, J.H.; Wi, S.H.; Kim, S.K.; Lim, H.B.; Hyun, T.K. Plant response to cold stress: Cold stress changes antioxidant metabolism in heading type Kimchi cabbage (Brassica rapa L. ssp. Pekinensis). Antioxidants 2022, 11, 700. [Google Scholar] [CrossRef]
- Kidokoro, S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional regulatory network of plant cold-stress responses. Trends Plant Sci. 2022, 27, 922–935. [Google Scholar] [CrossRef]
- Ponnaiah, M.; Gilard, F.; Gakière, B.; El-Maarouf-Bouteau, H.; Bailly, C. Regulatory actors and alternative routes for Arabidopsis seed germination are revealed using a pathway-based analysis of transcriptomic datasets. Plant J. 2019, 99, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, X.; Bürger, M.; Chory, J.; Wang, X. The role of ethylene in plant temperature stress response. Trends Plant Sci. 2023, 28, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Kumar, R.; Subramanian, S.; Doherty, C.J.; Jagadish, S.V.K. Auxin-cytokinin interplay shapes root functionality under low-temperature stress. Trends Plant Sci. 2022, 28, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Lv, S.; Zhang, Z.; Chen, Q.; Mai, J.; Wan, X.; Liu, P. Integrated metabolomics and transcriptomics analysis during seed germination of waxy corn under low temperature stress. BMC Plant Biol. 2023, 23, 190. [Google Scholar] [CrossRef]
- Meng, A.; Wen, D.; Zhang, C. Dynamic changes in seed germination under low-temperature stress in maize. Int. J. Mol. Sci. 2022, 23, 5495. [Google Scholar] [CrossRef]
- Peng, M.; Chang, Y.; Chu, G.; Wang, M. Low-temperature tolerance and transcriptome analyses during seed germination of Anabasis aphylla. J. Plant Interact. 2019, 14, 254–264. [Google Scholar] [CrossRef]
- Shi, Y.; Ding, Y.; Yang, S. Molecular regulation of CBF signaling in cold acclimation. Trends Plant Sci. 2018, 23, 623–637. [Google Scholar] [CrossRef]
- Liu, C.; Li, C.; Bing, H.; Zhao, J.; Li, L.; Sun, P.; Li, T.; Du, D.; Zhao, J.; Wang, X.; et al. Integrated physiological, transcriptomic, and metabolomic analysis reveals the mechanism of guvermectin promoting seed germination in direct-seeded rice under chilling stress. J. Agric. Food Chem. 2023, 71, 7348–7358. [Google Scholar] [CrossRef]
- Shen, Q.; Zhang, S.; Liu, S.; Chen, J.; Ma, H.; Cui, Z.; Zhang, X.; Ge, C.; Liu, R.; Li, Y.; et al. Comparative Transcriptome analysis provides insights into the seed germination in cotton in response to chilling stress. Int. J. Mol. Sci. 2020, 21, 2067. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, M.F. Plant cold acclimation: Freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 571–599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Z.; Xie, S.; Si, T.; Li, Y.; Zhu, J.K. Mutational evidence for the critical role of CBF transcription factors in cold acclimation in Arabidopsis. Plant Physiol. 2016, 171, 2744–2759. [Google Scholar] [CrossRef]
- Sinha, A.K.; Jaggi, M.; Raghuram, B.; Tuteja, N. Mitogen-activated protein kinase signaling in plants under abiotic stress. Plant Signal. Behav. 2011, 6, 196–203. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, P.; Si, T.; Hsu, C.C.; Wang, L.; Zayed, O.; Yu, Z.; Zhu, Y.; Dong, J.; Tao, W.A.; et al. MAP Kinase cascades regulate the cold response by modulating ICE1 protein stability. Dev. Cell 2017, 43, 618–629. [Google Scholar] [CrossRef]
