Applications and Trends of Machine Learning in Genomics and Phenomics for Next-Generation Breeding



Abstract
:1. Introduction
2. Genomics Applied to Breeding: What We Gain from Short Reads
3. Third Era of Generation Sequencing: The Impact on Plant Genetics
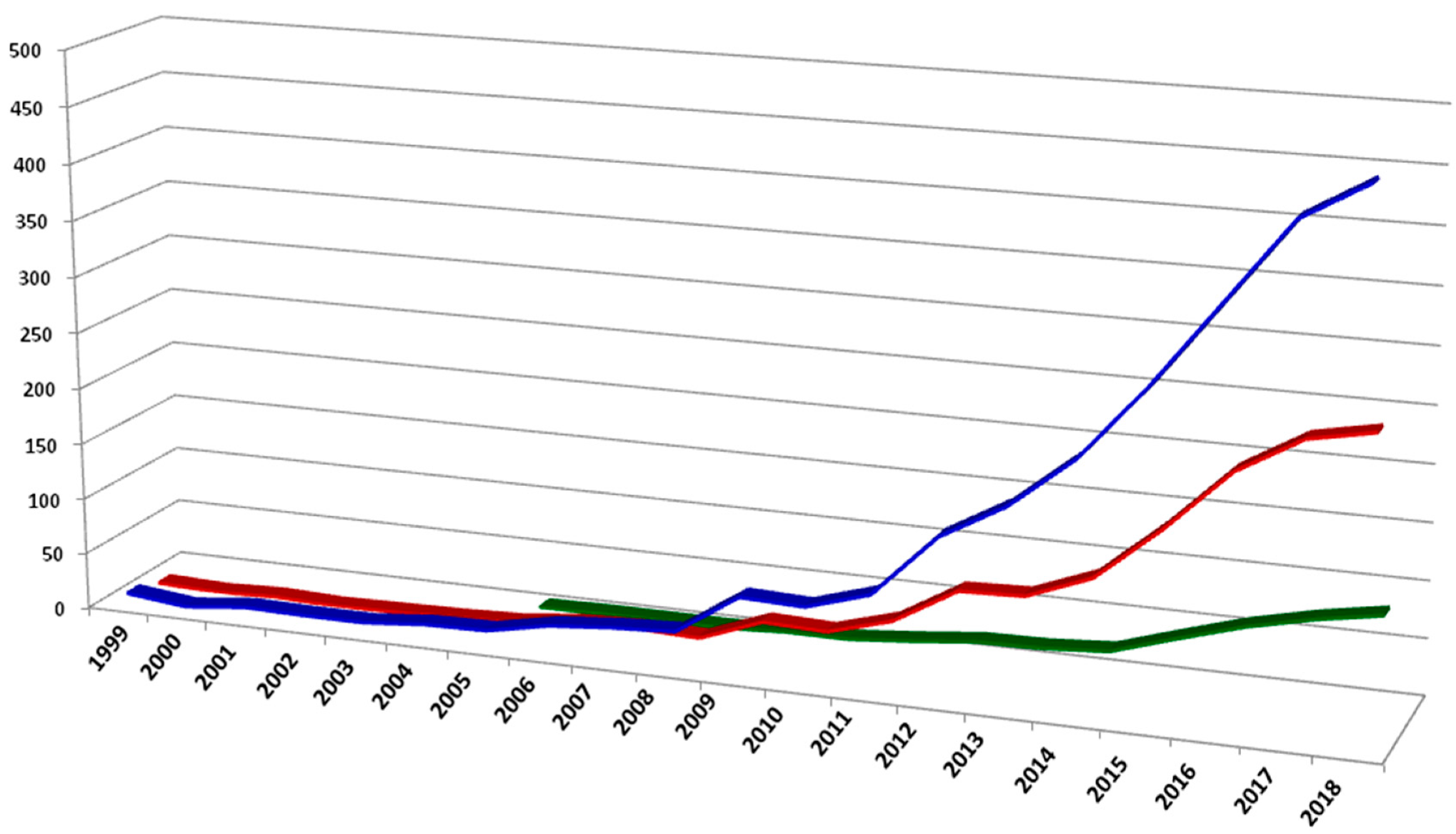
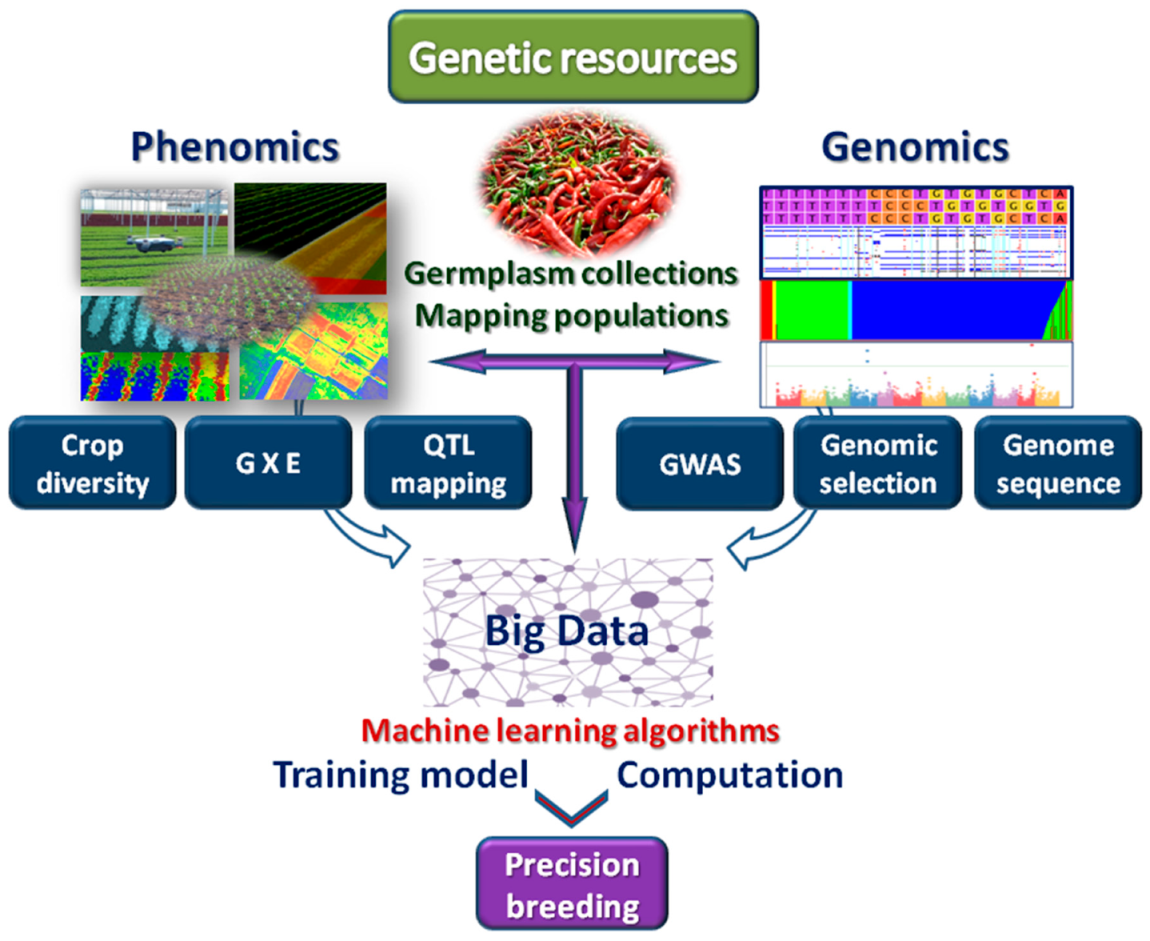
4. Machine Learning for Genomic Studies
5. Machine Learning for Plant Phenomics and Smart Agriculture
6. Machine Learning for Next-Generation Breeding
7. Machine Learning and Big Data Management
8. Genomics and Machine Learning: A Case Study to Predict Differentially Expressed miRNA
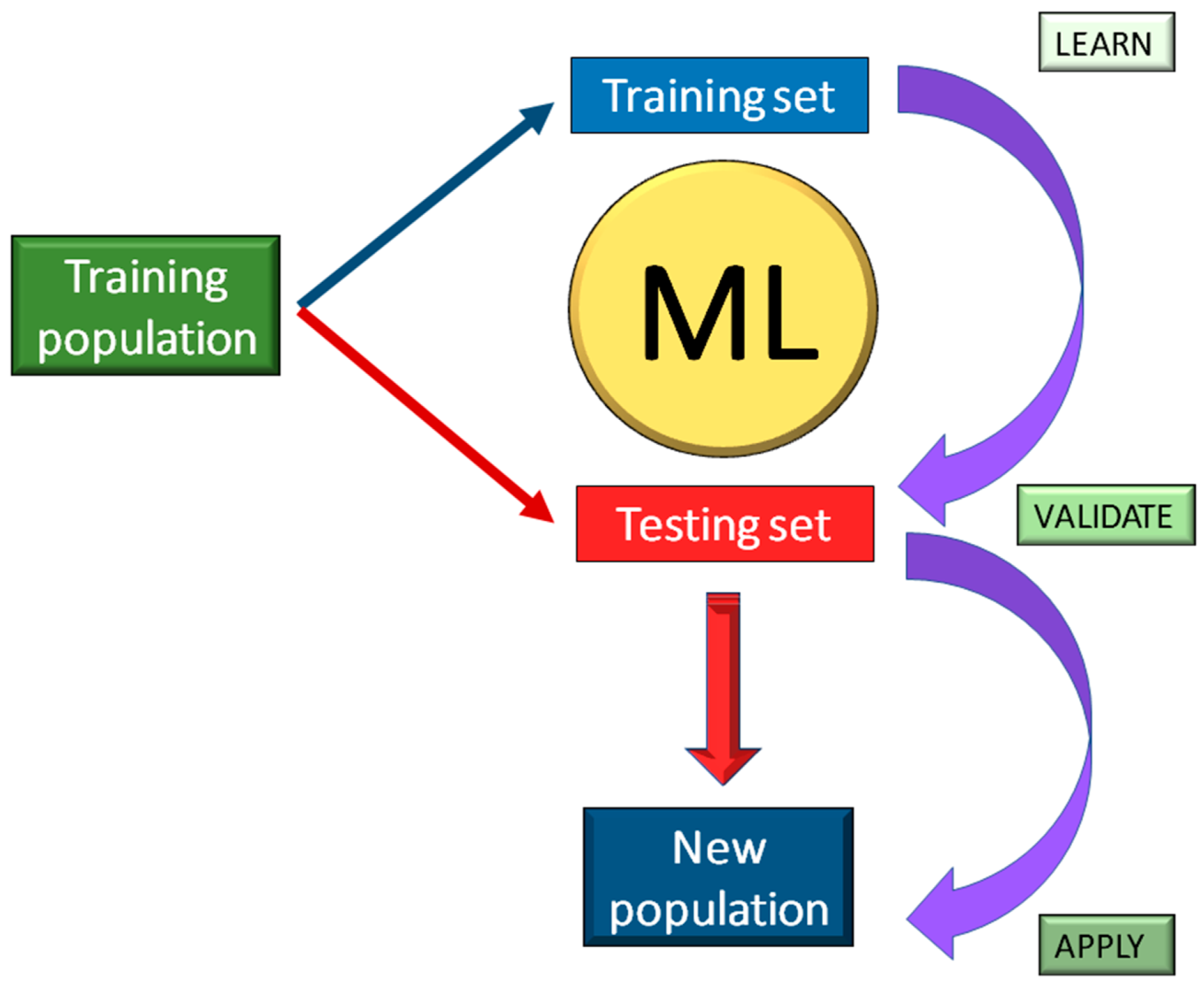
8.1. Materials and Methods
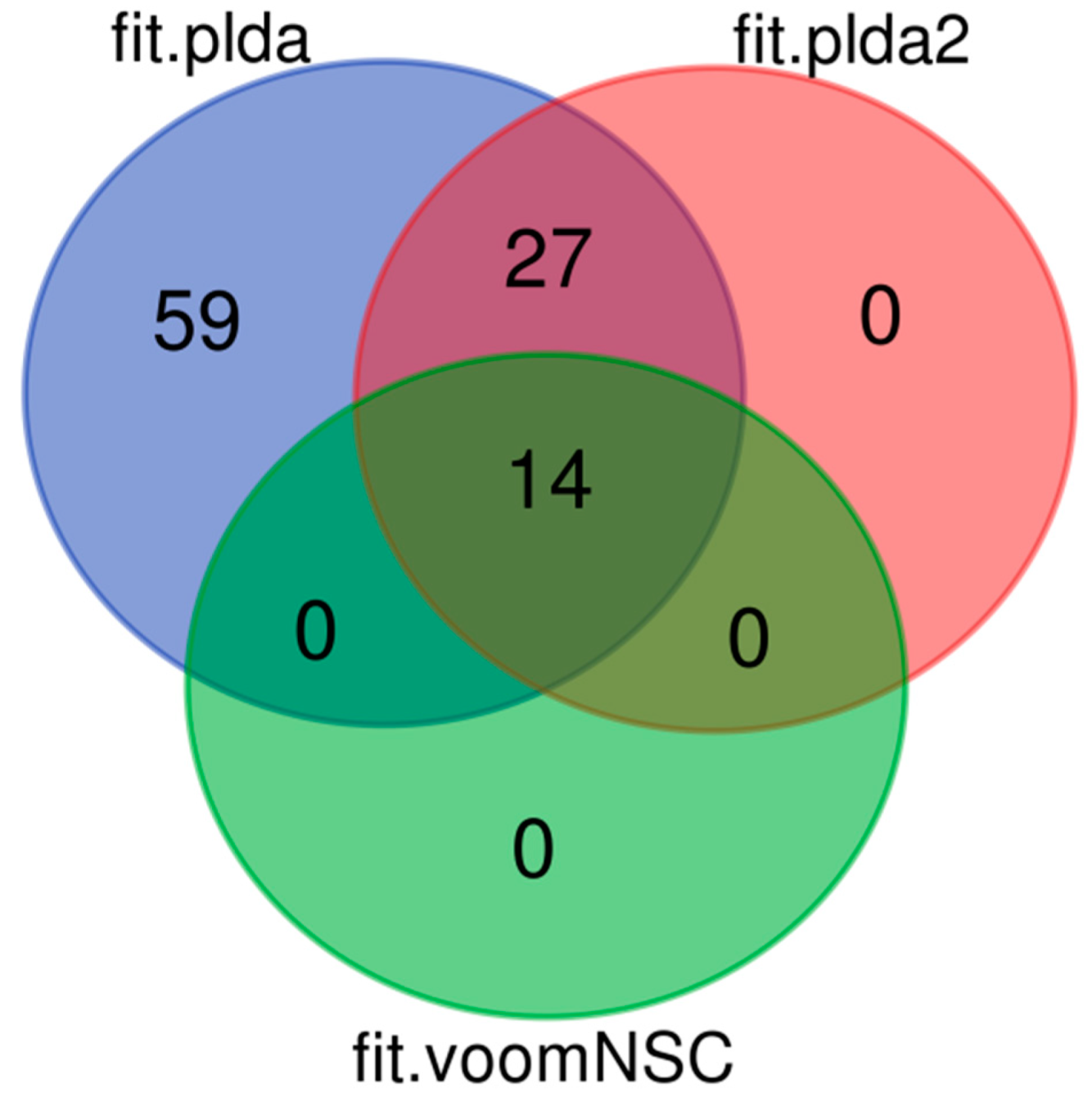
8.2. Results
9. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keating, B.A.; Herrero, M.; Carberry, P.S.; Gardner, J.; Cole, M.B. Food wedges: Framing the global food demand and supply challenge towards 2050. Glob. Food Secur. 2014, 3, 125–132. [Google Scholar] [CrossRef]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannarozzi, G.; Plaza-Wuthrich, S.; Esfeld, K.; Larti, S.; Wilson, Y.S.; Girma, D.; de