

Unexpected Classes of Aquaporin Channels Detected by Transcriptomic Analysis in Human Brain Are Associated with Both Patient Age and Alzheimer’s Disease Status

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Human Brain Atlas Database
2.3. Institute Aging, Dementia and TBI Database
2.4. Gene Probes
2.5. Statistical Analysis
2.5.1. Regression Model Analyses
2.5.2. Supervised Clustering Analyses
2.5.3. Differential Expression Analysis
2.5.4. Expression Analysis–Group Comparison
3. Results
3.1. Subject Population Characteristics
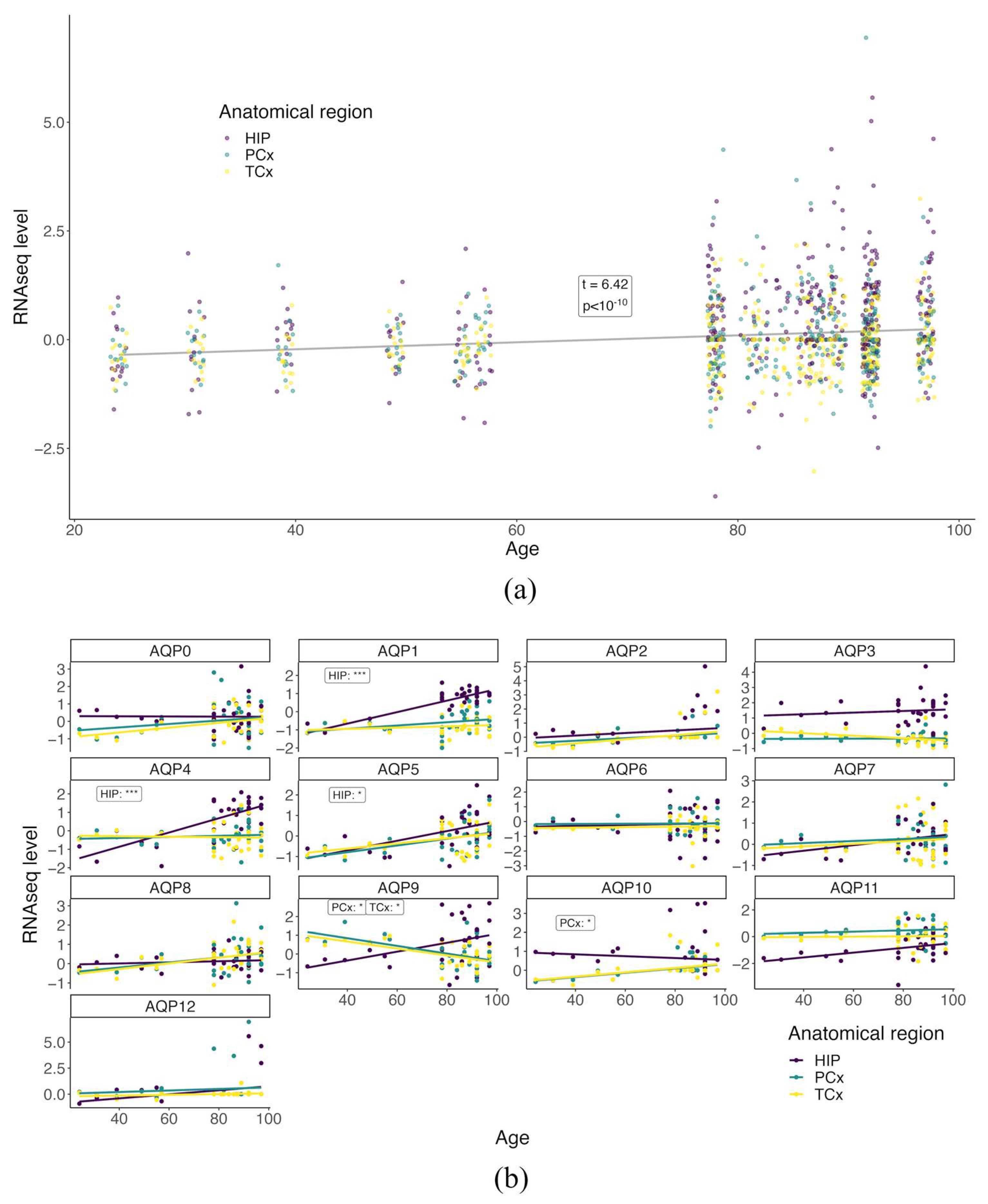
3.2. Baseline AQP Expression Profiles Differ with Age in the Healthy Brain
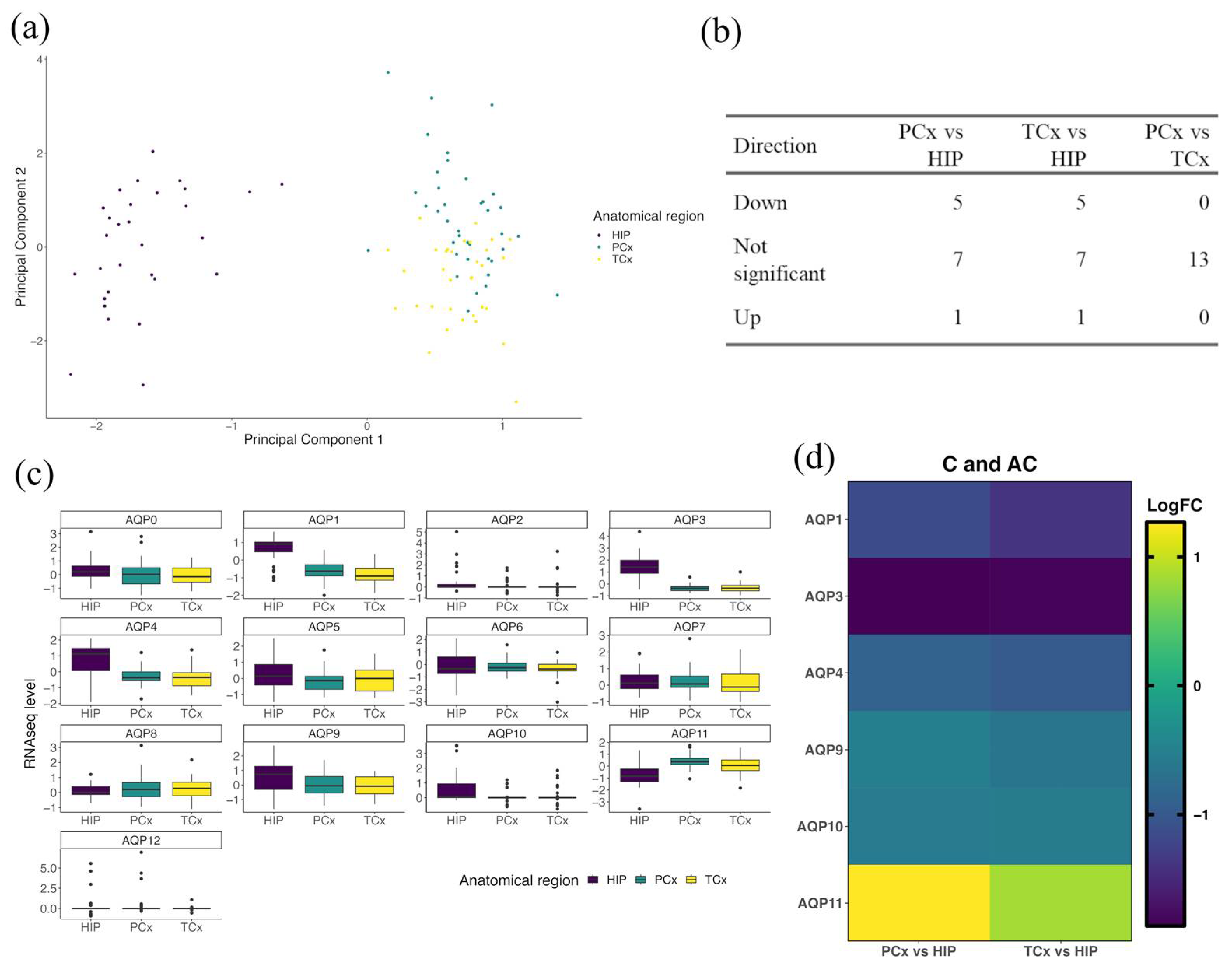
3.3. AQP Expression Profiles in the Hippocampus Differ from Those in Cortex in Healthy Brain
3.4. Age-Dependent Changes in AQP Expression Profiles Differ between the HIP and Cortical Regions in the Healthy Brain
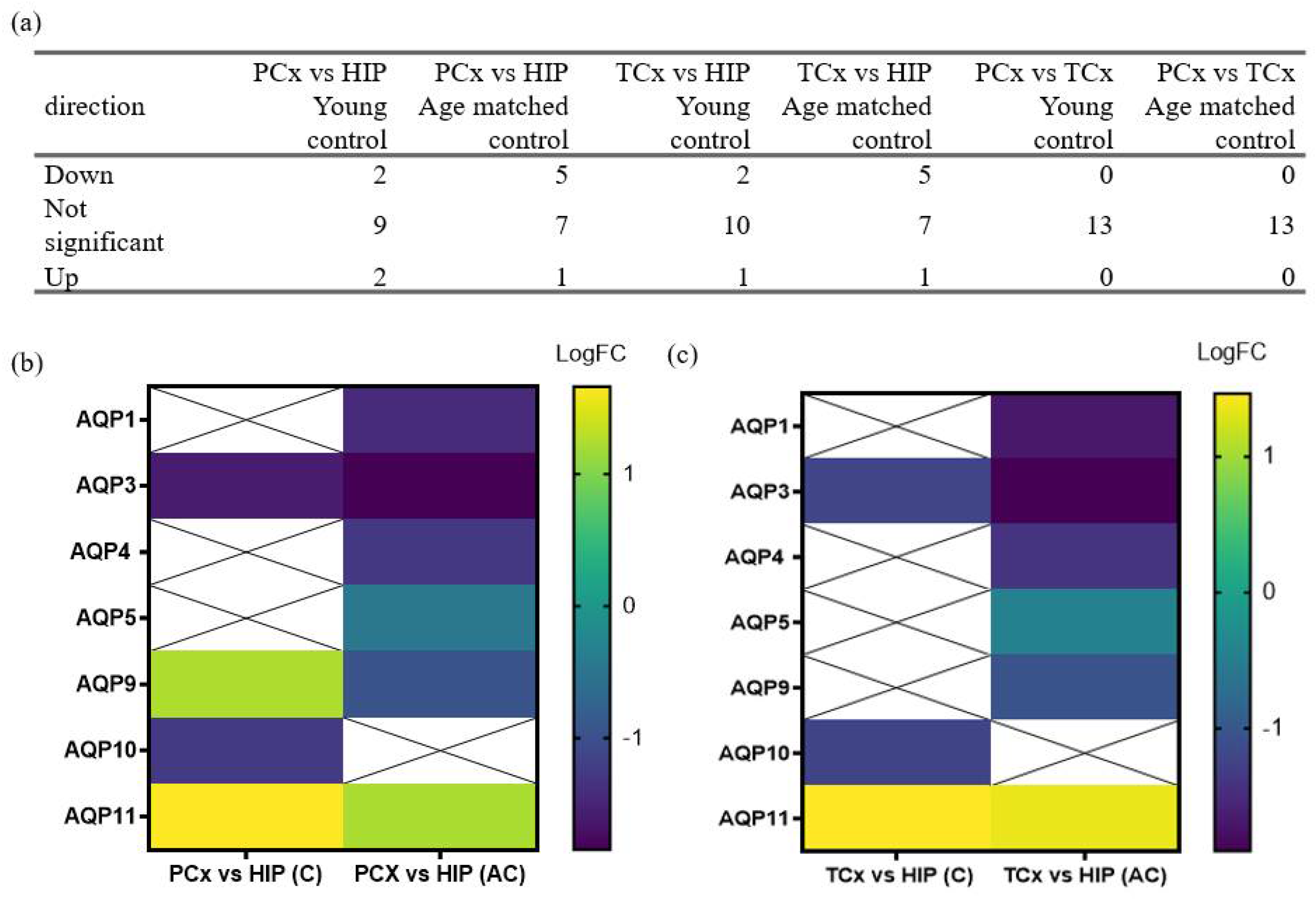
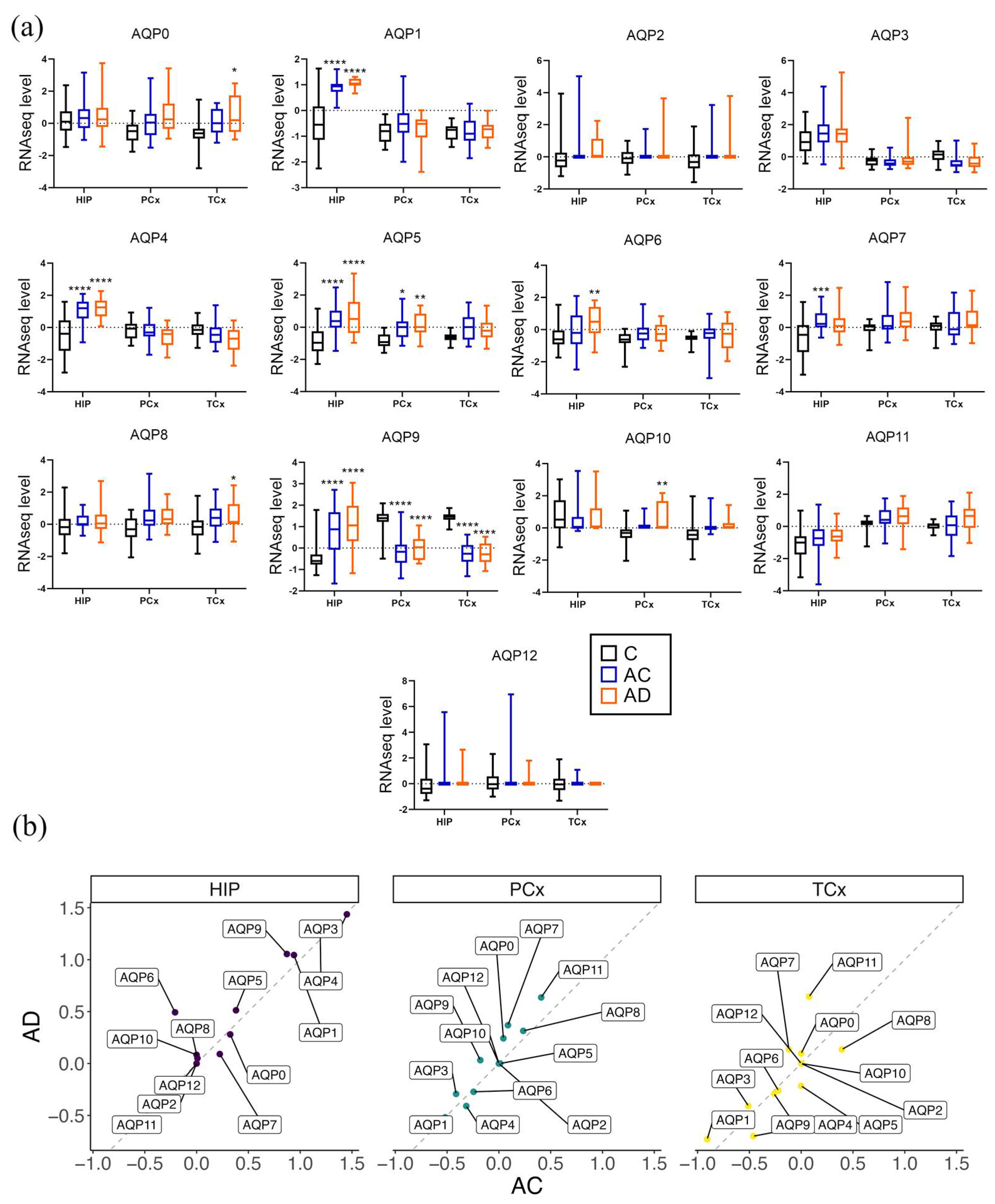
3.5. Regional Differences in Levels of AQP Transcripts Associated with Probable Alzheimer’s Disease
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Povova, J.; Ambroz, P.; Bar, M.; Pavukova, V.; Sery, O.; Tomaskova, H.; Janout, V. Epidemiological of and risk factors for Alzheimer’s disease: A review. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2012, 156, 108–114. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Pierrot, N.; Santos, S.F.; Feyt, C.; Morel, M.; Brion, J.P.; Octave, J.N. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J. Biol. Chem. 2006, 281, 39907–39914. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Coughlan, G.; Laczó, J.; Hort, J.; Minihane, A.-M.; Hornberger, M. Spatial navigation deficits—Overlooked cognitive marker for preclinical Alzheimer disease? Nat. Rev. Neurol. 2018, 14, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Amro, Z.; Yool, A.J.; Collins-Praino, L.E. The potential role of glial cells in driving the prion-like transcellular propagation of tau in tauopathies. Brain Behav. Immun. Health 2021, 14, 100242. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Wegiel, J. Spatial relationships between astrocytes and classical plaque components. Neurobiol. Aging 1991, 12, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef]
- Nedergaard, M. Garbage Truck of the Brain. Science 2013, 340, 1529–1530. [Google Scholar] [CrossRef] [Green Version]
