
Phylogeographic Pattern of Sargassum hemiphyllum var. chinense (Phaeophyceae, Ochrophyta) in Chinese Coastal Waters


Abstract
:1. Introduction
2. Results
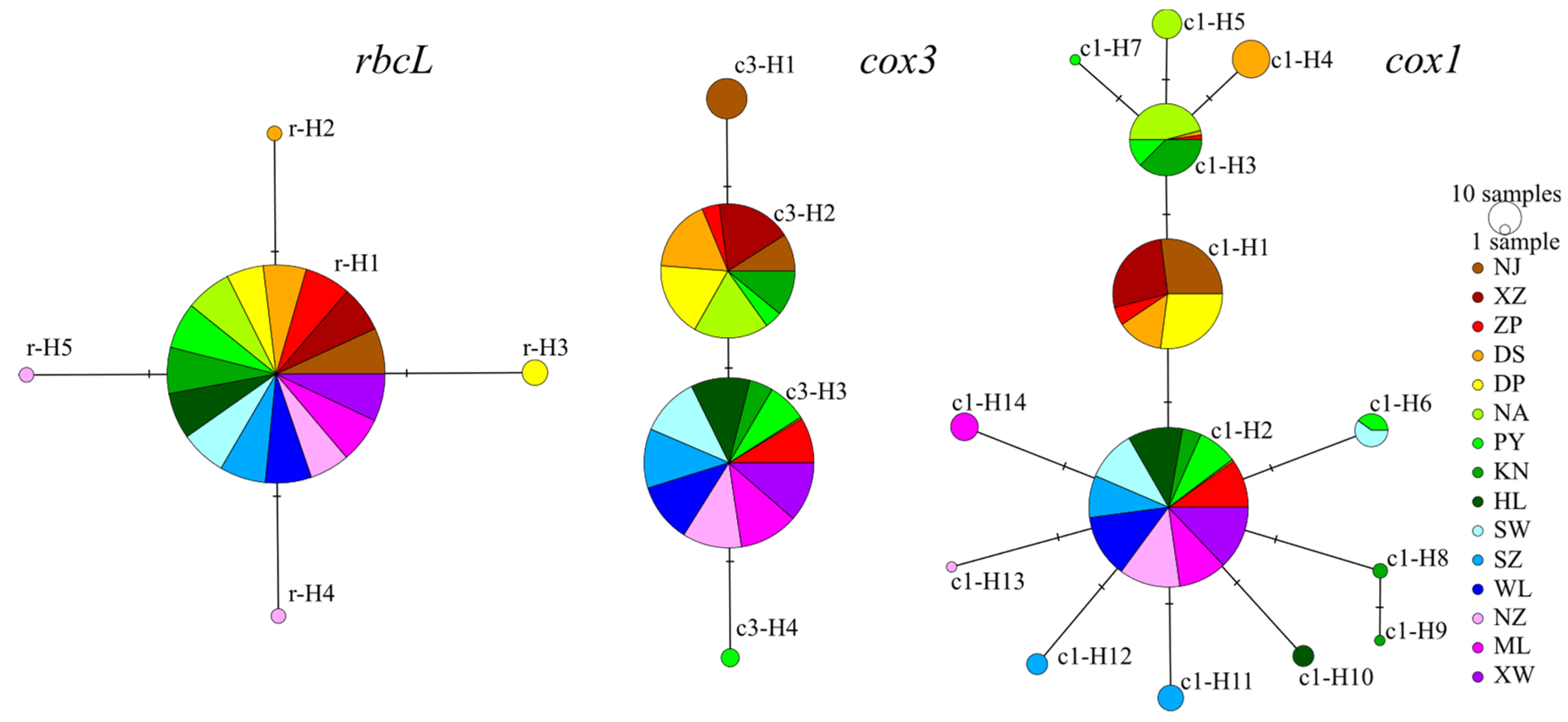
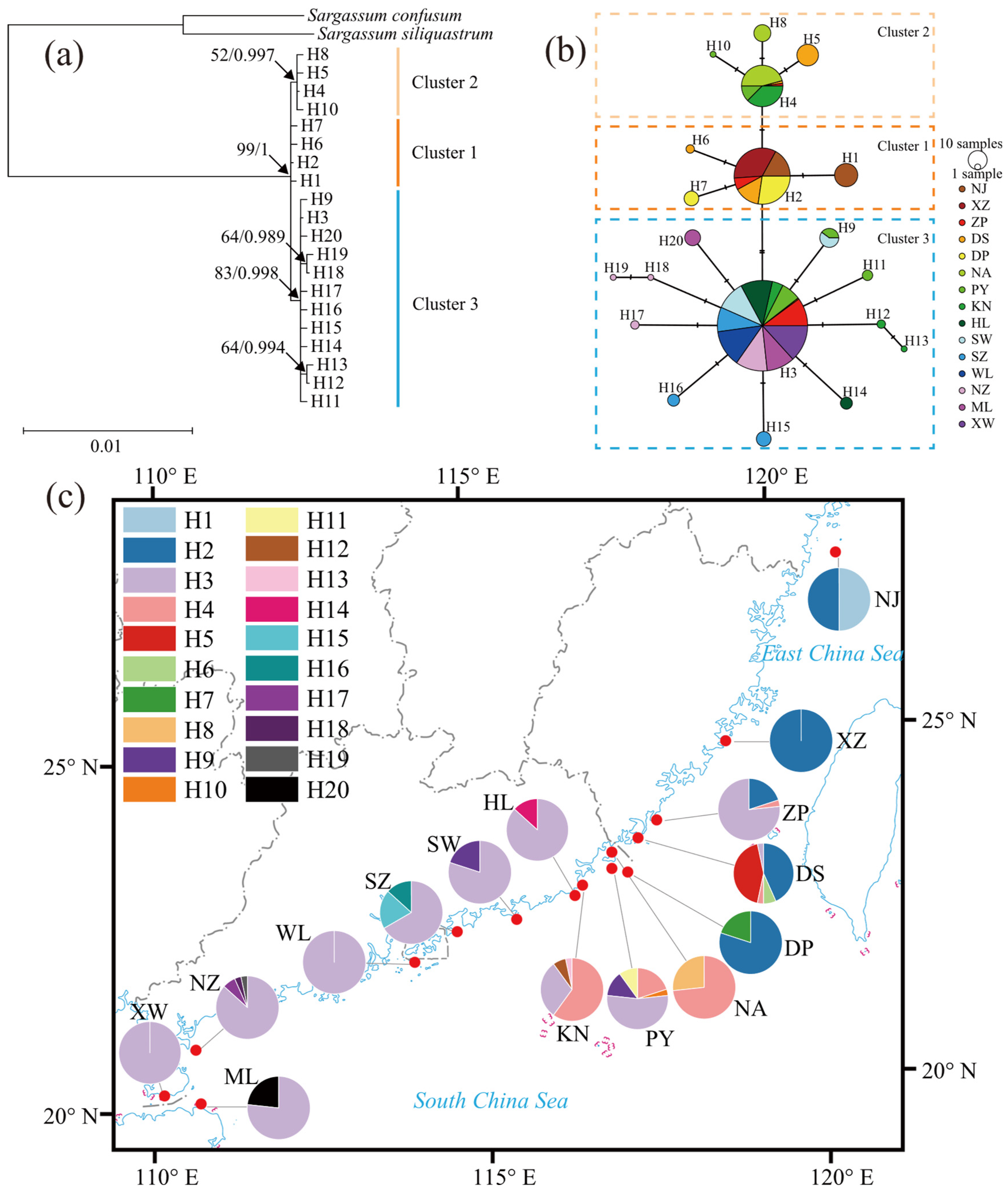
2.1. Genetic Diversity and Phylogenetic Analysis