- Ain, Q.T.; Siddique, K.; Bawazeer, S.; Ali, I.; Mazhar, M.; Rasool, R.; Mubeen, B.; Ullah, F.; Unar, A.; Jafar, T.H. Adaptive mechanisms in quinoa for coping in stressful environments: An update. PeerJ 2023, 11, e14832. [Google Scholar] [CrossRef]
- Hinojosa, L.; González, J.A.; Barrios-Masias, F.H.; Fuentes, F.; Murphy, K.M. Quinoa abiotic stress responses: A Review. Plants 2018, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Pihlanto, A.; Mattila, P.; Mäkinen, S.; Pajari, A.M. Bioactivities of alternative protein sources and their potential health benefits. Food Funct. 2017, 8, 3443–3458. [Google Scholar] [CrossRef]
- Bascuñán-Godoy, L.; Reguera, M.; Abdel-Tawab, Y.M.; Blumwald, E. Water deficit stress-induced changes in carbon and nitrogen partitioning in Chenopodium quinoa Willd. Planta 2016, 243, 591–603. [Google Scholar] [CrossRef]
- Iqbal, H.; Yaning, C.; Waqas, M.; Shareef, M.; Raza, S.T. Differential response of quinoa genotypes to drought and foliage-applied H2O2 in relation to oxidative damage, osmotic adjustment and antioxidant capacity. Ecotoxicol. Environ. Saf. 2018, 164, 344–354. [Google Scholar] [CrossRef]
- Gholami, S.; Dehaghi, M.A.; Rezazadeh, A.; Naji, A.M. Seed germination and physiological responses of quinoa to selenium priming under drought stress. Bragantia 2022, 81, e0722. [Google Scholar] [CrossRef]
- Mamedi, A.; Tavakkol, A.R.; Oveisi, M. Cardinal temperatures for seed germination of three quinoa (Chenopodium quinoa Willd.) cultivars. Iran. J. Field Crop Sci. 2017, 48, 89–100. [Google Scholar]
- Strenske, A.; Vasconcelos, E.S.D.; Egewarth, V.A.; Herzog, N.F.M.; Malavasi, M.D.M. Responses of quinoa (Chenopodium quinoa willd.) seeds stored under different germination temperatures. Acta Sci.-Agron. 2017, 39, 83–88. [Google Scholar] [CrossRef]
- González, J.A.; Gallardo, M.; Hilal, M.; Rosa, M.; Prado, F.E. Physiological responses of quinoa (Chenopodium quinoa Willd.) to drought and waterlogging stress: Dry matter partitioning. Bot. Stud. 2009, 50, 35–42. [Google Scholar]
- Rosa, M.; Hilal, M.; González, J.A.; Prado, F.E. Low-temperature effect on enzyme activities involved in sucrose-starch partitioning in salt-stressed and salt-acclimated cotyledons of quinoa (Chenopodium quinoa Willd.) seedlings. Plant Physiol. Biochem. 2009, 47, 300–307. [Google Scholar] [CrossRef]
- Jacobsen, S.E.; Monteros, C.; Corcuera, L.J.; Bravo, L.A.; Christiansen, J.L.; Mujica, A. Frost resistance mechanisms in quinoa (Chenopodium quinoa Willd.). Eur. J. Agron. 2007, 26, 471–475. [Google Scholar] [CrossRef]
- Pradhan, S.K.; Pandit, E.; Nayak, D.K.; Behera, L.; Mohapatra, T. Genes, pathways and transcription factors involved in seedling stage chilling stress tolerance in indica rice through RNA-Seq analysis. BMC Plant Biol. 2019, 19, 352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhang, H.; Ren, J.; Dong, J.; Zhao, X.; Wang, X.; Wang, J.; Zhong, C.; Zhao, S.; Liu, X.; et al. Comparative transcriptome-based mining and expression profiling of transcription factors related to cold tolerance in peanut. Int. J. Mol. Sci. 2020, 21, 1921. [Google Scholar] [CrossRef]
- Xie, H.; Wang, Q.; Zhang, P.; Zhang, X.; Huang, T.; Guo, Y.; Liu, J.; Li, L.; Li, H.; Qin, P. Transcriptomic and metabolomic analysis of the response of quinoa seedlings to low temperatures. Biomolecules 2022, 12, 977. [Google Scholar] [CrossRef]
