Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome


Abstract
:1. Introduction
2. Results
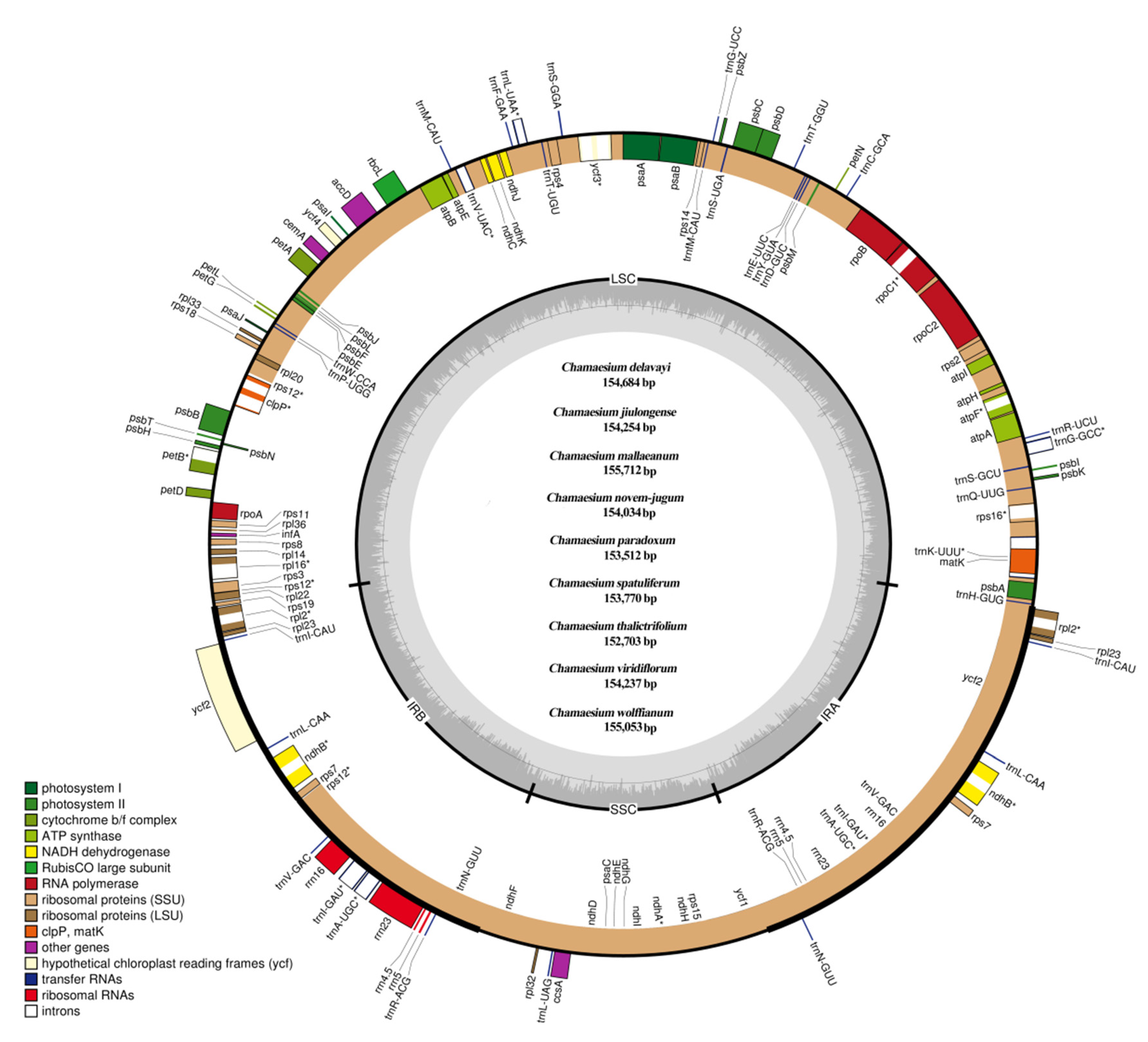
2.1. The Plastid Genome of Chamaesium Species
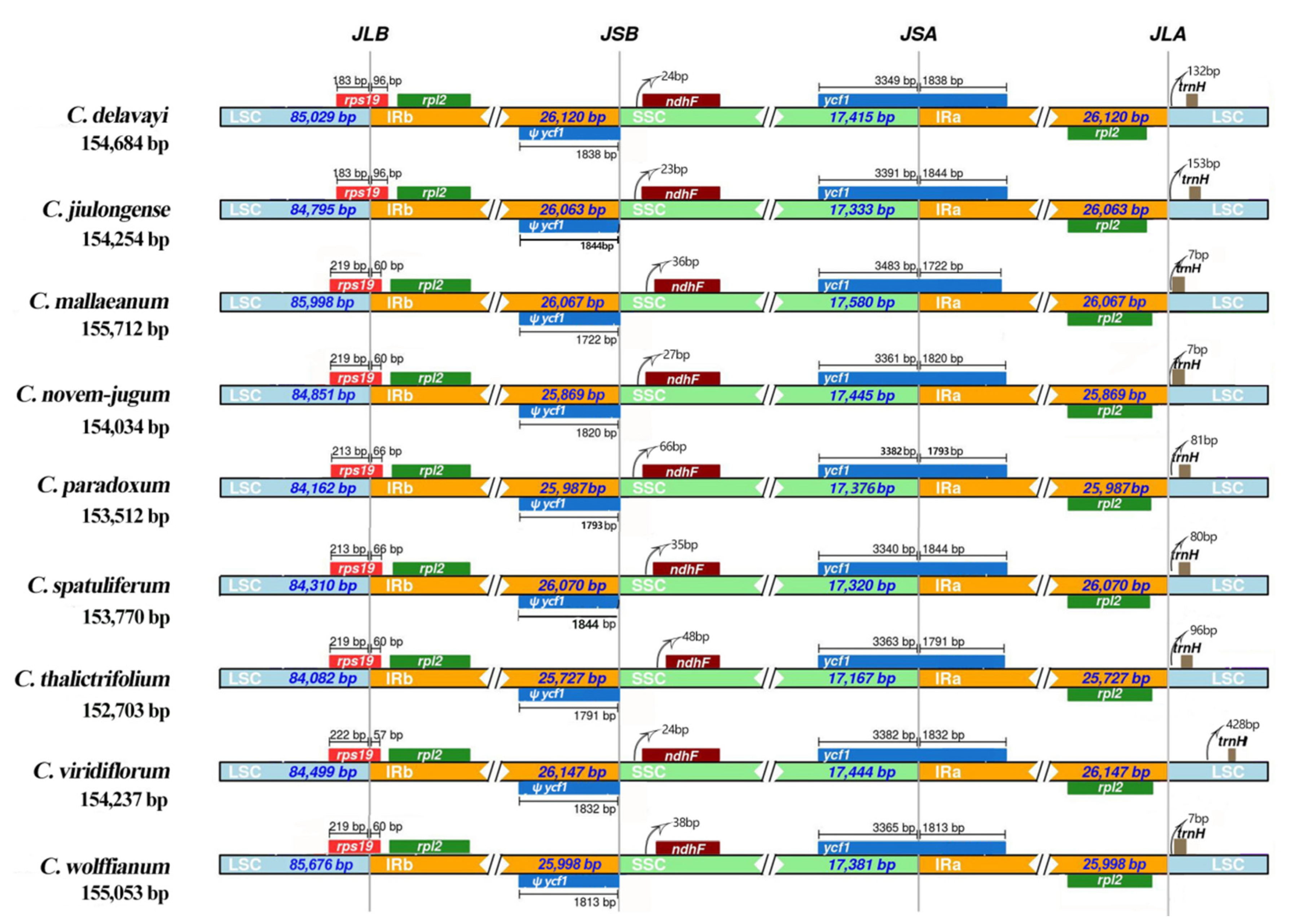
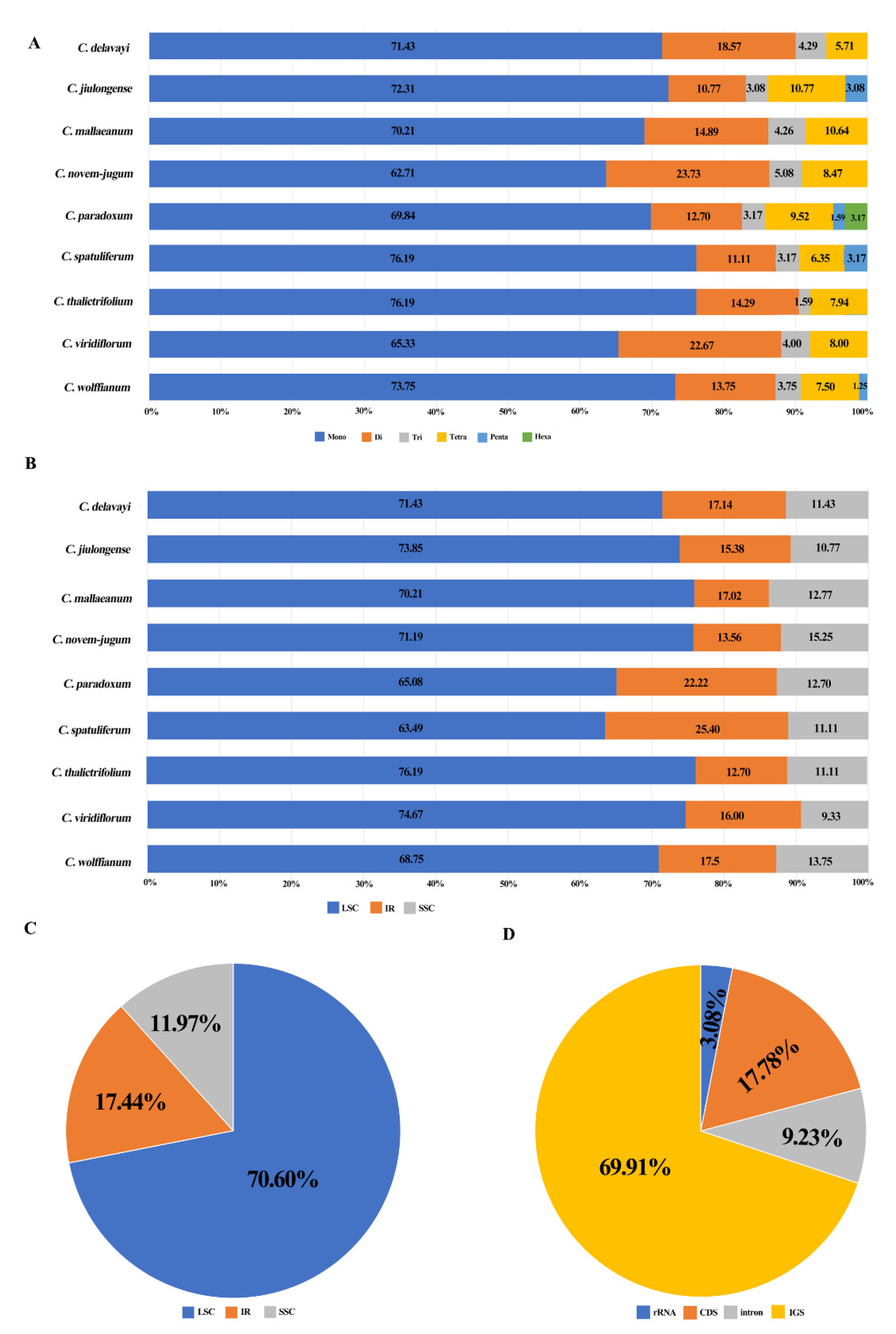
2.2. Contraction and Expansion of IRs and Simple Sequence Repeat (SSR) Analysis
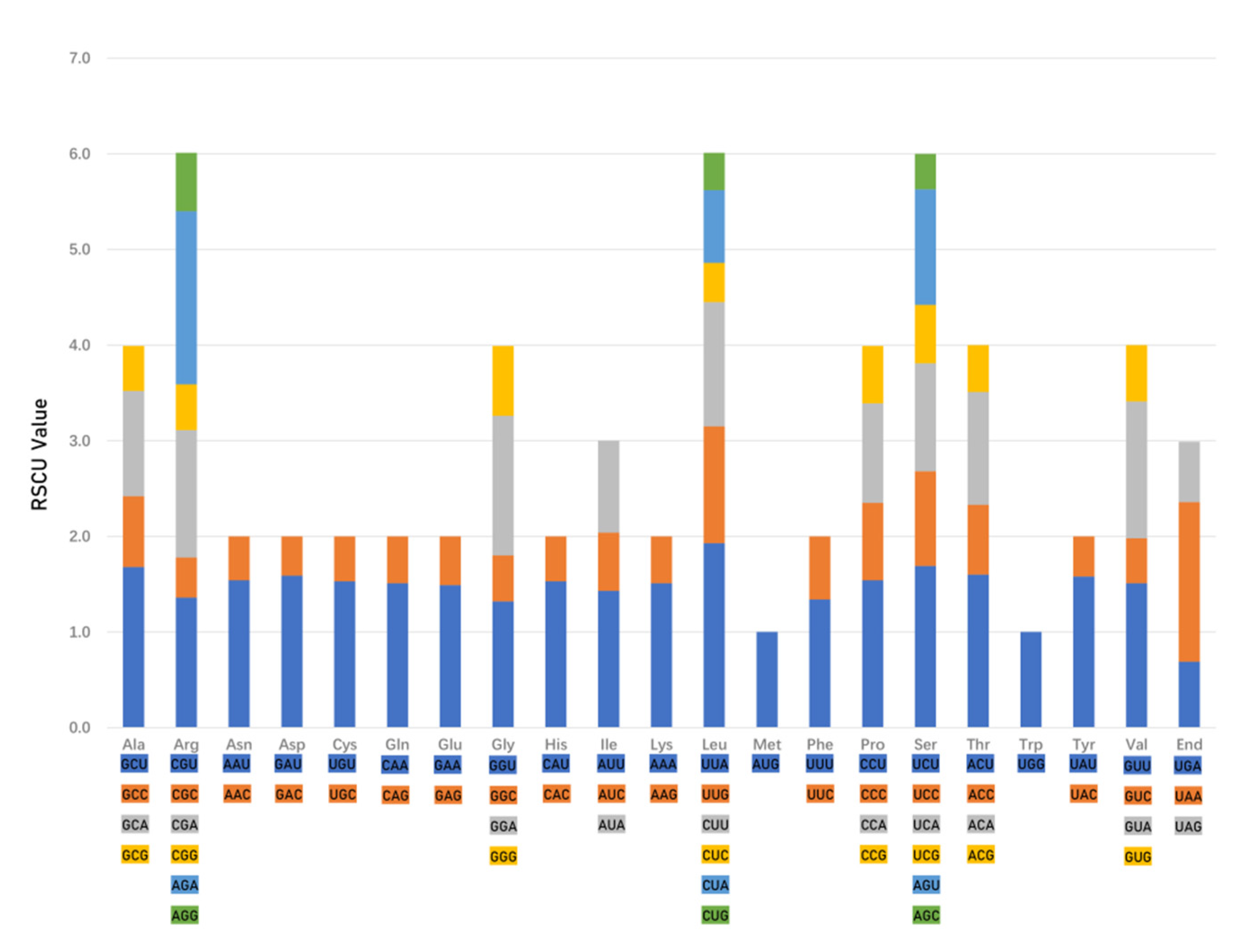
2.3. Codon Usage Bias of Chamaesium Species and Ka/Ks Ratios of Species Pairwise
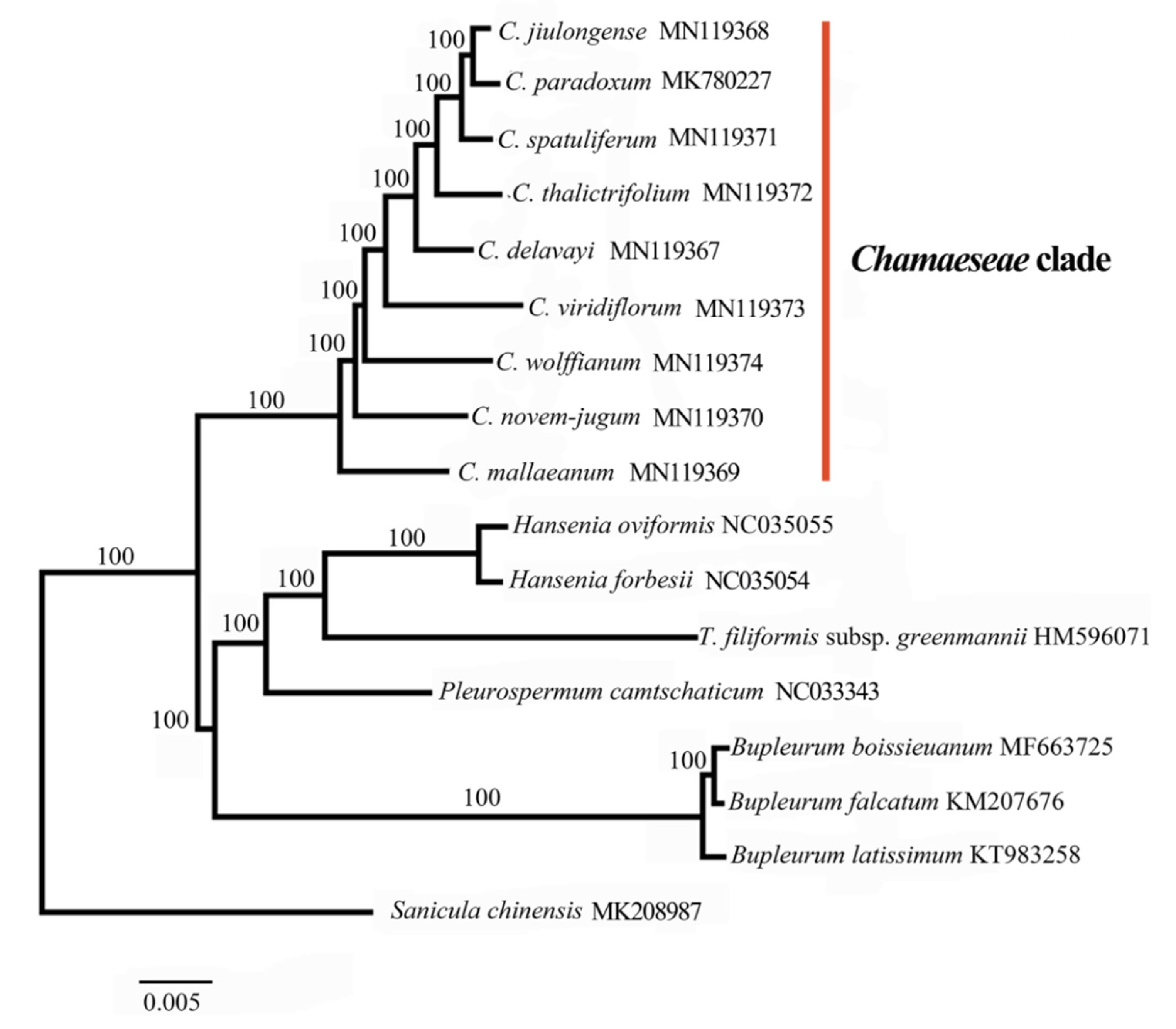
2.4. Phylogeny of Chamaesium
3. Discussion
3.1. The Fluctuations of IR Regions, Genes and Pseudogenes (ψs) in the Plastid Genome
3.2. Codon Usage Analysis, Ka/Ks and Selection Pressure
3.3. Phylogenetic Analysis
4. Materials and Methods
4.1. Plant Material and DNA Extraction
4.2. Illumina Sequencing, Assembly, and Annotation
4.3. Genome Annotation and Repeat Structure
4.4. Contraction and Expansion of IRs, GC Content and SSR
4.5. Phylogenetic Analyses
4.6. Codon Usage and Ka/Ks Ratios of Species Pairwise Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, S.L. Chamaesium. In Flora Reipublicae Popularis Sinicae; Shan, R.H., She, M.L., Eds.; Science Press: Beijing, China, 1979; Volume 55, pp. 124–133. [Google Scholar]
- Wu, Z.Y. Umbelliferae. In Flora Yunnanica; Science Press: Beijing, China, 1997; Volume 17, pp. 357–640. [Google Scholar]
- Pimenov, M.G.; Kljuykov, E.V.; Dickore, W.B.; Miehe, G. Four Himalayan Umbelliferae new to the flora of China, with critical notes on Tordyliopsis DC. and Keraymonia Farille. Willdenowia 2000, 30, 361–367. [Google Scholar] [CrossRef]
- Pimenov, M.G.; Kljuykov, E.V. What is Sium frigidum Hand.-Mazz. (Umbelliferae)? Feddes Repert. 2003, 114, 350–357. [Google Scholar] [CrossRef]