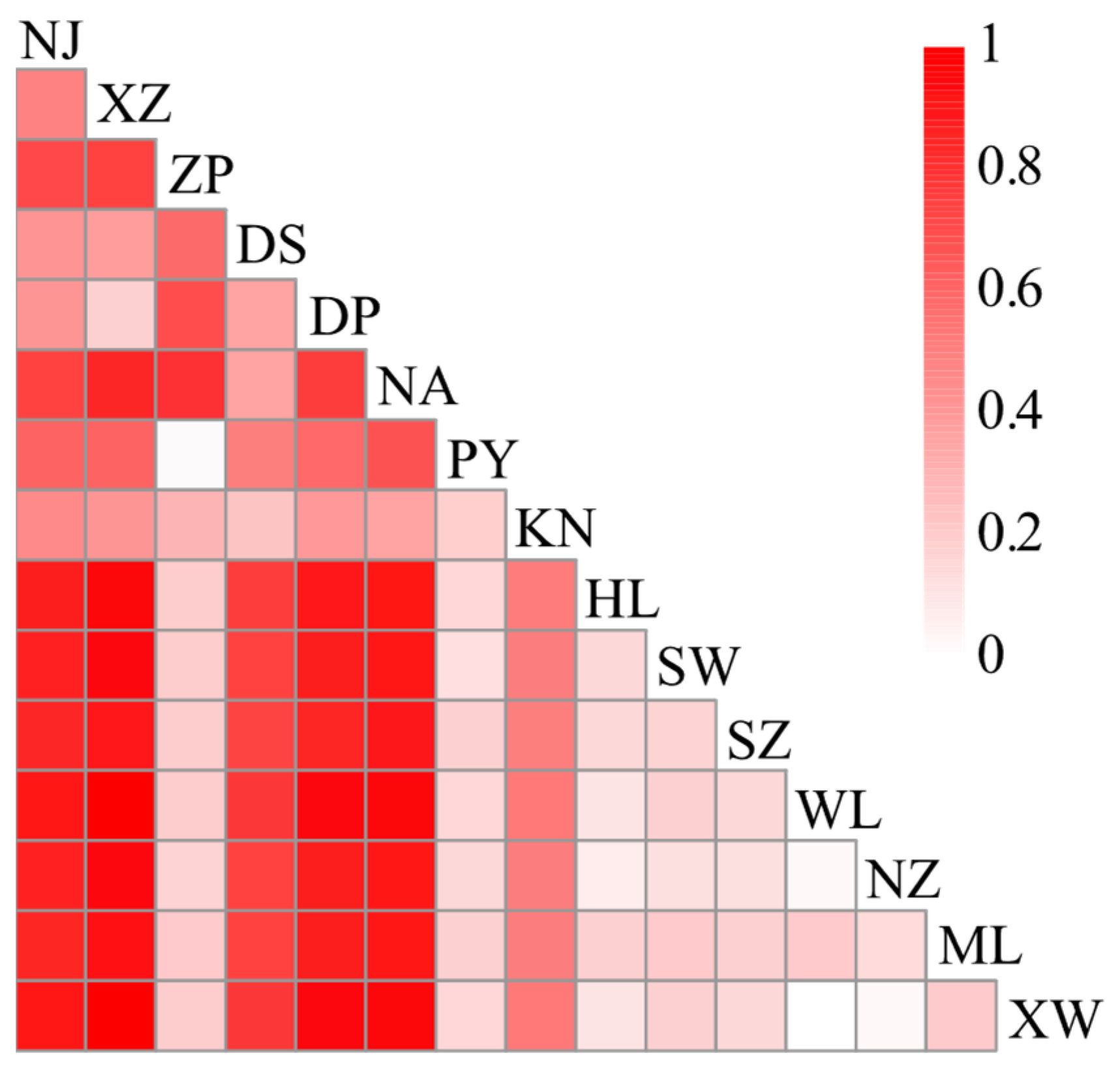
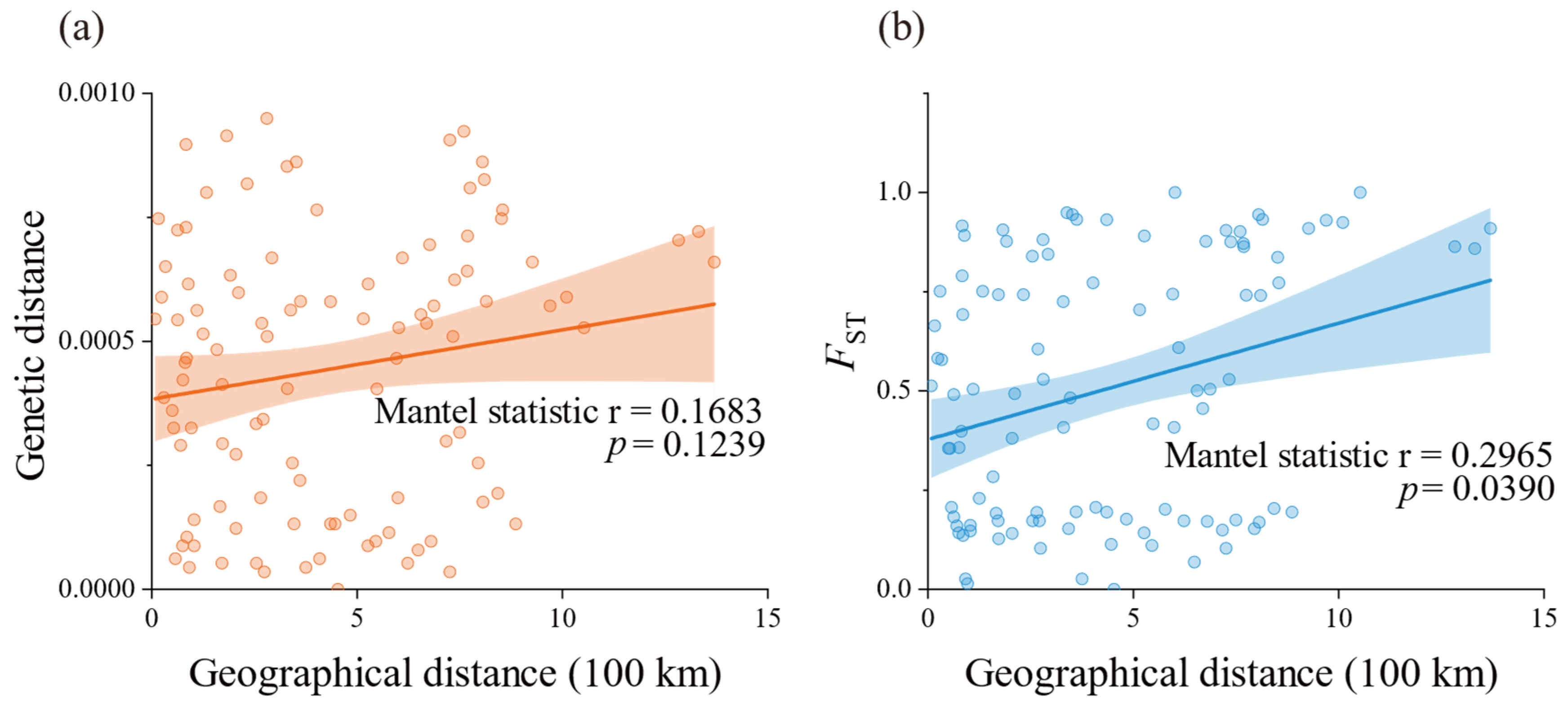
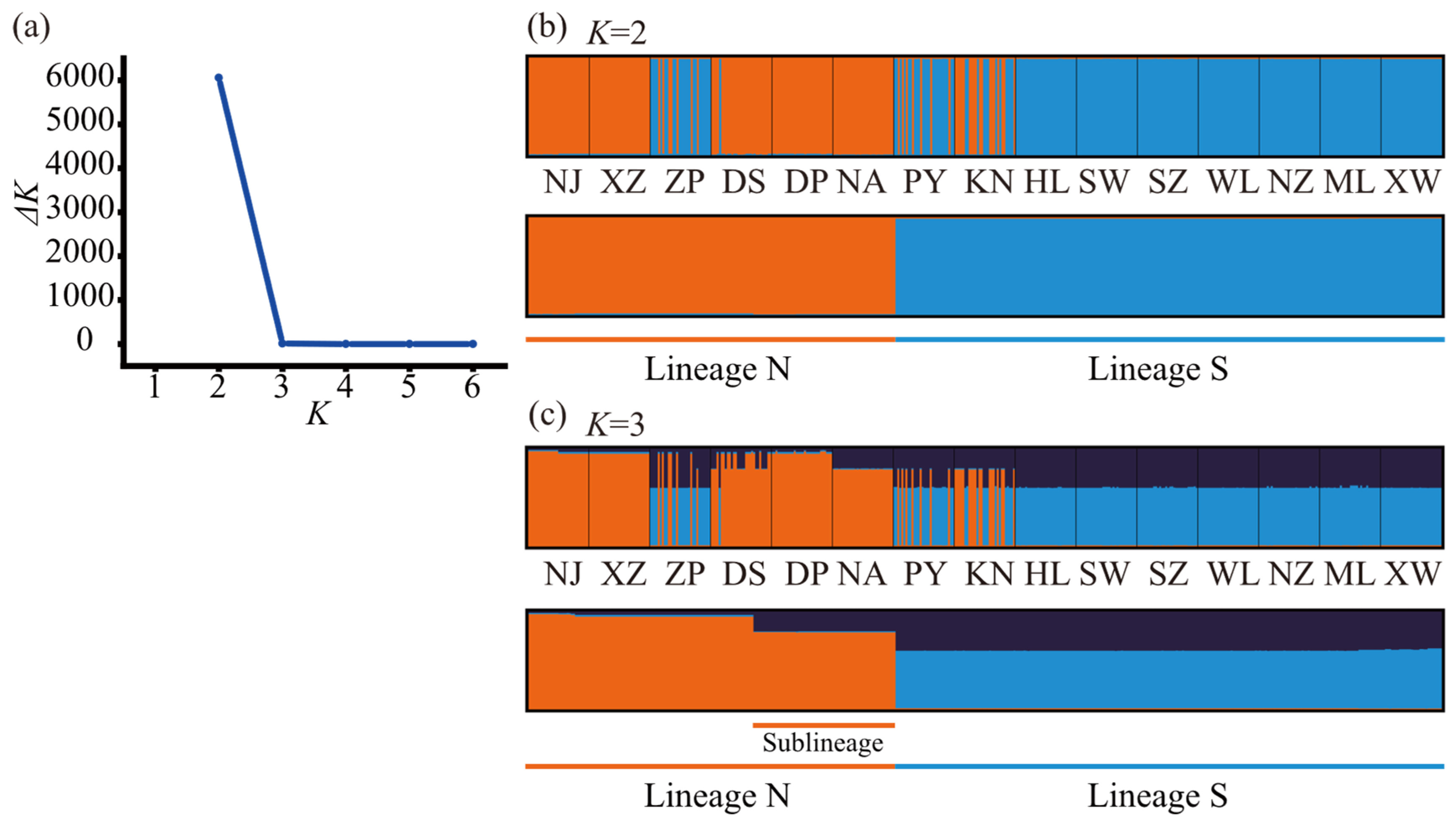
2.2. Population Genetic Structure
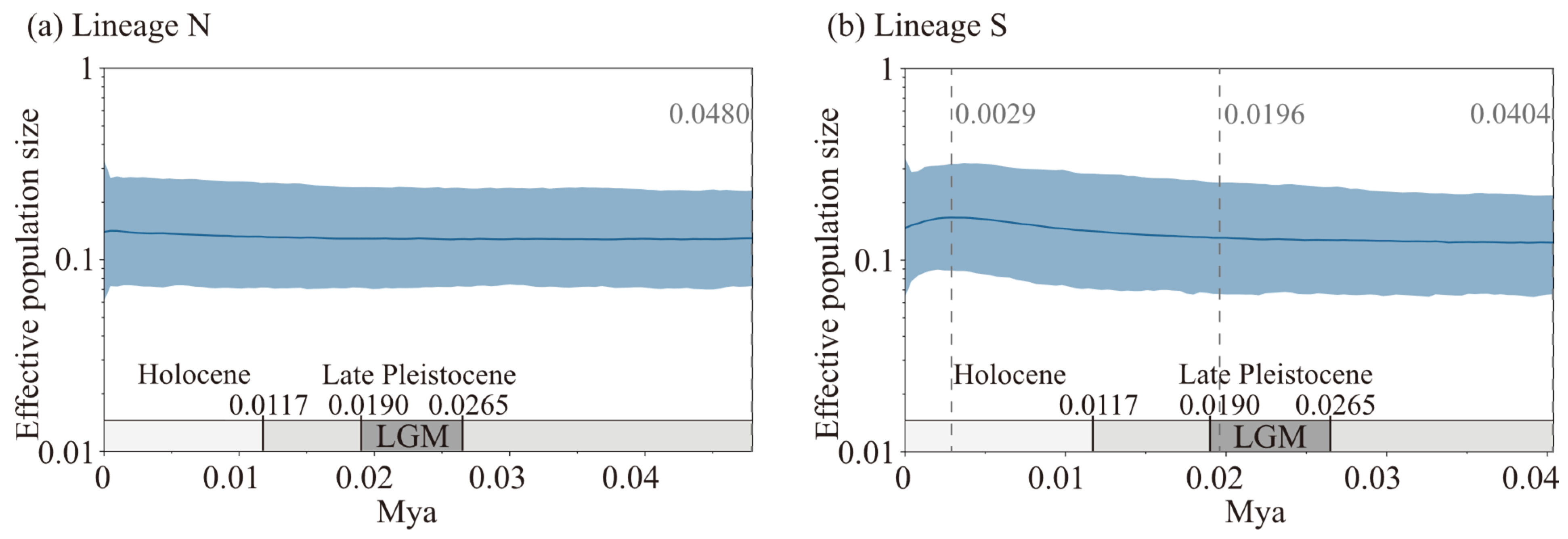
2.3. Divergence Time and Historical Demographic Dynamics
3. Discussion
3.1. Variability and Applicability of the Genetic Markers
3.2. Genetic Variability and Population Structure
3.3. Phylogeographic Structure and Demographic History
3.4. Insights of Species Conservation
4. Materials and Methods
4.1. Seaweed Collection and Molecular Procedures
4.2. Genetic Diversity, Phylogeny, and Population Genetic Structure
4.3. Divergence Time and Demographic History
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guiry, M.D.; Guiry, G.M. AlgaeBase. 2022. Available online: https://www.algaebase.org (accessed on 14 March 2025).