- Tang, Y.; Li, J.; Song, Q.; Cheng, Q.; Tan, Q.; Zhou, Q.; Nong, Z.; Lv, P. Transcriptome and WGCNA reveal hub genes in sugarcane tiller seedlings in response to drought stress. Sci. Rep. 2023, 13, 12823. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, G.; Wang, X.; Bai, Y.; Zhang, P.; Liu, J.; Li, L.; Huang, L.; Qin, P. Identifying core genes related to low-temperature stress resistance in quinoa seedlings based on WGCNA. Int. J. Mol. Sci. 2024, 25, 6885. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, Y.; Shi, Y.; Zhang, X.; Zhang, S.; Gong, Z.; Yang, S. MPK3- and MPK6-Mediated ICE1 phosphorylation negatively regulates ICE1 stability and freezing tolerance in Arabidopsis. Dev. Cell 2017, 43, 630–642.e4. [Google Scholar] [CrossRef]
- Hwarari, D.; Guan, Y.; Ahmad, B.; Movahedi, A.; Min, T.; Hao, Z.; Lu, Y.; Chen, J.; Yang, L. ICE-CBF-COR signaling cascade and its regulation in plants responding to cold stress. Int. J. Mol. Sci. 2022, 23, 1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Li, F.; Liu, H.; Yang, W.; Chong, K.; Xu, Y. OsMAPK3 phosphorylates OsbHLH002/OsICE1 and inhibits its ubiquitination to activate OsTPP1 and enhances rice chilling tolerance. Dev. Cell 2017, 43, 731–743.e5. [Google Scholar] [CrossRef]
- Yu, M.M.; Wang, R.; Xia, J.Q.; Li, C.; Xu, Q.H.; Cang, J.; Wang, Y.Y.; Zhang, D. JA-induced TaMPK6 enhanced the freeze tolerance of Arabidopsis thaliana through regulation of ICE-CBF-COR module and antioxidant enzyme system. Plant Sci. 2023, 329, 111621. [Google Scholar] [CrossRef]
- Barrero-Gil, J.; Salinas, J. Gene regulatory networks mediating cold acclimation: The CBF pathway. Adv. Exp. Med. Biol. 2018, 1081, 3–22. [Google Scholar] [PubMed]
- Lissarre, M.; Ohta, M.; Sato, A.; Miura, K. Cold-responsive gene regulation during cold acclimation in plants. Plant Signal. Behav. 2010, 5, 948–952. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, M.; Lee, J.H.; Lee, H.J.; Park, C.M. The unified ICE-CBF pathway provides a transcriptional feedback control of freezing tolerance during cold acclimation in Arabidopsis. Plant Mol. Biol. 2015, 89, 187–201. [Google Scholar] [CrossRef]
- An, J.P.; Li, R.; Qu, F.J.; You, C.X.; Wang, X.F.; Hao, Y.J. R2R3-MYB transcription factor MdMYB23 is involved in the cold tolerance and proanthocyanidin accumulation in apple. Plant J. 2018, 96, 562–577. [Google Scholar] [CrossRef]
- Liu, C.; Schläppi, M.R.; Mao, B.; Wang, W.; Wang, A.; Chu, C. The bZIP73 transcription factor controls rice cold tolerance at the reproductive stage. Plant Biotechnol. J. 2019, 17, 1834–1849. [Google Scholar] [CrossRef]
- Liu, X.; Wei, R.; Tian, M.; Liu, J.; Ruan, Y.; Sun, C.; Liu, C. Combined transcriptome and metabolome profiling provide insights into cold responses in rapeseed (Brassica napus L.) genotypes with contrasting cold-stress sensitivity. Int. J. Mol. Sci. 2022, 23, 13546. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Sun, B.; He, H.; Zhang, Y.; Tian, H.; Wang, B. Current understanding of bHLH transcription factors in plant abiotic stress tolerance. Int. J. Mol. Sci. 2021, 22, 4921. [Google Scholar] [CrossRef]
- Agarwal, M.; Hao, Y.; Kapoor, A.; Dong, C.H.; Fujii, H.; Zheng, X.; Zhu, J.K. A R2R3 type MYB transcription factor is involved in the cold regulation of CBF genes and in acquired freezing tolerance. J. Biol. Chem. 2006, 281, 37636–37645. [Google Scholar] [CrossRef]
