Fast Track to Discover Novel Promoters in Rice

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Summary of the Promoter Trap Line Analysis Process
2.2. Integration of Annotated Rice Genes from the Rice Genome Annotation Project
2.3. Identification of Tissue/Organ-Preferred Genes in Rice Using Meta-Expression Data
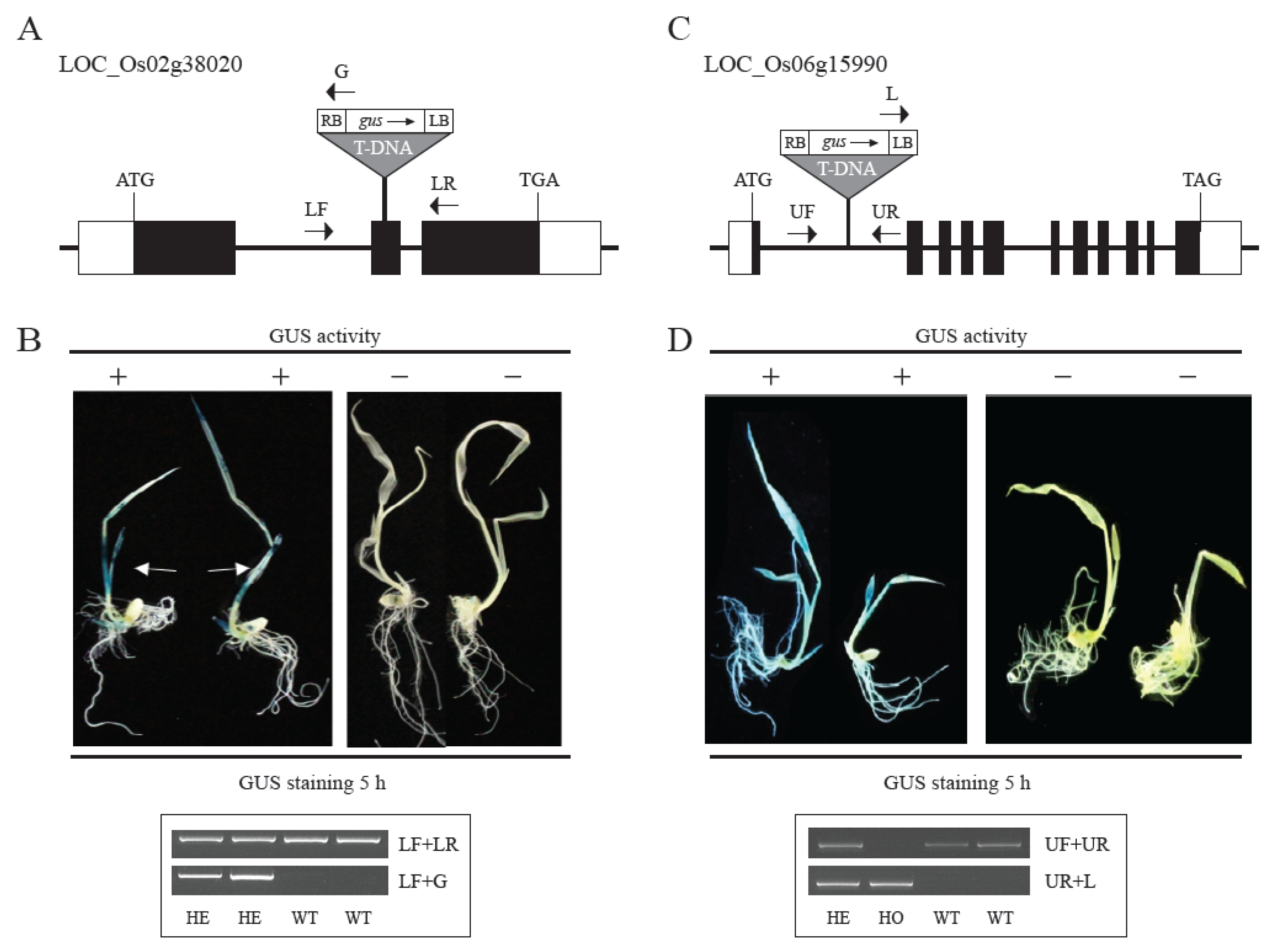
2.4. Validation of Promoters of Tissue/Organ-Preferential Genes Using the Promoter Trap System and Genotyping
2.5. Abiotic Stress Analyses of Rice Genes via Meta-Expression Data
2.6. Evaluation of Promoter Trap Lines through a Literature Search
2.7. Validation of Drought-Inducible Genes Using the GUS Reporter System and qRT-PCR
3. Materials and Methods
3.1. Integration of Whole Rice Genes from Public Data Source
3.2. Collection of Transcriptome Data
3.3. Classification of Organ-Preferential or Abiotic Stress-Responsive Gene Groups
3.4. Histochemical GUS Assay
3.5. Quantitative Real-Time PCR (qRT-PCR) Analysis
3.6. Analysis of Promoter Trap Lines via Literature Search
4. Conclusions and Future Prospects
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jung, K.H.; Hur, J.; Ryu, C.H.; Choi, Y.; Chung, Y.Y.; Miyao, A.; Hirochika, H.; An, G. Characterization of a rice chlorophyll-deficient mutant using the T-DNA gene-trap system. Plant Cell Physiol. 2003, 44, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Chandran, A.K.N.; An, G.; Lee, C.; Jung, K.H. Genome-wide analysis of root hair-preferential genes in rice. Rice 2018, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Oo, M.M.; Kim, B.; Koh, H.-J.; Oh, S.A.; Yi, G.; An, G.; Park, S.K.; Jung, K.-H. Genome-wide analyses of late pollen-preferred genes conserved in various rice cultivars and functional identification of a gene involved in the key processes of late pollen development. Rice 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.-H.; Kim, S.-R.; Giong, H.-K.; Nguyen, M.X.; Koh, H.-J.; An, G. Genome-wide identification and functional analysis of genes expressed ubiquitously in rice. Mol. Plant 2015, 8, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.-H.; Nalini Chandran, A.K.; Park, J.-C.; Gho, Y.-S.; Lee, S.-W.; An, G.; Jung, K.-H. OsPhyB-Mediating Novel Regulatory Pathway for Drought Tolerance in Rice Root Identified by a Global RNA-Seq Transcriptome Analysis of Rice Genes in Response to Water Deficiencies. Front. Plant Sci. 2017, 8, 580. [Google Scholar] [CrossRef]