- Alallam, B.; Abd Kadir, E.; Dewi, F.R.P.; Yong, Y.K.; Lim, V. Extraction and characterization of sodium alginate from native Malaysian brown seaweed Sargassum polycystum. Int. J. Biol. Macromol. 2025, 287, 138552. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Hu, G.; Lv, F.; Shi, Z.; Dong, W.; Qi, Y.; Wang, C.; Tong, H.; Huang, F.; Yan, P. Sargafusal, a rare norsesquiterpenoid from the edible brown alga Sargassum fusiforme. Phytochem. Lett. 2025, 66, 86–90. [Google Scholar] [CrossRef]
- Melchor-Martínez, E.M.; Reyes, A.G.; Morreeuw, Z.P.; Flores-Contreras, E.A.; Araújo, R.G.; Ramírez-Gamboa, D.; Sosa-Hernández, J.E.; Iqbal, H.M.N.; González-Meza, G.M.; Bonaccorso, A.D.; et al. Comparative study on the valorization of Sargassum from the Mexican Caribbean coast and Gulf of California as an ingredient on healthy diets for shrimp farming. Aquac. Rep. 2023, 32, 101709. [Google Scholar] [CrossRef]
- Tagliapietra, B.L.; Salvador-Reyes, R.; Pinto, C.C.; de Souza, S.M.; Pallone, J.A.L.; de Araújo Bezerra, J.; Moreira Mar, J.; Aparecido Sanches, E.; Clerici, M.T.P.S. Nutritional and techno-functional properties of the brown seaweed Sargassum filipendula. Food Res. Int. 2024, 191, 114728. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Q.; Luo, D.; Xu, Z.; Sun, P.; Guo, J.; Wang, H.; Liu, Z.; Yang, Y. Local effects of Sargassum beds on the seawater carbonate system and plankton community. Algal Res. 2025, 85, 103860. [Google Scholar] [CrossRef]
- Jung, S.M.; Lee, J.H.; Han, S.H.; Jeon, W.B.; Kim, G.Y.; Kim, S.; Kim, S.; Lee, H.-R.; Hwang, D.S.; Jung, S.; et al. A new approach to the restoration of seaweed beds using Sargassum fulvellum. J. Appl. Phycol. 2020, 32, 2575–2581. [Google Scholar] [CrossRef]
- Cheung-Wong, R.W.Y.; Kotta, J.; Hemraj, D.A.; Russell, B.D. Persistence in a tropical transition zone? Sargassum forests alternate seasonal growth forms to maintain productivity in warming waters at the expense of annual biomass production. Sci. Total Environ. 2022, 851, 158154. [Google Scholar] [CrossRef]
- Matsui, A.; Kawamura, M.; Nozawa, S.; Takeyama, M.; Inoue, N. Comparison of Sargassum community succession between artificial and natural reefs in Wakasa Bay, Japan. Landsc. Ecol. Eng. 2025, 21, 205–219. [Google Scholar] [CrossRef]
- Ajisaka, T.; Nang, H.Q.; Dinh, N.H.; Lu, B.R.; Chiang, Y.M.; Yoshida, T. Sargassum hemiphyllum (Turner) C. Agardh var. chinense J. Agardh from Vietnam, the Chinese mainland, and Taiwan (Fucales, Phaeophyta). In Taxonomy of Economic Seaweeds with Reference to Some Pacific Species; Abbott, I.A., Ed.; California Sea Grant College Program, University of California: La Jolla, CA, USA, 1997; Volume 6, pp. 37–50. [Google Scholar]
- Huang, C. A Taxonomic Revision of the Genus Sargassum (Fucales, Phaeophyceae) in the Yellow Sea and the East China Sea. Master’s Thesis, University of Chinese Academy of Sciences, Beijing, China, 2017. [Google Scholar]
- Jia, X.; Chen, W.; Liu, T.; Chen, Z. Organellar Genomes of Sargassum hemiphyllum var. chinense Provide Insight into the Characteristics of Phaeophyceae. Int. J. Mol. Sci. 2024, 25, 8584. [Google Scholar] [CrossRef]
- Cheang, C.C.; Chu, K.H.; Ang, P.O. Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Mol. Ecol. 2010, 19, 2933–2948. [Google Scholar] [CrossRef]
- Cheang, C.C.; Chu, K.H.; Ang, P.O. Morphological and genetic variation in the populations of Sargassum hemiphyllum (Phaeophyceae) in the northwestern Pacific. J. Phycol. 2008, 44, 855–865. [Google Scholar] [CrossRef]
- Li, X.; Wang, K.; Zhang, S.; Feng, M. Distribution and Flora of Seaweed Beds in the Coastal Waters of China. Sustainability 2021, 13, 3009. [Google Scholar] [CrossRef]
- Han, T.; Shi, R.; Qi, Z.; Huang, H. The overgrowth of epiphytic Ulva prolifera during seedling cultivation of Sargassum hemiphyllum can be mitigated by regulating nitrogen availability. Aquaculture 2021, 543, 736930. [Google Scholar] [CrossRef]
- Han, T.; Song, P.; Shi, R.; Qi, Z.; Li, J.; Huang, H. Optimal nutrient availability could alleviate diatom Cylindrotheca closterium fouling during seedling cultivation of Sargassum hemiphyllum. Aquaculture 2022, 552, 738020. [Google Scholar] [CrossRef]
- Yu, Z.; Robinson, S.M.C.; Xia, J.; Sun, H.; Hu, C. Growth, bioaccumulation and fodder potentials of the seaweed Sargassum hemiphyllum grown in oyster and fish farms of South China. Aquaculture 2016, 464, 459–468. [Google Scholar] [CrossRef]