Castro, E.; Chanyalew, S.; Blosch, R.; Farinelli, L.; et al. Genome and transcriptome sequencing identifies breeding targets in the orphan crop tef (Eragrostis tef). BMC Genom. 2014, 15, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Boil. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drovandi, C.C.; Holmes, C.; McGree, J.M.; Mengersen, K.; Richardson, S.; Ryan, E.G. Principles of experimental design for big data analysis. Stat. Sci. 2017, 3, 385–404. [Google Scholar] [CrossRef] [Green Version]
- Cobb, J.N.; Biswas, P.S.; Platten, J.D. Back to the future: Revisiting MAS as a tool for modern plant breeding. Theor. Appl. Genet. 2018, 132, 647–667. [Google Scholar] [CrossRef] [Green Version]
- Bedre, R.; Irigoyen, S.; Petrillo, E.; Mandadi, K.K. New Era in Plant Alternative Splicing Analysis Enabled by Advances in High-Throughput Sequencing (HTS) Technologies. Front. Plant Sci. 2019, 10, 740. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Poorter, H.; Dumschott, K.; Bolger, M.E.; Arend, D.; Osorio, S.; Gundlach, H.; Mayer, K.F.X.; Lange, M.; Scholz, U.; et al. Computational aspects underlying genome to phenome analysis in plants. Plant J. 2018, 97, 182–198. [Google Scholar] [CrossRef]