- She, M.L.; Pu, F.D.; Pan, Z.H.; Watson, M.F.; Cannon, J.F.M.; Holmes-Smith, I.; Kljuykov, E.V.; Phillippe, L.R.; Pimenov, M.G. Apiaceae (Umbelliferae). In Flora of China; Wu, Z.Y., Raven, R.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2005; Volume 15, pp. 1–205. [Google Scholar]
- Pimenov, M.G. Updated checklist of Chinese Umbelliferea: Nomenclature, synonymy, typification, distribution. Turczaninowia 2017, 20, 106–239. [Google Scholar]
- Guo, X.L.; Xie, C.; Bai, J.; He, X.J. Chamaesium jiulongense sp. nov (Apiaceae), from Sichuan, China. Nord. J. Bot. 2017, 35, 676–680. [Google Scholar] [CrossRef]
- Guo, X.L.; Wang, C.B.; Wen, J.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Chamaesium (Apiaceae: Apioideae) inferred from ITS, cpDNA and morphological characters. Phytotaxa 2018, 376, 1. [Google Scholar] [CrossRef]
- Zhou, J.; Peng, H.; Downie, S.R.; Liu, Z.W.; Gong, X.A. Molecular phylogeny of Chinese Apiaceae subfamily Apioideae inferred from nuclear ribosomal DNA internal transcribed spacer sequences. Taxon 2008, 57, 402–416. [Google Scholar]
- Zhou, J.; Gong, X.; Downie, S.R.; Peng, H. Towards a more robust molecular phylogeny of Chinese Apiaceae subfamily Apioideae: Additional evidence from nrDNA ITS and cpDNA intron (rpl16 and rps16) sequences. Molecul. Phylogenet. Evol. 2009, 53, 56–68. [Google Scholar] [CrossRef]
- Marechal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; de Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzym. 2005, 395, 348–384. [Google Scholar]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 35, pp. 103–126. [Google Scholar]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Huang, J.; Chen, R.; Li, X. Comparative Analysis of the Complete