- Kam, Y.L.K.; Ang, P.O. Phenology and experimental evaluation of temperature as a triggering factor for reproduction in Sargassum hemiphyllum. J. Appl. Phycol. 2016, 28, 2459–2470. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Chen, J.; Luo, B.; Zhu, F.; Li, R.; Wang, Z.; Cheong, K.-L.; Zhong, S. Fucoidan from Sargassum hemiphyllum (Turner) C. Agardh inhibits human hepatoma cell (HepG2) through cell cycle arrest and apoptotic induction. Food Biosci. 2024, 62, 105051. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: Retrospect and prospect. J. Biogeogr. 2009, 36, 3–15. [Google Scholar] [CrossRef]
- Kobayashi, H.; Haino, Y.; Iwasaki, T.; Tezuka, A.; Nagano, A.J.; Shimada, S. ddRAD-seq based phylogeographic study of Sargassum thunbergii (Phaeophyceae, Heterokonta) around Japanese coast. Mar. Environ. Res. 2018, 140, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Hu, Z.-M.; Gao, X.; Sun, Z.-M.; Choi, H.-G.; Duan, D.-L.; Endo, H. Oceanic currents drove population genetic connectivity of the brown alga Sargassum thunbergii in the north-west Pacific. J. Biogeogr. 2017, 44, 230–242. [Google Scholar] [CrossRef]
- Le Cam, S.; Daguin-Thiébaut, C.; Bouchemousse, S.; Engelen, A.H.; Mieszkowska, N.; Viard, F. A genome-wide investigation of the worldwide invader Sargassum muticum shows high success albeit (almost) no genetic diversity. Evol. Appl. 2020, 13, 500–514. [Google Scholar] [CrossRef]
- Hu, Z.-M.; Zhang, J.; Lopez-Bautista, J.; Duan, D.-L. Asymmetric genetic exchange in the brown seaweed Sargassum fusiforme (Phaeophyceae) driven by oceanic currents. Mar. Biol. 2013, 160, 1407–1414. [Google Scholar] [CrossRef]
- Hu, Z.-M.; Li, J.-J.; Sun, Z.-M.; Gao, X.; Yao, J.-T.; Choi, H.-G.; Endo, H.; Duan, D.-L. Hidden diversity and phylogeographic history provide conservation insights for the edible seaweed Sargassum fusiforme in the Northwest Pacific. Evol. Appl. 2017, 10, 366–378. [Google Scholar] [CrossRef]
- Lin, L.; Wang, F.; Wu, M.; Wang, S. ddRAD Sequencing-Based Scanning of Genetic Variants in Sargassum fusiforme. J. Mar. Sci. Eng. 2022, 10, 958. [Google Scholar] [CrossRef]
- Hu, Z.-M.; Uwai, S.; Yu, S.-H.; Komatsu, T.; Ajisaka, T.; Duan, D.-L. Phylogeographic heterogeneity of the brown macroalga Sargassum horneri (Fucaceae) in the northwestern Pacific in relation to late Pleistocene glaciation and tectonic configurations. Mol. Ecol. 2011, 20, 3894–3909. [Google Scholar] [CrossRef]
- Li, J.-J.; Huang, S.-H.; Liu, Z.-Y.; Bi, Y.-X. Climate-Driven Range Shifts of Brown Seaweed Sargassum horneri in the Northwest Pacific. Front. Mar. Sci. 2020, 7, 570881. [Google Scholar] [CrossRef]
- Li, J.-J.; Liu, Z.-Y.; Zhong, Z.-H.; Zhuang, L.-C.; Bi, Y.-X.; Qin, S. Limited Genetic Connectivity Among Sargassum horneri (Phaeophyceae) Populations in the Chinese Marginal Seas Despite Their High Dispersal Capacity. J. Phycol. 2020, 56, 994–1005. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, W.; Zhuang, M.; Wu, T.; Zhao, C.; Dai, W.; Zhang, J. Population Genetic Structure of Sargassum horneri, the Dominant Species of Golden Tide in the Yellow Sea. J. Mar. Sci. Eng. 2024, 12, 900. [Google Scholar] [CrossRef]
- Chan, S.W.; Cheang, C.C.; Yeung, C.W.; Chirapart, A.; Gerung, G.; Ang, P. Recent expansion led to the lack of genetic structure of Sargassum aquifolium populations in Southeast Asia. Mar. Biol. 2014, 161, 785–795. [Google Scholar] [CrossRef]
- Ng, P.-K.; Chiou, Y.-S.; Liu, L.; Sun, Z.; Shimabukuro, H.; Lin, S.-M. Phylogeography and genetic connectivity of the marine macro-alga Sargassum ilicifolium (Phaeophyceae, Ochrophyta) in the northwestern Pacific. J. Phycol. 2018, 55, 7–24. [Google Scholar] [CrossRef]
- Hu, Z.-M.; Kantachumpoo, A.; Liu, R.-Y.; Sun, Z.-M.; Yao, J.-T.; Komatsu, T.; Uwai, S.; Duan, D.-L. A late Pleistocene marine glacial refugium in the south-west of Hainan Island, China: Phylogeographical insights from the brown alga Sargassum polycystum. J. Biogeogr. 2018, 45, 355–366. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, S.; Yan, C.; Draisma, S.G.A.; Kantachumpoo, A.; Li, Z.; Lin, Y.; Zhu, J.; Zhang, J.; Duan, D. Influence of Indo-Pacific ocean currents on the distribution and demographic patterns of the brown seaweed Sargassum polycystum in tropical east Asia. Front. Mar. Sci. 2022, 9, 895554. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, J.; Du, Y.; Zhang, S.; Liu, L.; Draisma, S.G.A.; Duan, D. Marine conditions in Andaman Sea shape the unique genetic structure of Sargassum plagiophyllum C. Agardh. J. Appl. Phycol. 2024, 36, 501–511. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, J.; Song, X.; Choi, H.-G.; Gao, X.; Duan, D.; Hu, Z. Low genetic diversity in the endangered marine alga Silvetia siliquosa (Ochrophyta: Fucaceae) and the implication to conservation. J. Oceanol. Limnol. 2022, 40, 216–225. [Google Scholar] [CrossRef]