- Joshi, D.C.; Chaudhari, G.V.; Sood, S.; Kant, L.; Pattanayak, A.; Zhang, K.; Fan, Y.; Janovska, D.; Meglic, V.; Zhou, M. Revisiting the versatile buckwheat: Reinvigorating genetic gains through integrated breeding and genomics approach. Planta 2019, 250, 783–801. [Google Scholar] [CrossRef]
- Lobos, G.A.; Camargo, A.V.; Del Pozo, A.; Araus, J.L.; Ortiz, R.; Doonan, J.H. Plant phenotyping and phenomics for plant breeding. Front. Plant Sci. 2017, 8, 2181. [Google Scholar] [CrossRef] [Green Version]
- Das Choudhury, S.; Samal, A.; Awada, T. Leveraging image analysis for High-Throughput plant phenotyping. Front. Plant Sci. 2019, 10, 508. [Google Scholar] [CrossRef] [PubMed]
- Van Emon, J. Omics revolution in agricultural research. J. Agric. Food. Chem. 2016, 64, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Argueso, C.T.; Assmann, S.M.; Birnbaum, K.D.; Chen, S.; Dinneny, J.R.; Doherty, C.J.; Eveland, A.L.; Friesner, J.; Greenlee, V.R.; Law, J.A.; et al. Directions for research and training in plant omics: Big Questions and Big