- Kruse, E.; Uehlein, N.; Kaldenhoff, R. The aquaporins. Genome Biol. 2006, 7, 206. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, I.; Bill, R.M.; Kayingo, I.; Prior, B.A. Microbial MIP channels. Trends Microbiol. 2000, 8, 33–38. [Google Scholar] [CrossRef]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118, 3225–3232. [Google Scholar] [CrossRef] [Green Version]
- Agre, P.; Sasaki, S.; Chrispeels, M.J. Aquaporins: A family of water channel proteins. Am. J. Physiol. 1993, 265, F461. [Google Scholar] [CrossRef]
- Yool, A.J.; Stamer, W.D.; Regan, J.W. Forskolin stimulation of water and cation permeability in aquaporin1 water channels. Science 1996, 273, 1216–1218. [Google Scholar] [CrossRef]
- Varadaraj, K.; Kumari, S.S. Lens aquaporins function as peroxiporins to facilitate membrane transport of hydrogen peroxide. Biochem. Biophys Res. Commun. 2020, 524, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.; Pimpão, C.; Mósca, A.F.; Coxixo, A.S.; Lopes, D.; da Silva, I.V.; Pedersen, P.A.; Antunes, F.; Soveral, G. Human Aquaporin-5 Facilitates Hydrogen Peroxide Permeation Affecting Adaption to Oxidative Stress and Cancer Cell Migration. Cancers 2019, 11, 932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellavio, G.; Martinotti, S.; Patrone, M.; Ranzato, E.; Laforenza, U. Aquaporin-6 May Increase the Resistance to Oxidative Stress of Malignant Pleural Mesothelioma Cells. Cells 2022, 11, 1892. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Farinelli, G.; Galli, M.; Aiuti, A.; Sitia, R. AQP8 transports NOX2-generated H2O2 across the plasma membrane to promote signaling in B cells. J. Leukoc. Biol. 2016, 100, 1071–1079. [Google Scholar] [CrossRef]
- Frühbeck, G.; Balaguer, I.; Méndez-Giménez, L.; Valentí, V.; Becerril, S.; Catalán, V.; Gómez-Ambrosi, J.; Silva, C.; Salvador, J.; Calamita, G.; et al. Aquaporin-11 Contributes to TGF-β1-induced Endoplasmic Reticulum Stress in Human Visceral Adipocytes: Role in Obesity-Associated Inflammation. Cells 2020, 9, 1403. [Google Scholar] [CrossRef]
- Hara-Chikuma, M.; Verkman, A.S. Physiological roles of glycerol-transporting aquaporins: The aquaglyceroporins. Cell. Mol. Life Sci. 2006, 63, 1386–1392. [Google Scholar] [CrossRef]
- Watanabe, S.; Moniaga, C.S.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9 facilitates membrane transport of hydrogen peroxide in mammalian cells. Biochem. Biophys Res. Commun. 2016, 471, 191–197. [Google Scholar] [CrossRef]