- Kumar, M.; Gho, Y.S.; Jung, K.H.; Kim, S.R. Genome-wide identification and analysis of genes, conserved between japonica and indica rice cultivars, that respond to low-temperature stress at the vegetative growth stage. Front. Plant Sci. 2017, 8, 1120. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.J.; Kim, Y.J.; Chandran, A.K.N.; Jung, K.H. Infrastructures of systems biology that facilitate functional genomic study in rice. Rice 2019, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.J.; Jung, K.H. Comparative Analysis of Flanking Sequence Tags of T-DNA/Transposon Insertional Mutants and Genetic Variations of Fast-neutron Treated Mutants in Rice. J. Plant Biol. 2018, 61, 80–84. [Google Scholar] [CrossRef]
- Chandran, A.; Jung, K.-H. Resources for systems biology in rice. J. Plant Biol. 2014, 57, 80–92. [Google Scholar] [CrossRef]
- Jeong, D.H.; An, S.; Kang, H.-G.; Moon, S.; Han, J.-J.; Park, S.; Lee, H.S.; An, K.; An, G. T-DNA insertional mutagenesis for activation tagging in rice. Plant Physiol. 2002, 130, 1636–1644. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, P.; Laule, O.; Schmitz, J.; Hruz, T.; Bleuler, S.; Gruissem, W. Genevestigator transcriptome meta-analysis and biomarker search using rice and barley gene expression databases. Mol. Plant 2008, 1, 851–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.J.; Jung, K.H. Rice tissue-specific promoters and condition-dependent promoters for effective translational application. J. Integr. Plant Biol. 2015, 57, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.J.; Yoo, Y.H.; Park, S.A.; Moon, S.; Kim, S.R.; An, G.; Jung, K.H. Genome-wide identification and extensive analysis of rice-endosperm preferred genes using reference expression database. J. Plant Biol. 2017, 60, 249–258. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, A.K.N.; Yoo, Y.H.; Cao, P.; Sharma, R.; Sharma, M.; Dardick, C.; Ronald, P.C.; Jung, K.H. Updated Rice Kinase Database RKD 2.0: Enabling transcriptome and functional analysis of rice kinase genes. Rice 2016, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.H.; Hong, W.J.; Jung, K.H. A systematic view exploring the role of chloroplasts in plant abiotic stress responses. BioMed Res. Int. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Yao, W.; Li, G.; Yu, Y.; Ouyang, Y. funRiceGenes dataset for comprehensive understanding and application of rice functional genes. Gigascience 2018, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR Rice Genome Annotation Resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.; Jung, K.H.; Choi, D.; Hwang, D.; Zhu, J.; Ronald, P.C. The rice Oligonucleotide array database: An atlas of rice gene expression. Rice 2012, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.V.; Vo, K.T.X.; Hong, W.J.; Jung, K.H.; Jeon, J.S. Defense Response to Pathogens Through Epigenetic Regulation in Rice. J. Plant Biol. 2018, 61, 1–10. [Google Scholar] [CrossRef]