Data. Plant Direct 2019, 3, e00133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, V.; D’Agostino, N.; Garrison, E.; Albrechtsen, A.; Meisner, J.M.; Facchiano, A.; Cardi, T.; Tripodi, P. Genomic diversity and novel genome-wide association with fruit morphology in Capsicum, from 746k polymorphic sites. Sci. Rep. 2019, 9, 10067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruana, B.M.; Pembleton, L.W.; Constable, F.; Rodoni, B.; Slater, A.T.; Cogan, N.O.I. Validation of genotyping by sequencing using transcriptomics for diversity and application of genomic selection in tetraploid potato. Front. Plant Sci. 2019, 10, 670. [Google Scholar] [CrossRef] [Green Version]
- Sim, S.C.; Durstewitz, G.; Plieske, J.; Wieseke, R.; Ganal, M.W.; Van Deynze, A.; Hamilton, J.P.; Buell, C.R.; Causse, M.; Wijeratne, S.; et al. Development of a large SNP genotyping array and generation of high-density genetic maps in tomato. PLoS ONE 2012, 7, e40563. [Google Scholar] [CrossRef]
- FAOSTAT. Food and Agriculture Organization of the United Nations. 2017. Available online: http://faostat3.fao.org/home/ (accessed on 24 December 2019).