- Yakata, K.; Hiroaki, Y.; Ishibashi, K.; Sohara, E.; Sasaki, S.; Mitsuoka, K.; Fujiyoshi, Y. Aquaporin-11 containing a divergent NPA motif has normal water channel activity. Biochim. Biophys Acta 2007, 1768, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Itoh, T.; Rai, T.; Kuwahara, M.; Ko, S.B.; Uchida, S.; Sasaki, S.; Ishibashi, K. Identification of a novel aquaporin, AQP12, expressed in pancreatic acinar cells. Biochem. Biophys Res. Commun. 2005, 330, 832–838. [Google Scholar] [CrossRef]
- Oshio, K.; Watanabe, H.; Song, Y.; Verkman, A.S.; Manley, G.T. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 76–78. [Google Scholar] [CrossRef]
- Suzuki, R.; Okuda, M.; Asai, J.; Nagashima, G.; Itokawa, H.; Matsunaga, A.; Fujimoto, T.; Suzuki, T. Astrocytes co-express aquaporin-1, -4, and vascular endothelial growth factor in brain edema tissue associated with brain contusion. Acta Neurochir. Suppl. 2006, 96, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potokar, M.; Jorgačevski, J.; Zorec, R. Astrocyte Aquaporin Dynamics in Health and Disease. Int. J. Mol. Sci. 2016, 17, 1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, E.; Barrachina, M.; Rodriguez, A.; Torrejon-Escribano, B.; Boada, M.; Hernandez, I.; Sanchez, M.; Ferrer, I. Aquaporin expression in the cerebral cortex is increased at early stages of Alzheimer disease. Brain Res. 2007, 1128, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Huysseune, S.; Kienlen-Campard, P.; Hebert, S.; Tasiaux, B.; Leroy, K.; Devuyst, O.; Brion, J.P.; De Strooper, B.; Octave, J.N. Epigenetic control of aquaporin 1 expression by the amyloid precursor protein. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 4158–4167. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Arima, K.; Mizusawa, H.; Satoh, J. Close association of water channel AQP1 with amyloid-beta deposition in Alzheimer disease brains. Acta Neuropathol. 2008, 116, 247–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moftakhar, P.; Lynch, M.D.; Pomakian, J.L.; Vinters, H.V. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 1201–1209. [Google Scholar] [CrossRef] [Green Version]