- Oak, J.H.; Suh, Y.; Lee, I.K. Phylogenetic relationships of Sargassum subgenus Bactrophycus (Sargassaceae, Phaeophyceae) inferred from rDNA ITS sequences. Algae 2002, 17, 235–247. [Google Scholar] [CrossRef]
- Kendrick, G.A.; Walker, D.I. Dispersal of propagules of Sargassum spp. (Sargassaceae: Phaeophyta): Observations of local patterns of dispersal and consequences for recruitment and population structure. J. Exp. Mar. Biol. Ecol. 1995, 192, 273–288. [Google Scholar] [CrossRef]
- Yamasaki, M.; Aono, M.; Ogawa, N.; Tanaka, K.; Imoto, Z.; Nakamura, Y. Drifting algae and fish: Implications of tropical Sargassum invasion due to ocean warming in western Japan. Estuar. Coast. Shelf Sci. 2014, 147, 32–41. [Google Scholar] [CrossRef]
- Zhong, K.-L.; Hiraoka, M.; Gao, X.; Russell, B.; Hu, Z.-M.; Chen, W.; Kim, J.-H.; Yotsukura, N.; Endo, H.; Oka, N.; et al. Environmental gradients influence geographic differentiation and low genetic diversity of morphologically similar Ulva species in the Northwest Pacific. Bot. Mar. 2024, 67, 139–151. [Google Scholar] [CrossRef]
- Li, J.; Chen, Z.; Sun, Z.; Chen, W. Genetic Diversity and Profile of Red Algae Pterocladiella capillacea (Gelidiales, Rhodophyta) along the Coast of China. Diversity 2024, 16, 389. [Google Scholar] [CrossRef]
- Zhong, K.-L.; Song, X.-H.; Choi, H.-G.; Satoshi, S.; Weinberger, F.; Draisma, S.G.A.; Duan, D.-L.; Hu, Z.-M. MtDNA-Based Phylogeography of the Red Alga Agarophyton vermiculophyllum (Gigartinales, Rhodophyta) in the Native Northwest Pacific. Front. Mar. Sci. 2020, 7, 366. [Google Scholar] [CrossRef]
- Motomura, T.; Nagasato, C.; Kimura, K. Cytoplasmic inheritance of organelles in brown algae. J. Plant Res. 2010, 123, 185–192. [Google Scholar] [CrossRef]
- Tseng, C.K.; Lu, B.R. Flora Algarum Marinarum Sinicarum Tomus III Phaeophyta No. II Fucales; Science Press: Beijing, China, 2000. [Google Scholar]
- Ota, H. Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu Archipelago, Japan, with special reference to their paleogeographical implications. Popul. Ecol. 1998, 40, 189–204. [Google Scholar] [CrossRef]
- Wang, P. Response of Western Pacific marginal seas to glacial cycles: Paleoceanographic and sedimentological features. Mar. Geol. 1999, 156, 5–39. [Google Scholar] [CrossRef]
- Letouzey, J.; Kimura, M. Okinawa Trough genesis: Structure and evolution of a backarc basin developed in a continent. Mar. Pet. Geol. 1985, 2, 111–130. [Google Scholar] [CrossRef]
- Comes, H.P.; Kadereit, J.W. The effect of Quaternary climatic changes on plant distribution and evolution. Trends Plant Sci. 1998, 3, 432–438. [Google Scholar] [CrossRef]
- Maggs, C.A.; Castilho, R.; Foltz, D.; Henzler, C.; Jolly, M.T.; Kelly, J.; Olsen, J.; Perez, K.E.; Stam, W.; Väinölä, R.; et al. Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 2008, 89, S108–S122. [Google Scholar] [CrossRef]
- Provan, J.; Bennett, K.D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 2008, 23, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-M.; Duan, D.-L. Insufficient geographical sampling could severely influence phylogeographic interpretations. Mar. Biol. 2013, 160, 1521–1522. [Google Scholar] [CrossRef]
- Harley, C.D.G.; Anderson, K.M.; Demes, K.W.; Jorve, J.P.; Kordas, R.L.; Coyle, T.A.; Graham, M.H. Effects of Climate Change on Global Seaweed Communities. J. Phycol. 2012, 48, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Du, X.-K. Will climate change cause Sargassum beds in temperate waters to expand or contract? Evidence from the range shift pattern of Sargassum. Mar. Environ. Res. 2024, 200, 106659. [Google Scholar] [CrossRef] [PubMed]
- Duarte, B.; Martins, I.; Rosa, R.; Matos, A.R.; Roleda, M.Y.; Reusch, T.B.H.; Engelen, A.H.; Serrão, E.A.; Pearson, G.A.; Marques, J.C.; et al. Climate Change Impacts on Seagrass Meadows and Macroalgal Forests: An Integrative Perspective on Acclimation and Adaptation Potential. Front. Mar. Sci. 2018, 5, 190. [Google Scholar] [CrossRef]