- Hirakawa, H.; Shirasawa, K.; Ohyama, A.; Fukuoka, H.; Aoki, K.; Rothan, C.; Sato, S.; Isobe, S.; Tabata, S. Genome-wide SNP genotyping to infer the effects on gene functions in tomato. DNA Res. 2013, in press. [Google Scholar] [CrossRef] [Green Version]
- Gonda, I.; Ashrafi, H.; Lyon, D.A.; Strickler, S.R.; Hulse-Kemp, A.M.; Ma, Q.; Sun, H.; Stoffel, K.; Powell, A.F.; Futrell, S.; et al. Sequencing-based bin map construction of a tomato mapping population, facilitating high-resolution quantitative trait loci detection. Plant Genome 2019, 12, 180010. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Liu, C.; Chen, K. Assessment of Genetic Differentiation and Linkage Disequilibrium in Solanum pimpinellifolium using genome-wide high-density SNP markers. G3 Genes Genomes Genet. 2019, 9, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Barchi, L.; Acquadro, A.; Alonso, D.; Aprea, G.; Bassolino, L.; Demurtas, O.; Ferrante, P.; Gramazio, P.; Mini, P.; Portis, E.; et al. Single Primer Enrichment Technology (SPET) for High-Throughput Genotyping in Tomato and Eggplant Germplasm. Front. Plant Sci. 2019, 10, 1005. [Google Scholar] [CrossRef] [Green Version]
- Devran, Z.; Kahveci, E.; Ozkaynak, E.; Studholme, D.J.; Tor, M. Development of molecular markers tightly linked to Pvr4 gene in pepper using next-generation sequencing. Mol. Breed. 2015, 35, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastien, M.; Boudhrioua, C.; Fortin, G.; Belzile, F. Exploring the potential and limitations of genotyping-by-sequencing for SNP discovery and genotyping in tetraploid potato. Genome 2018, 61, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khlestkin, V.K.; Rozanova, I.V.; Efimov, V.M.; Khlestkina, E.K. Starch phosphorylation associated SNPs found by genome-wide association studies in potato (Solanum tuberosum L.). BMC Genet. 2019, 20 (Suppl. 1). [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oladzad, A.; Porch, T.; Rosas, J.C.; Mafi Moghaddam, S.; Beaver, J.; Beebe, S.; Burridge, J.; Jochua, C.N.; Miguel, M.A.; Miklas, P.N.; et al. Single and multi-trait GWAS identify genetic factors associated with production traits in common bean under abiotic stress environments. G3 (Bethesda) 2019, 9, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Gupta, S.; Bandhiwal, N.; Kumar, T.; Bharadwaj, C.; Bhatia, S. High-density linkage map construction and mapping of seed trait QTLs in chickpea (Cicer arietinum L.) using Genotyping-by-Sequencing (GBS). Sci. Rep. 2015, 5, 17512. [Google Scholar] [CrossRef] [Green Version]
- Amalraj, A.; Taylor, J.; Bithell, S.; Li, Y.; Moore, K.; Hobson, K.; Sutton, T. Mapping resistance to Phytophthora root rot identifies independent loci from cultivated (Cicer arietinum L.) and wild (Cicer echinospermum PH Davis) chickpea. Theor. Appl. Genet. 2018, 132, 1017–1033. [Google Scholar] [CrossRef]
- Saintenac, C.; Lee, W.S.; Cambon, F.; Rudd, J.J.; King, R.C.; Marande, W.; Powers, S.J.; Berges, H.; Phillips, A.L.; Uauy, C.; et al. Wheat receptor-kinase-like protein Stb6 controls gene-for-gene resistance to fungal pathogen Zymoseptoria tritici. Nat. Genet. 2018, 50, 368–374. [Google Scholar] [CrossRef]
- Huang, B.E.; George, A.W.; Forrest, K.L.; Kilian, A.; Hayden, M.J.; Morell, M.K.; Cavanagh, C.R. A multiparent advanced generation inter-cross population for genetic analysis in wheat. Plant Biotechnol. J. 2012, 10, 826–839. [Google Scholar] [CrossRef]
- Mackay, I.J.; Bansept-Basler, P.; Barber, T.; Bentley, A.R.; Cockram, J.; Gosman, N.; Greenland, A.J.; Horsnell, R.; Howells, R.; O’Sullivan, D.M.; et al. An eight-parent multiparent advanced generation inter-cross population for winter-sown wheat: Creation, properties, and validation. G3 (Bethesda) 2014, 4, 1603. [Google Scholar] [CrossRef] [Green Version]