- Tait, M.J.; Saadoun, S.; Bell, B.A.; Papadopoulos, M.C. Water movements in the brain: Role of aquaporins. Trends Neurosci. 2008, 31, 37–43. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Xu, H.; Feng, W.; Su, D.; Xiao, M. Deletion of aquaporin-4 aggravates brain pathology after blocking of the meningeal lymphatic drainage. Brain Res. Bull. 2018, 143, 83–96. [Google Scholar] [CrossRef]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliff, J.J.; Goldman, S.A.; Nedergaard, M. Implications of the discovery of brain lymphatic pathways. Lancet Neurol. 2015, 14, 977–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaut, J.; Hirt, L.; Granziera, C.; Bogousslavsky, J.; Magistretti, P.J.; Regli, L. Astrocyte-specific expression of aquaporin-9 in mouse brain is increased after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2001, 21, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaut, J.; Regli, L. Distribution and possible roles of aquaporin 9 in the brain. Neuroscience 2004, 129, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Shin, L.; Basi, N.; Jeremic, A.; Lee, J.S.; Cho, W.J.; Chen, Z.; Abu-Hamdah, R.; Oupicky, D.; Jena, B.P. Involvement of vH(+)-ATPase in synaptic vesicle swelling. J. Neurosci. Res. 2010, 88, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, N.; Yoneda, K.; Asai, K.; Sobue, K.; Tada, T.; Fujita, Y.; Katsuya, H.; Fujita, M.; Aihara, N.; Mase, M.; et al. Alterations in the expression of the AQP family in cultured rat astrocytes during hypoxia and reoxygenation. Brain Res. Mol. Brain Res. 2001, 90, 26–38. [Google Scholar] [CrossRef]
- Gorelick, D.A.; Praetorius, J.; Tsunenari, T.; Nielsen, S.; Agre, P. Aquaporin-11: A channel protein lacking apparent transport function expressed in brain. BMC Biochem. 2006, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Guillozet-Bongaarts, A.; Gibbons, L.E.; Postupna, N.; Renz, A.; Beller, A.E.; Sunkin, S.M.; Ng, L.; Rose, S.E.; Smith, K.A.; et al. Neuropathological and transcriptomic characteristics of the aged brain. eLife 2017, 6, 31126. [Google Scholar] [CrossRef]
- Villareal, D.T.; Grant, E.; Miller, J.P.; Storandt, M.; McKeel, D.W.; Morris, J.C. Clinical outcomes of <em>possible</em> versus <em>probable</em> Alzheimer’s disease. Neurology 2003, 61, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Shively, S.; Scher, A.I.; Perl, D.P.; Diaz-Arrastia, R. Dementia resulting from traumatic brain injury: What is the pathology? Arch. Neurol. 2012, 69, 1245–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, G., 3rd; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohart, F.; Gautier, B.; Singh, A.; KA, L.C. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Owasil, R.; O’Neill, R.; Keable, A.; Nimmo, J.; MacGregor Sharp, M.; Kelly, L.; Saito, S.; Simpson, J.E.; Weller, R.O.; Smith, C.; et al. The Pattern of AQP4 Expression in the Ageing Human Brain and in Cerebral Amyloid Angiopathy. Int. J. Mol. Sci. 2020, 21, 1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Murale, D.P.; Kim, Y.K.; Lee, J.S. Crosstalk between Oxidative Stress and Tauopathy. Int. J. Mol. Sci. 2019, 20, 1959. [Google Scholar] [CrossRef] [Green Version]
- Montiel, V.; Bella, R.; Michel, L.Y.M.; Esfahani, H.; De Mulder, D.; Robinson, E.L.; Deglasse, J.P.; Tiburcy, M.; Chow, P.H.; Jonas, J.C.; et al. Inhibition of aquaporin-1 prevents myocardial remodeling by blocking the transmembrane transport of hydrogen peroxide. Sci. Transl. Med. 2020, 12, aay2176. [Google Scholar] [CrossRef]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Møller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindhu Kumari, S.; Gupta, N.; Shiels, A.; FitzGerald, P.G.; Menon, A.G.; Mathias, R.T.; Varadaraj, K. Role of Aquaporin 0 in lens biomechanics. Biochem. Biophys Res. Commun. 2015, 462, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.H.T.; Bråthe, A.; Hassel, B. Neuronal uptake and metabolism of glycerol and the neuronal expression of mitochondrial glycerol-3-phosphate dehydrogenase. J. Neurochem. 2003, 85, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Hirt, L.; Price, M.; Mastour, N.; Brunet, J.F.; Barrière, G.; Friscourt, F.; Badaut, J. Increase of aquaporin 9 expression in astrocytes participates in astrogliosis. J. Neurosci. Res. 2018, 96, 194–206. [Google Scholar] [CrossRef]
- Gotfryd, K.; Mósca, A.F.; Missel, J.W.; Truelsen, S.F.; Wang, K.; Spulber, M.; Krabbe, S.; Hélix-Nielsen, C.; Laforenza, U.; Soveral, G.; et al. Human adipose glycerol flux is regulated by a pH gate in AQP10. Nat. Commun. 2018, 9, 4749. [Google Scholar] [CrossRef] [Green Version]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [Green Version]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, M.M.; Blazer-Yost, B. Channels and Transporters in Astrocyte Volume Regulation in Health and Disease. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2022, 56, 12–30. [Google Scholar] [CrossRef]
- Pike, C.J. Sex and the development of Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | n | Age (yrs) | Education | Braak Stage |