Chloroplast Genome of Four Known Ziziphus Species. Genes 2017, 8, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.Y.; Guo, X.L.; He, X.J.; Yu, Y.; Zhou, S.D. The complete chloroplast genome of Chamaesium paradoxum. Mitochondrial DNA B 2019, 4, 2069–2070. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Wang, Y.; Zhan, D.F.; Jia, X.; Mei, W.L.; Dai, H.F.; Chen, X.T.; Peng, S.Q. Complete Chloroplast Genome Sequence of Aquilaria sinensis (Lour.) Gilg and Evolution Analysis within the Malvales Order. Front. Plant Sci. 2016, 7, 280. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Dong, W.P.; Li, W.Q.; Lu, Y.Z.; Xie, X.M.; Jin, X.B.; Shi, J.P.; He, K.L.; Suo, Z.L. Comparative Analysis of Six Lagerstroemia Complete Chloroplast Genomes. Front. Plant Sci. 2017, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium Species inSection Daghestanica and Adaptive Evolution of Allium (Amaryllidaceae, Allioideae) Species Revealed by the Chloroplast Complete Genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef] [Green Version]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef]
- Li, X.W.; Gao, H.H.; Wang, Y.T.; Song, J.Y.; Henry, R.; Wu, H.Z.; Hu, Z.G.; Yao, H.; Luo, H.M.; Luo, K.; et al. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. China Life Sci. 2013, 56, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Liu, H.; Hu, J.; Liang, Y.; Liang, J.; Wuyun, T.; Tan, X. Five complete chloroplast genome sequences from diospyros: Genome organization and comparative analysis. PLoS ONE 2016, 11, e0159566. [Google Scholar] [CrossRef] [Green Version]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.-J.; Jung, J.D.; Park, H.-W.; Kim, J.-H.; Cha, H.W.; Min, S.R.; Jeong, W.