- Yao, D.; Chen, Z.; Zhu, J.; Zou, X.; Mu, D.; Bao, S. Genetic evolution analysis of Sargassum based on cox I, ITS and rbcL gene sequences. Mol. Plant Breed. 2022, 20, 1153–1162. [Google Scholar] [CrossRef]
- Kawai, H.; Hanyuda, T.; Draisma, S.G.A.; Müller, D.G. Molecular phylogeny of Discosporangium Mesarthrocarpum (phaeophyceae) with a reinstatement of the order Discosporangiales. J. Phycol. 2007, 43, 186–194. [Google Scholar] [CrossRef]
- Ni-Ni-Win; Hanyuda, T.; Arai, S.; Uchimura, M.; Abbott, I.A.; Kawai, H. Three new records of Padina in Japan based on morphological and molecular markers. Phycol. Res. 2008, 56, 288–300. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.-Y.; Gao, F.; Jakovlić, I.; Lei, H.-P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.-T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J. STRUCTURESELECTOR: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Hoarau, G.; Coyer, J.A.; Veldsink, J.H.; Stam, W.T.; Olsen, J.L. Glacial refugia and recolonization pathways in the brown seaweed Fucus serratus. Mol. Ecol. 2007, 16, 3606–3616. [Google Scholar] [CrossRef]
- Uwai, S.; Kogame, K.; Yoshida, G.; Kawai, H.; Ajisaka, T. Geographical genetic structure and phylogeography of the Sargassum horneri/filicinum complex in Japan, based on the mitochondrial cox3 haplotype. Mar. Biol. 2009, 156, 901–911. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site | Code | Coordinates | Nh/Neh | Hd | π (×10−2) |
|---|---|---|---|---|---|
| Nanji Isl., Wenzhou | NJ | 27.442 N, 121.066 E | 2/1 | 0.517 ± 0.024 | 0.014 ± 0.001 |
| Xiaozuo, Huian | XZ | 24.959 N, 119.024 E | 1/0 | 0 | 0 |
| Liuao, Zhangpu | ZP | 23.915 N, 117.774 E | 3/0 | 0.384 ± 0.093 | 0.021 ± 0.005 |
| Nanyu Isl., Dongshan | DS | 23.722 N, 117.524 E | 5/2 | 0.639 ± 0.052 | 0.034 ± 0.004 |
| Dingpeng Isl., Nanao | DP | 23.287 N, 117.301 E | 2/1 | 0.331 ± 0.089 | 0.009 ± 0.002 |
| Shenao Bay, Nanao | NA | 23.482 N, 117.110 E | 2/1 | 0.405 ± 0.078 | 0.011 ± 0.002 |
| Pingyu Isl., Nanao | PY | 23.332 N, 117.079 E | 5/2 | 0.669 ± 0.074 | 0.042 ± 0.007 |
| Keniaowei, Huilai | KN | 23.018 N, 116.569 E | 4/2 | 0.563 ± 0.072 | 0.046 ± 0.006 |
| Zishen, Huilai | HL | 22.966 N, 116.517 E | 2/1 | 0.239 ± 0.092 | 0.006 ± 0.002 |
| Zhelang, Shanwei | SW | 22.656 N, 115.570 E | 2/0 | 0.331 ± 0.089 | 0.009 ± 0.002 |
| Yangmeikeng, Shenzhen | SZ | 22.548 N, 114.574 E | 3/2 | 0.515 ± 0.087 | 0.015 ± 0.003 |
| Wailingding Isl., Zhuhai | WL | 22.106 N, 114.022 E | 1/0 | 0 | 0 |
| Naozhou Isl., Zhanjiang | NZ | 20.917 N, 110.637 E | 4/3 | 0.251 ± 0.102 | 0.009 ± 0.004 |
| Mulan Bay, Wenchang | ML | 20.149 N, 110.690 E | 2/1 | 0.370 ± 0.084 | 0.010 ± 0.002 |
| Sitang, Xuwen | XW | 20.230 N, 110.149 E | 1/0 | 0 | 0 |
| Total | 20/16 | 0.692 ± 0.020 | 0.042 ± 0.001 |
| Among Populations/Lineages | Within Populations/Lineages | ΦST | |||
|---|---|---|---|---|---|
| d.f. | % var | d.f. | % var | ||
| Populations | |||||
| rbcL | 14 | 10.34 | 435 | 89.66 | 0.10345 *** |
| cox3 | 14 | 77.29 | 435 | 22.71 | 0.77293 *** |
| cox1 | 14 | 62.87 | 435 | 37.13 | 0.62867 *** |
| cox3–cox1–rbcL | 14 | 66.25 | 435 | 33.75 | 0.66252 *** |
| Lineages | |||||
| cox3–cox1–rbcL | 1 | 79.22 | 448 | 20.78 | 0.79218 *** |
| Tajima’s D | Fu’s FS | |
|---|---|---|
| Lineage N | −0.47261 (p = 0.31860) | −1.64597 (p = 0.25790) |
| Lineage S | −1.90073 (p = 0.00090) | −13.79697 (p = 0.00000) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Chen, W.; Du, H. Phylogeographic Pattern of Sargassum hemiphyllum var. chinense (Phaeophyceae, Ochrophyta) in Chinese Coastal Waters. Plants 2025, 14, 1269. https://doi.org/10.3390/plants14091269
Chen Z, Chen W, Du H. Phylogeographic Pattern of Sargassum hemiphyllum var. chinense (Phaeophyceae, Ochrophyta) in Chinese Coastal Waters. Plants. 2025; 14(9):1269. https://doi.org/10.3390/plants14091269
Chicago/Turabian StyleChen, Zepan, Weizhou Chen, and Hong Du. 2025. "Phylogeographic Pattern of Sargassum hemiphyllum var. chinense (Phaeophyceae, Ochrophyta) in Chinese Coastal Waters" Plants 14, no. 9: 1269. https://doi.org/10.3390/plants14091269
APA StyleChen, Z., Chen, W., & Du, H. (2025). Phylogeographic Pattern of Sargassum hemiphyllum var. chinense (Phaeophyceae, Ochrophyta) in Chinese Coastal Waters. Plants, 14(9), 1269. https://doi.org/10.3390/plants14091269