|---|---|---|---|---|
| Young control (C) | 6 | Range: 24–57 Median: 44 (31, 55) | N/A | N/A |
| Aged control (AC) | 29 | Range: 78–99 **** Median: 86 (78.5, 89) | Median: 15 (12, 16) | Median: 3 (2, 3.5) |
| Probable Alzheimer’s disease (AD) | 11 | Range: 79–99+ **** Median: 87 (85–91.5) | Median: 14 (12, 16) | Median: 5 (2, 6) * |
| (i) PCx | Gene | logFC | AveExpr | t | p Value | adj. p Val |
|---|---|---|---|---|---|---|
| Higher expression | ||||||
| AQP11 | 1.2660 | −0.1312 | 6.8948 | <0.001 | <0.001 | |
| Lower expression | ||||||
| AQP3 | −1.8606 | 0.2854 | −13.497 | <0.001 | <0.001 | |
| AQP1 | −1.1530 | −0.2480 | −8.046 | <0.001 | <0.001 | |
| AQP4 | −0.8933 | 0.0305 | −4.396 | <0.001 | 1 × 10−4 | |
| AQP10 | −0.5890 | 0.2891 | −3.164 | 0.0022 | 0.0058 | |
| AQP9 | −0.5273 | 0.1916 | −2.441 | 0.017 | 0.0368 | |
| No significant difference | ||||||
| AQP5 | −0.3550 | −0.0021 | −1.954 | 0.0544 | 0.0911 | |
| AQP2 | −0.3983 | 0.2462 | −1.940 | 0.0561 | 0.0911 | |
| AQP0 | −0.2684 | 0.0776 | −1.362 | 0.1773 | 0.2561 | |
| AQP8 | 0.1785 | 0.2422 | 1.0513 | 0.2964 | 0.3854 | |
| AQP12 | 0.1078 | 0.2901 | 0.3748 | 0.7088 | 0.8377 | |
| AQP7 | 0.0299 | 0.2040 | 0.1616 | 0.8721 | 0.9448 | |
| AQP6 | 0.0129 | −0.2175 | 0.0623 | 0.9505 | 0.9505 | |
| (ii) TCx | Gene | logFC | AveExpr | t | p Value | adj. p Val |
| Higher expression | ||||||
| AQP11 | 0.8442 | -0.1312 | 4.6514 | <0.001 | <0.001 | |
| Lower expression | ||||||
| AQP3 | −1.8251 | 0.2854 | −13.39 | <0.001 | <0.001 | |
| AQP1 | −1.3989 | −0.2480 | −9.876 | <0.001 | <0.001 | |
| AQP4 | −0.9932 | 0.0305 | −4.945 | <0.001 | <0.001 | |
| AQP9 | −0.6515 | 0.1916 | −3.051 | 0.0031 | 0.0082 | |
| AQP10 | −0.5451 | 0.2891 | −2.963 | 0.0041 | 0.0088 | |
| No significant difference | ||||||
| AQP0 | −0.4008 | 0.0776 | −2.057 | 0.0431 | 0.0801 | |
| AQP5 | −0.3258 | −0.0021 | −1.814 | 0.0737 | 0.1197 | |
| AQP2 | −0.3557 | 0.2462 | −1.753 | 0.0837 | 0.1208 | |
| AQP12 | −0.3756 | 0.2901 | −1.321 | 0.1904 | 0.2475 | |
| AQP8 | 0.1857 | 0.2422 | 1.106 | 0.2721 | 0.3216 | |
| AQP6 | −0.2120 | −0.2175 | −1.039 | 0.302 | 0.3271 | |
| AQP7 | −0.0685 | 0.2040 | −0.375 | 0.7086 | 0.7086 | |
| (i) C | Gene | logFC | AveExpr | t | p Value | adj. p Val |
|---|---|---|---|---|---|---|
| Higher expression | ||||||
| AQP11 | 1.6950 | −0.1312 | 3.9866 | 1 × 10−4 | 8 × 10−4 | |