-J.; Liu, J.R. The complete chloroplast genome sequences of Solanum tuberosum and comparative analysis with Solanaceae species identified the presence of a 241-bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Rep. 2006, 25, 1369–1379. [Google Scholar] [CrossRef]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcate, H.R.; Calie, P.J.; Boore, J.L.; Jason, R.K. The complete chloroplast genome sequence of Pelargonium xhortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef]
- Hirao, T.; Watanabe, A.; Manabu, K.; Kondo, T.; Takata, K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: Diversified genomic structure of coniferous species. BMC Plant Biol. 2008, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [Green Version]
- Plunkett, G.M.; Downie, S.R. Expansion and Contraction of the Chloroplast Inverted Repeat in Apiaceae Subfamily Apioideae. Syst. Bot. 2000, 25, 648–667. [Google Scholar] [CrossRef]
- Hansen, D.R.; Dastidar, S.G.; Cai, Z.; Penaflor, C.; Kuehl, J.V.; Boore, J.L.; Jason, R.K. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early diverging angiosperms: Buxus (buxaceae), chloranthus (chloranthaceae), dioscorea (dioscoreaceae), and illicium (schisandraceae). mol phylogenet evol. Mol. Phylogenetics Evol. 2007, 45, 547–563. [Google Scholar] [CrossRef]
- Peery, R. Understanding Angiosperm Genome Interactions and Evolution: Insights from Sacred Lotus (Nelumbo Nucifera) and the Carrot Family (Apiaceae). Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champagne, IL, USA, 2015. [Google Scholar]