- Milner, S.G.; Maccaferri, M.; Huang, B.E.; Mantovani, P.; Massi, A.; Fras-caroli, E.; Tuberosa, R.; Salvi, S. A multiparental cross population for mapping QTL for agronomic traits in durum wheat (Triticum turgidum ssp. durum). Plant Biotechnol. J. 2015, 14, 735–748. [Google Scholar] [CrossRef] [Green Version]
- Dixon, L.E.; Greenwood, J.R.; Bencivenga, S.; Zhang, P.; Cockram, J.; Mellers, G.; Ramm, K.; Cavanagh, C.; Swain, S.M.; Boden, S.A. TEOSINTE BRANCHED 1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum L.). Plant Cell 2018, 30, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, S.; Dreisigacker, S.; Lopes, M.; Chavez, P.; Reynolds, M.P. Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor. Appl. Genet. 2015, 128, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Maccaferri, M.; Bulli, P.; Rynearson, S.; Tuberosa, R.; Chen, X.; Pumphrey, M. Genome-wide association mapping for seedling and field resistance to Puccinia striiformis. sp. tritici in elite durum wheat. Theor. Appl. Genet. 2017, 130, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Tian, Z.; Rao, Y.; Dong, G.; Yang, Y.; Huang, L.; Leng, Y.; Xu, J.; Sun, C.; Zhang, G.; et al. Rational design of high-yield and superior-quality rice. Nat. Plants 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2017, 7, e32253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkoski, J.E.; Poland, J.; Jannink, J.-L.; Sorrells, M.E. Imputation of unordered markers and the impact on genomic selection accuracy. G3 (Bethesda) 2013, 3, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spindel, J.E.; Begum, H.; Akdemir, D.; Collard, B.; Redona, E.; Jannink, J.L.; McCouch, S. Genome-wide prediction models that incorporate de-novo GWAS are a powerful new tool for tropical rice improvement. Heredity 2016, 116, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, R.; Yu, J. Prospects for genome wide selection for quantitative trait in maize. Crop Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Gen. 2011, 4, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-based fourth-generation DNA sequencing technology. Genom. Proteom. Bioinform. 2015, 13, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Park, S.T.; Kim, J. Trends in next-generation sequencing and a new era for whole genome sequencing. Int. Neurourol. J. 2016, 20, S76–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heng, J.B.; Ho, C.; Kim, T.; Timp, R.; Aksimentiev, A.; Grinkova, Y.V.; Sligar, S.; Schulten, K.; Timp, G. Sizing DNA using a nanometer-diameter pore. Biophys. J. 2004, 87, 2905–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabil, G.; Rubio, B.; Bert, P.F. De novo phased assembly of the Vitis riparia grape genome. Sci. Data 2019, 6, 127. [Google Scholar] [CrossRef] [Green Version]
- Minoche, A.E.; Dohm, J.C.; Himmelbauer, H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biol. 2011, 12, R112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J. Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 2018, 20, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.E.; Staber, C.; Zeitlinger, J.; Hawley, R.S. Highly contiguous genome assemblies of 15 Drosophila species generated using nanopore sequencing. G3 (Bethesda) 2018, 8, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Boweden, R.; Davies, R.W.; Heger, A.; Pagnamenta, A.T.; De Cesare, M.; Oikkonen, L.E.; Parkes, D.; Freeman, C.; Dhalla, F.; Patel, S.Y.; et al. Sequencing of human genomes with nanopore technology. Nat. Commun. 2019, 10, 1869. [Google Scholar] [CrossRef]
- Miga, K.H. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2018, 51, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Wittenberg, A. PromethION Sequencing of Complex Plant Genomes. Presentation. Available online: https://nanoporetech.com/resource-centre/talk/promethion-sequencing-complex-plant-genomes (accessed on 24 December 2019).