| AQP9 | 1.2396 | 0.1916 | 2.7069 | 0.0079 | 0.0257 | |
| Lower expression | ||||||
| AQP3 | −1.5951 | 0.2854 | −4.4059 | <0.001 | 3 × 10−3 | |
| AQP10 | −1.2611 | 0.2891 | −3.1242 | 0.0023 | 0.01 | |
| No significant difference | ||||||
| AQP0 | −0.7967 | 0.0776 | −1.6654 | 0.0988 | 0.2568 | |
| AQP4 | 0.6127 | 0.0305 | 1.5691 | 0.1196 | 0.2591 | |
| AQP7 | 0.4365 | 0.2040 | 1.0265 | 0.307 | 0.5701 | |
| AQP2 | −0.3546 | 0.2462 | −0.7644 | 0.4463 | 0.7253 | |
| AQP6 | 0.2345 | −0.2175 | 0.5093 | 0.6116 | 0.8834 | |
| AQP12 | 0.1776 | 0.2901 | 0.2705 | 0.7873 | 0.9189 | |
| AQP8 | −0.0834 | 0.2422 | −0.2207 | 0.8258 | 0.9189 | |
| AQP5 | 0.0477 | −0.0021 | 0.1169 | 0.9072 | 0.9189 | |
| AQP1 | −0.0299 | −0.2480 | −0.1020 | 0.9189 | 0.9189 | |
| (ii) AC | Gene | logFC | AveExpr | t | p Value | adj. p Val |
| Higher expression | ||||||
| AQP11 | 1.2161 | −0.1312 | 6.0634 | <0.001 | <0.001 | |
| Lower expression | ||||||
| AQP3 | −1.8470 | 0.2854 | −10.815 | <0.001 | <0.001 | |
| AQP1 | −1.4439 | −0.2480 | −10.439 | <0.001 | <0.001 | |
| AQP4 | −1.2894 | 0.0305 | −7.0001 | <0.001 | <0.001 | |
| AQP9 | −0.9655 | 0.1916 | −4.4691 | <0.001 | 1 × 10−4 | |
| AQP5 | −0.4671 | −0.0021 | −2.4253 | 0.017 | 0.0368 | |
| No significant difference | ||||||
| AQP10 | −0.3984 | 0.2891 | −2.0920 | 0.0388 | 0.0721 | |
| AQP2 | −0.3731 | 0.2462 | −1.7051 | 0.0911 | 0.148 | |
| AQP8 | 0.2499 | 0.2422 | 1.4010 | 0.1641 | 0.2371 | |
| AQP0 | −0.0976 | 0.0776 | −0.4325 | 0.6663 | 0.8245 | |
| AQP12 | 0.1206 | 0.2901 | 0.3895 | 0.6977 | 0.8245 | |
| AQP7 | −0.0571 | 0.2040 | −0.2847 | 0.7764 | 0.8411 | |
| AQP6 | −0.0128 | −0.2175 | −0.0587 | 0.9533 | 0.9533 | |
| (i) C | Gene | logFC | AveExpr | t | p Value | adj. p Val |
|---|---|---|---|---|---|---|
| Higher expression | ||||||
| AQP11 | 1.4622 | −0.1312 | 3.4391 | 8 × 10−4 | 0.0074 | |
| Lower expression | ||||||
| AQP3 | −1.2113 | 0.2854 | −3.3457 | 0.0011 | 0.0074 | |
| AQP10 | −1.2381 | 0.2891 | −3.0671 | 0.0027 | 0.0119 | |
| No significant difference | ||||||
| AQP9 | 1.0602 | 0.1916 | 2.3150 | 0.0225 | 0.0732 | |
| AQP4 | 0.7552 | 0.0305 | 1.9340 | 0.0558 | 0.1209 | |
| AQP0 | −0.9250 | 0.0776 | −1.9335 | 0.0558 | 0.1209 | |
| AQP2 | −0.5220 | 0.2462 | −1.1253 | 0.263 | 0.4884 | |
| AQP7 | 0.2543 | 0.2040 | 0.5980 | 0.5511 | 0.8955 | |
| AQP8 | −0.1370 | 0.2422 | −0.3624 | 0.7178 | 0.9279 | |