- Ko, D.K.; Brandizzi, F. Network-based approaches for understanding gene regulation and function in plants. Plant J. 2020, 104, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, H.; Wang, L.; Zhao, Q.; Wang, D.; Zhang, T. Cold acclimation alleviates cold stress-induced PSII inhibition and oxidative damage in tobacco leaves. Plant Signal. Behav. 2022, 17, 2013638. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, Q.; Wang, Z.; Li, J.; Liu, S.; Chang, R.; Chen, H.; Liu, G. Integrated analysis of transcriptomics and metabolomics of peach under cold stress. Front. Plant Sci. 2023, 14, 1153902. [Google Scholar] [CrossRef]
- Serrat, X.; Quello, A.; Manikan, B.; Lino, G.; Nogués, S. Comparative salt-stress responses in salt-tolerant (Vikinga) and salt-sensitive (Regalona) quinoa varieties. Physiological, anatomical and biochemical perspectives. Agronomy 2024, 14, 3003. [Google Scholar] [CrossRef]
- Jensen, C.; Jacobsen, S.E.; Andersen, M.; Nunez, N.; Andersen, S.; Rasmussen, L.; Mogensen, V.O. Leaf gas exchange and water relation characteristics of field quinoa (Chenopodium quinoa Willd.) during soil drying. Eur. J. Agron. 2000, 13, 11–25. [Google Scholar] [CrossRef]
- Liang, G.; Li, Y.; Wang, P.; Jiao, S.; Wang, H.; Mao, J.; Chen, B. VaAPL1 promotes starch synthesis to constantly contribute to soluble sugar accumulation, improving low temperature tolerance in Arabidopsis and tomato. Front. Plant Sci. 2022, 13, 920424. [Google Scholar] [CrossRef]
- Bao, Q.; Wu, Y.; Wang, Y.; Zhang, Y. Comparative Transcriptomic analysis reveals transcriptional differences in the response of quinoa to salt and alkali stress responses. Agronomy 2024, 14, 1596. [Google Scholar] [CrossRef]
- Wang, C.; Lu, C.; Wang, J.; Liu, X.; Wei, Z.; Qin, Y.; Zhang, H.; Wang, X.; Wei, B.; Lv, W.; et al. Molecular mechanisms regulating glucose metabolism in quinoa (Chenopodium quinoa Willd.) seeds under drought stress. BMC Plant Biol. 2024, 24, 796. [Google Scholar] [CrossRef] [PubMed]
- Sohag, A.A.M.; Tahjib-Ul-Arif, M.; Afrin, S.; Khan, M.K.; Hannan, M.A.; Skalicky, M.; Mortuza, M.G.; Brestic, M.; Hossain, M.A.; Murata, Y. Insights into nitric oxide-mediated water balance, antioxidant defence and mineral homeostasis in rice (Oryza sativa L.) under chilling stress. Nitric Oxide-Biol. Chem. 2020, 100–101, 7–16. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, R.; Liang, X.; Li, J.; Song, Y.; Yi, K.; Nie, W.; Ma, L.; Li, J.; Li, M.; Wang, X.; et al. Comparative Transcriptomic Analyses Reveal Key Pathways in Response to Cold Stress at the Germination Stage of Quinoa (Chenopodium quinoa Willd.) Seeds. Plants 2025, 14, 1212. https://doi.org/10.3390/plants14081212
Fu R, Liang X, Li J, Song Y, Yi K, Nie W, Ma L, Li J, Li M, Wang X, et al. Comparative Transcriptomic Analyses Reveal Key Pathways in Response to Cold Stress at the Germination Stage of Quinoa (Chenopodium quinoa Willd.) Seeds. Plants. 2025; 14(8):1212. https://doi.org/10.3390/plants14081212
Chicago/Turabian StyleFu, Rao, Xiaoyan Liang, Jiajia Li, Yanjing Song, Kuihua Yi, Wenjing Nie, Lan Ma, Junlin Li, Meng Li, Xiangyu Wang, and et al. 2025. "Comparative Transcriptomic Analyses Reveal Key Pathways in Response to Cold Stress at the Germination Stage of Quinoa (Chenopodium quinoa Willd.) Seeds" Plants 14, no. 8: 1212. https://doi.org/10.3390/plants14081212
APA StyleFu, R., Liang, X., Li, J., Song, Y., Yi, K., Nie, W., Ma, L., Li, J., Li, M., Wang, X., Zhang, H., & Zhang, H. (2025). Comparative Transcriptomic Analyses Reveal Key Pathways in Response to Cold Stress at the Germination Stage of Quinoa (Chenopodium quinoa Willd.) Seeds. Plants, 14(8), 1212. https://doi.org/10.3390/plants14081212