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct Determination of Diploid Genome Sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Lind, A.L.; Lai, Y.Y.Y.; Mostovoy, Y.; Holloway, A.K.; Iannucci, A.; Mak, A.C.Y.; Fondi, M.; Orlandini, V.; Eckalbar, W.L.; Milan, M.; et al. Genome of the Komodo dragon reveals adaptations in the cardiovascular and chemosensory systems of monitor lizards. Nat. Ecol. Evol. 2019, 3, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Sedlazeck, F.J.; Lam, T.W.; Schatz, M.C. A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nat. Commun. 2019, 10, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. Creating a universal SNP and small indel variant caller with deep neural networks. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Schrider, D.R.; Kern, A.D. Supervised machine learning for population genetics: A new paradigm. Trends Genet. 2018, 34, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Vara, C.; Paytuví-Gallart, A.; Cuartero, Y.; Le Dily, F.; Garcia, F.; Salvà-Castro, J.; Gómez-H, L.; Julià, E.; Moutinho, C.; Aiese Cigliano, R.; et al. Three-dimensional genomic structure and cohesin occupancy correlate with transcriptional activity during spermatogenesis. Cell Rep. 2019, 28, 352–367. [Google Scholar] [CrossRef]
- Tripodi, P.; Massa, D.; Venezia, A.; Cardi, T. Sensing Technologies for Precision Phenotyping in Vegetable Crops: Current Status and Future Challenges. Agronomy 2018, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Shan, C. Learning local binary patterns for gender classification on real-world face images. Pattern Recognit. Lett. 2012, 33, 431–437. [Google Scholar] [CrossRef]
- Gaonkar, B.; Davatzikos, C. Analytic estimation of statistical significance maps for support vector machine based multi-variate image analysis and classification. Neuroimage 2013, 10, 78270–78283. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, 29. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, S.; Blystone, D.; Singh, A.K.; Ganapathysubramanian, B.; Singh, A.; Sarkar, S. An explainable deep machine vision framework for plant stress phenotyping. Proc. Natl. Acad. Sci. USA 2018, 115, 4613–4618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, U.; Chang, S.; Putra, G.A.; Kim, H.; Kim, D.H. An automated, high-throughput plant phenotyping system using machine learning-based plant segmentation and image analysis. PLoS ONE 2018, 13, e0196615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.; Carriquiry, A.L.; Beavis, W.D. Parametric and nonparametric statistical methods for genomic selection of traits with additive and epistatic genetic architectures. G3 (Bethesda) 2014, 4, 1027–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CASSAVABASE. Available online: https://cassavabase.org/solgs (accessed on 24 December 2019).
- NCBI Website. Available online: https://www.ncbi.nlm.nih.gov/sra (accessed on 24 December 2019).
- AMAZON Website. Available online: http://aws.amazon.com/1000genomes (accessed on 24 December 2019).
- Esposito, S.; Aversano, R.; D’amelia, V.; Villano, C.; Alioto, D.; Mirouze, M.; Carputo, D. Dicer-like and RNA-dependent RNA polymerase gene family identification and annotation in the cultivated Solanum tuberosum and its wild relative S. commersonii. Planta 2018, 248, 729–743. [Google Scholar] [CrossRef]
- Carputo, D.; Castaldi, L.; Caruso, I.; Aversano, R.; Monti, L.; Frusciante, L. Resistance to frost and tuber soft rot in near-pentaploid Solanum tuberosum–S. commersonii hybrids. Breed. Sci. 2007, 57, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Folgado, R.; Panis, B.; Sergeant, K.; Renaut, J.; Swennen, R.; Hausman, J.F. Differential protein expression in response to abiotic stress in two potato species: Solanum commersonii Dun. and Solanum tuberosum L. Int. J. Mol. Sci. 2013, 14, 4912–4933. [Google Scholar] [CrossRef] [Green Version]
- Puigvert, M.; Guarischi-Sousa, R.; Zuluaga, P.; Coll, N.S.; Macho, A.P.; Setubal, J.C.; Valls, M. Transcriptomes of Ralstonia solanacearum during root colonization of Solanum commersonii. Front. Plant Sci. 2017, 8, 370. [Google Scholar] [CrossRef] [Green Version]
- Aversano, R.; Contaldi, F.; Ercolano, M.R.; Grosso, V.; Iorizzo, M.; Tatino, F.; Xumerle, L.; Molin, A.D.; Avanzato, C.; Ferrarini, A.; et al. The Solanum commersonii genome sequence provides insights into adaptation to stress conditions and genome evolution of wild potato relatives. Plant Cell 2015, 27, 954–968. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, L.A.; Murphy, P.R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunkar, R.; Zhou, X.; Zheng, Y.; Zhang, W.; Zhu, J.K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Zhang, M.; Zhao, Y.; He, X.; Ding, C.; Wang, S.; Feng, Y.; Song, X.; Li, P.; Wang, B. Identification of salt tolerance-related microRNAs and their targets in Maize (Zea mays L.) using high-throughput sequencing and degradome analysis. Front. Plant Sci. 2017, 8, 864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Liu, Y.; Liu, Z.; Kong, D.; Duan, M.; Luo, L. Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 2010, 61, 4157–4168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kumari, S.; Zhang, L.; Zheng, Y.; Ware, D. Characterization of miRNAs in response to short-term waterlogging in three inbred lines of Zea mays. PLoS ONE 2012, 7, e39786. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, G.; Zhang, W. UV-B responsive microRNA genes in Arabidopsis thaliana. Mol. Syst. Biol. 2007, 3, 103. [Google Scholar] [CrossRef]