- Sun, Y.; Moore, M.J.; Zhang, S.; Soltis, P.S.; Soltis, D.E.; Zhao, T.; Meng, A.; Li, X.; Li, J.; Wang, H. Phylogenomic and structural analyses of 18 complete plastomes across all families of early-diverging eudicots, including an angiosperm-wide analysis of ir gene content evolution. Mol. Phylogenetics Evol. 2016, 96, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xie, D.F.; Guo, X.L.; Zheng, Z.Y.; He, X.J.; Zhou, S.D. Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship. Plants 2020, 9, 543. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Zheng, D.; Liu, Y.-J.; Fang, G.; Frankish, A.; Carriero, N.; Robilotto, R.; Cayting, P.; Gerstein, M. Comparative analysis of processed ribosomal protein pseudogenes in four mammalian genomes. Genome Biol. 2009, 10, R2. [Google Scholar] [CrossRef] [Green Version]
- Sisu, C.; Pei, B.; Leng, J.; Frankish, A.; Zhang, Y.; Balasubramanian, S.; Harte, R.; Wang, D.; Rutenberg-Schoenberg, M.; Clark, W.; et al. Comparative analysis of pseudogenes across three phyla. Proc. Natl. Acad. Sci. USA 2014, 111, 13361–13366. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Harrison, P.M.; Liu, Y.; Gerstein, M. Millions of years of evolution preserved: A comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 2003, 13, 2541–2558. [Google Scholar] [CrossRef]
- Zou, C.; Lehti-Shiu, M.D.; Thibaud-Nissen, F.; Prakash, T.; Buell, C.R.; Shiu, S.H. Evolutionary and expression sig-natures of pseudogenes in Arabidopsis and rice. Plant Physiol. 2009, 151, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Linneweber, C.; Maier, R.M.; Alcaraz, J.-P.; Cottet, A.; Herrmann, R.G.; Mache, R. The plastid chromosome of spinach (Spinacia oleracea): Complete nucleotide sequence and gene organization. Plant Mol. Biol. 2001, 45, 307–315. [Google Scholar] [CrossRef]