- Yu, G.H.; Huang, S.C.; He, R.; Li, Y.Z.; Cheng, X.G. Transgenic Rice Overexperessing a Tomato Mitochondrial Phosphate Transporter, SlMPT3;1, Promotes Phosphate Uptake and Increases Grain Yield. J. Plant Biol. 2018, 61, 383–400. [Google Scholar] [CrossRef]
- Jain, M.; Nijhawan, A.; Tyagi, A.K.; Khurana, J.P. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2006, 345, 646–651. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Expression Pattern | Locus_ID | T-DNA Line No. | Putative Function | DOI References a |
|---|---|---|---|---|---|
| Anatomy | Leaf/Flag leaf/Shoot | LOC_Os03g20700 | 9-07117 | Magnesium chelatase | 10.1093/pcp/pcg064 |
| Root (root hair) | LOC_Os05g45900 | 3A-00457 | endonuclease/exonuclease/phosphatase family domain-containing protein | 10.1186/s12284-018-0241-2 | |
| LOC_Os10g42750 | 2B-60199 | CSLD1, cellulose synthase-like family D | |||
| LOC_Os12g02240 | 4A-50567 | expressed protein | |||
| Seed/Embryo/Endosperm | LOC_Os05g05790 | 1A-10540 | double-stranded RNA binding motif-containing protein | 10.1104/pp.014357 | |
| Anther/Pollen | LOC_Os11g20384 | 1A-13819 | SacI homology domain-containing protein | 10.1186/s12284-018-0219-0 | |
| LOC_Os07g17310 | 2D-41188 | B12D protein | |||
| Ubiquitous | LOC_Os03g01910 | 4A-04197 | transcription factor BTF3 | 10.1016/j.molp.2014.10.013 | |
| LOC_Os03g08010 | 5A-00191 | elongation factor Tu | |||
| LOC_Os04g42090 | 3A-05916 | CPuORF7, conserved peptide uORF-containing transcript | |||
| LOC_Os06g30750 | 2D-00098 | reticulon domain-containing protein | |||
| Abiotic stress | Drought | LOC_Os04g52290 | 3A-03417 | PPR repeat domain-containing protein | 10.3389/fpls.2017.00580 |
| LOC_Os07g02710 | 3A-13738 | expressed protein | |||
| Cold | LOC_Os01g31370 | 3A-50649 | glycosyltransferase | 10.3389/fpls.2017.01120 | |
| LOC_Os03g49830 | 1C-08613 | expressed protein |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, Y.-H.; Kim, Y.-J.; Moon, S.; Gho, Y.-S.; Hong, W.-J.; Kim, E.-J.; Jiang, X.; Jung, K.-H. Fast Track to Discover Novel Promoters in Rice. Plants 2020, 9, 125. https://doi.org/10.3390/plants9010125
Yoo Y-H, Kim Y-J, Moon S, Gho Y-S, Hong W-J, Kim E-J, Jiang X, Jung K-H. Fast Track to Discover Novel Promoters in Rice. Plants. 2020; 9(1):125. https://doi.org/10.3390/plants9010125
Chicago/Turabian StyleYoo, Yo-Han, Yu-Jin Kim, Sunok Moon, Yun-Shil Gho, Woo-Jong Hong, Eui-Jung Kim, Xu Jiang, and Ki-Hong Jung. 2020. "Fast Track to Discover Novel Promoters in Rice" Plants 9, no. 1: 125. https://doi.org/10.3390/plants9010125