| AQP1 | −0.0974 | −0.2480 | −0.3323 | 0.7403 | 0.9279 | |
| AQP5 | 0.1005 | −0.0021 | 0.2461 | 0.8061 | 0.9279 | |
| AQP12 | −0.0745 | 0.2901 | −0.1134 | 0.9099 | 0.9279 | |
| AQP6 | −0.0418 | −0.2175 | −0.0908 | 0.9279 | 0.9279 | |
| (ii) AC | Gene | logFC | AveExpr | t | p Value | adj. p Val |
| Higher expression | ||||||
| AQP11 | 0.7296 | −0.1312 | 3.6732 | 4 × 10−4 | 0.001 | |
| Lower expression | ||||||
| AQP1 | −1.7035 | −0.2480 | −12.435 | <0.001 | <0.001 | |
| AQP3 | −1.9022 | 0.2854 | −11.247 | <0.001 | <0.001 | |
| AQP4 | −1.4108 | 0.0305 | −7.7335 | <0.001 | <0.001 | |
| AQP9 | −1.0480 | 0.1916 | −4.8983 | <0.001 | <0.001 | |
| AQP5 | −0.4470 | −0.0021 | −2.3440 | 0.0209 | 0.0454 | |
| No significant difference | ||||||
| AQP10 | −0.3690 | 0.2891 | −1.9567 | 0.053 | 0.0984 | |
| AQP8 | 0.2502 | 0.2422 | 1.4164 | 0.1596 | 0.2227 | |
| AQP12 | −0.4251 | 0.2901 | −1.3858 | 0.1687 | 0.2227 | |
| AQP2 | −0.2985 | 0.2462 | −1.3772 | 0.1713 | 0.2227 | |
| AQP0 | −0.2390 | 0.0776 | −1.0695 | 0.2873 | 0.3395 | |
| AQP6 | −0.2083 | −0.2175 | −0.9685 | 0.335 | 0.3629 | |
| AQP7 | −0.1479 | 0.2040 | −0.7444 | 0.4583 | 0.4583 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amro, Z.; Ryan, M.; Collins-Praino, L.E.; Yool, A.J. Unexpected Classes of Aquaporin Channels Detected by Transcriptomic Analysis in Human Brain Are Associated with Both Patient Age and Alzheimer’s Disease Status. Biomedicines 2023, 11, 770. https://doi.org/10.3390/biomedicines11030770
Amro Z, Ryan M, Collins-Praino LE, Yool AJ. Unexpected Classes of Aquaporin Channels Detected by Transcriptomic Analysis in Human Brain Are Associated with Both Patient Age and Alzheimer’s Disease Status. Biomedicines. 2023; 11(3):770. https://doi.org/10.3390/biomedicines11030770
Chicago/Turabian StyleAmro, Zein, Matthew Ryan, Lyndsey E. Collins-Praino, and Andrea J. Yool. 2023. "Unexpected Classes of Aquaporin Channels Detected by Transcriptomic Analysis in Human Brain Are Associated with Both Patient Age and Alzheimer’s Disease Status" Biomedicines 11, no. 3: 770. https://doi.org/10.3390/biomedicines11030770
APA StyleAmro, Z., Ryan, M., Collins-Praino, L. E., & Yool, A. J. (2023). Unexpected Classes of Aquaporin Channels Detected by Transcriptomic Analysis in Human Brain Are Associated with Both Patient Age and Alzheimer’s Disease Status. Biomedicines, 11(3), 770. https://doi.org/10.3390/biomedicines11030770