- Esposito, S.; Aversano, R.; Bradeen, J.M.; Di Matteo, A.; Villano, C.; Carputo, D. Deep-sequencing of Solanum commersonii small RNA libraries reveals riboregulators involved in cold stress response. Plant Biol. 2019, in press. [Google Scholar] [CrossRef]
- Dong, C.H.; Pei, H. Over-expression of miR397 improves plant tolerance to cold stress in Arabidopsis thaliana. J. Plant Biol. 2014, 57, 209–217. [Google Scholar] [CrossRef]
- Song, J.B.; Gao, S.; Wang, Y.; Li, B.W.; Zhang, Y.L.; Yang, Z.M. miR394 and its target gene LCR are involved in cold stress response in Arabidopsis. Plant Gene 2016, 5, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.T.; Sun, X.L.; Hoshino, Y.; Yu, Y.; Jia, B.; Sun, Z.W.; Sun, M.Z.; Duan, X.B.; Zhu, Y.M. MicroRNA319 positively regulates cold tolerance by targeting OsPCF6 and OsTCP21 in rice (Oryza sativa L.). PLoS ONE 2014, 9, e91357. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Luan, Y.; Zhai, J. Sp-miR396a-5p acts as a stress-responsive genes regulator by conferring tolerance to abiotic stresses and susceptibility to Phytophthora nicotianae infection in transgenic tobacco. Plant Cell Rep. 2015, 34, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Burd, S.; Lers, A. miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015, 84, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Goksuluk, D.; Zararsiz, G.; Korkmaz, S.; Eldem, V.; Klaus, B.; Ozturk, A.; Karaagaoglu, A.E. MLSeq: Machine Learning Interface to RNA-Seq Data. Comput. Methods Programs Biomed. 2019, 175, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Oono, Y.; Seki, M.; Satou, M.; Iida, K.; Akiyama, K.; Sakurai, T.; Fujita, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Monitoring expression profiles of Arabidopsis genes during cold acclimation and deacclimation using DNA microarrays. Funct. Integr. Genom. 2006, 6, 212–234. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Zeng, Y.; Liu, L.; Huang, Y.; Zhao, E.; Zhang, F. Overexpression of the halophyte Kalidium foliatum H+-pyrophosphatase gene confers salt and drought tolerance in Arabidopsis thaliana. Mol. Biol. Rep. 2012, 39, 7989–7996. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kabbage, M.; Liu, W.; Dickman, M.B. Aspartyl Protease-Mediated cleavage of BAG6 is necessary for autophagy and fungal resistance in plants. Plant Cell 2016, 28, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, L.; Gupta, S.; Mishra, S.K.; Pandey, G.; Kumar, S.; Chauhan, P.S.; Chakrabarty, D.; Nautiyal, C.S. Elucidation of complex nature of peg induced drought-stress response in rice root using comparative proteomics approach. Front. Plant Sci. 2016, 7, 1466. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Data | S | R | S | R | S | S | R | S | S | R | R | Model Accuracy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SVM | S | R | S | R | S | S | R | R | S | R | R | 0.89 |
| NSC | S | R | S | R | S | S | R | S | S | R | R | 0.96 |
| PLDA | S | R | S | R | S | S | R | S | S | R | R | 0.93 |
| PLDA2 | S | R | S | R | S | S | R | S | S | R | R | 0.96 |
| VoomDLDA | S | R | S | R | S | S | R | S | S | R | R | 0.95 |
| VoomNSC | S | R | S | R | S | S | R | S | S | R | R | 0.96 |
| VoomNBLDA | S | R | S | R | S | S | R | S | S | R | R | 0.95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, S.; Carputo, D.; Cardi, T.; Tripodi, P. Applications and Trends of Machine Learning in Genomics and Phenomics for Next-Generation Breeding. Plants 2020, 9, 34. https://doi.org/10.3390/plants9010034
Esposito S, Carputo D, Cardi T, Tripodi P. Applications and Trends of Machine Learning in Genomics and Phenomics for Next-Generation Breeding. Plants. 2020; 9(1):34. https://doi.org/10.3390/plants9010034
Chicago/Turabian StyleEsposito, Salvatore, Domenico Carputo, Teodoro Cardi, and Pasquale Tripodi. 2020. "Applications and Trends of Machine Learning in Genomics and Phenomics for Next-Generation Breeding" Plants 9, no. 1: 34. https://doi.org/10.3390/plants9010034
APA StyleEsposito, S., Carputo, D., Cardi, T., & Tripodi, P. (2020). Applications and Trends of Machine Learning in Genomics and Phenomics for Next-Generation Breeding. Plants, 9(1), 34. https://doi.org/10.3390/plants9010034