- Goremykin, V.V.; Hirsch-Ernst, K.I.; Wölfl, S.; Hellwig, F.H. Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Mol. Biol. Evol. 2003, 20, 1499–1505. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.N.; Zhao, Z.L.; Ni, L.H. Prospect: Identification of medicinal plant based on plastid gene ycf15. Chin. Tradit. Herb. Drugs 2017, 48, 3201–3217. [Google Scholar]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xue, Q. Codon usage in the chloroplast genome of rice (Oryza sativa L. ssp. japonica). Acta Agron. Sin. 2004, 30, 1220–1224. [Google Scholar]
- Zhou, M.; Long, W.; Li, X. Analysis of synonymous codon usage in chloroplast genome of Populus alba. For. Res. 2008, 19, 293–297. [Google Scholar] [CrossRef]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of mungbean (vigna radiata) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2010, 17, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Du, Y.; Bi, Y.; Chen, X.; Yang, F.; Xue, J.; Zhang, X. The complete chloroplast genome of Lilium cernuum: Genome structure and evolution. Conserv. Genet. Resour. 2016, 8, 375–378. [Google Scholar] [CrossRef]
- Morton, B.R. Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J. Mol. Evol. 1998, 46, 449–459. [Google Scholar] [CrossRef]
- Maier, R.M.; Neckermann, K.; Igloi, G.L.; Kössel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995, 251, 614–628. [Google Scholar] [CrossRef]
- Meade, J.C.; Shah, P.H.; Lushbaugh, W.B. Trichomonas vaginalis: Analysis of Codon Usage. Exp. Parasitol. 1997, 87, 73–74. [Google Scholar] [CrossRef]
- Meng, J.; Li, X.P.; Li, H.T.; Yang, J.B.; Wang, H.; He, J. Comparative analysis of the complete chloroplast genomes of four aconitum medicinal species. Molecules 2018, 23, 1015. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xing, H.; Yuan, Y.; Wang, X.; Saeed, M.; Tao, J.; Feng, W.; Zhang, G.; Song, X.; Sun, X. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef] [Green Version]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2018, 18, 486. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution. Sci. Am. 1979, 5, 98–100. [Google Scholar] [CrossRef]
- Makalowski, W.; Boguski, M.S. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2,820 orthologous rodent and human sequences. Proc. Natl. Acad. Sci. USA 1998, 95, 9407–9412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, gkw955. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. Organellar genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression datasets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. Irscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting est databases for the development and characterization of gene-derived ssr-markers in barley (hordeum vulgarel.). Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhang, Y.; Zhang, Z.; Jiang, Z.; Yu, J. KaKs_Calculator2.0: A tool kit incorporating gamma- series methods and sliding windows trategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Name of Genes |
|---|---|
| Self-replication | |
| transfer RNAs | trnA-UGC *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, |
| trnG-GCC, trnG-UCC, trnH-GUG, trnI-CAU *, trnI-GAU *, trnK-UUU, | |
| trnL-CAA *, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU *, trnP-UGG, | |
| trnQ-UUG, trnR-ACG *, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, | |
| trnT-GGU, trnT-UGU, trnV-GAC *, trnV-UAC, trnW-CCA, trnY-GUA | |
| ribosomal RNAs | rrn4.5 *, rrna5 *, rrn16 *, rrn23 * |
| RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 |
| Small subunit of ribosomal proteins (SSU) | rps2, rps3, rps4, rps7 *, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19 * (rps19, ψrps19) |
| Large subunit of ribosomal proteins (LSU) | rpl2 *, rpl14, rpl16, rpl20, rpl22, rpl23 *, rpl32, rpl33, rpl36 |
| Genes for photosynthesis | |
| Subunits of NADH-dehydrogenase | ndhA, ndhB *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Subunits of cytochrome b/f complex | petA, petB, petD, petG, petL, petN |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI |
| Large subunit of rubisco | rbcL |
| Other genes | |
| Translational initiation factor | infA |
| Protease | clpP |
| Maturase | matK |
| Subunit of Acetyl-CoA-carboxylase | accD |
| Envelope membrane protein | cemA |
| C-type cytochrome synthesis gene | ccsA |
| Genes of unknown function | |
| Hypothetical chloroplast reading frames (ycf) | ycf1 * (ycf1,ψycf1), ycf2 *, ycf3, ycf4, ψycf15 * |
| Total | 133 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.-L.; Zheng, H.-Y.; Price, M.; Zhou, S.-D.; He, X.-J. Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome. Plants 2020, 9, 965. https://doi.org/10.3390/plants9080965
Guo X-L, Zheng H-Y, Price M, Zhou S-D, He X-J. Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome. Plants. 2020; 9(8):965. https://doi.org/10.3390/plants9080965
Chicago/Turabian StyleGuo, Xian-Lin, Hong-Yi Zheng, Megan Price, Song-Dong Zhou, and Xing-Jin He. 2020. "Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome" Plants 9, no. 8: 965. https://doi.org/10.3390/plants9080965
APA StyleGuo, X.-L., Zheng, H.-Y., Price, M., Zhou, S.-D., & He, X.-J. (2020). Phylogeny and Comparative Analysis of Chinese Chamaesium Species Revealed by the Complete Plastid Genome. Plants, 9(8), 965. https://doi.org/10.3390/plants9080965