
Trem2 Enhances Demyelination in the Csf1r+/− Mouse Model of Leukoencephalopathy
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Mouse Strains, Breeding and Maintenance
2.3. Gene Expression in Mouse Brains
2.4. Ultrastructural Studies
2.5. Immunohistochemistry and Data Analysis
2.6. Macrophage Preparation and Myelin Challenge
2.7. Cytokine Measurements
2.8. Statistical Analyses
3. Results
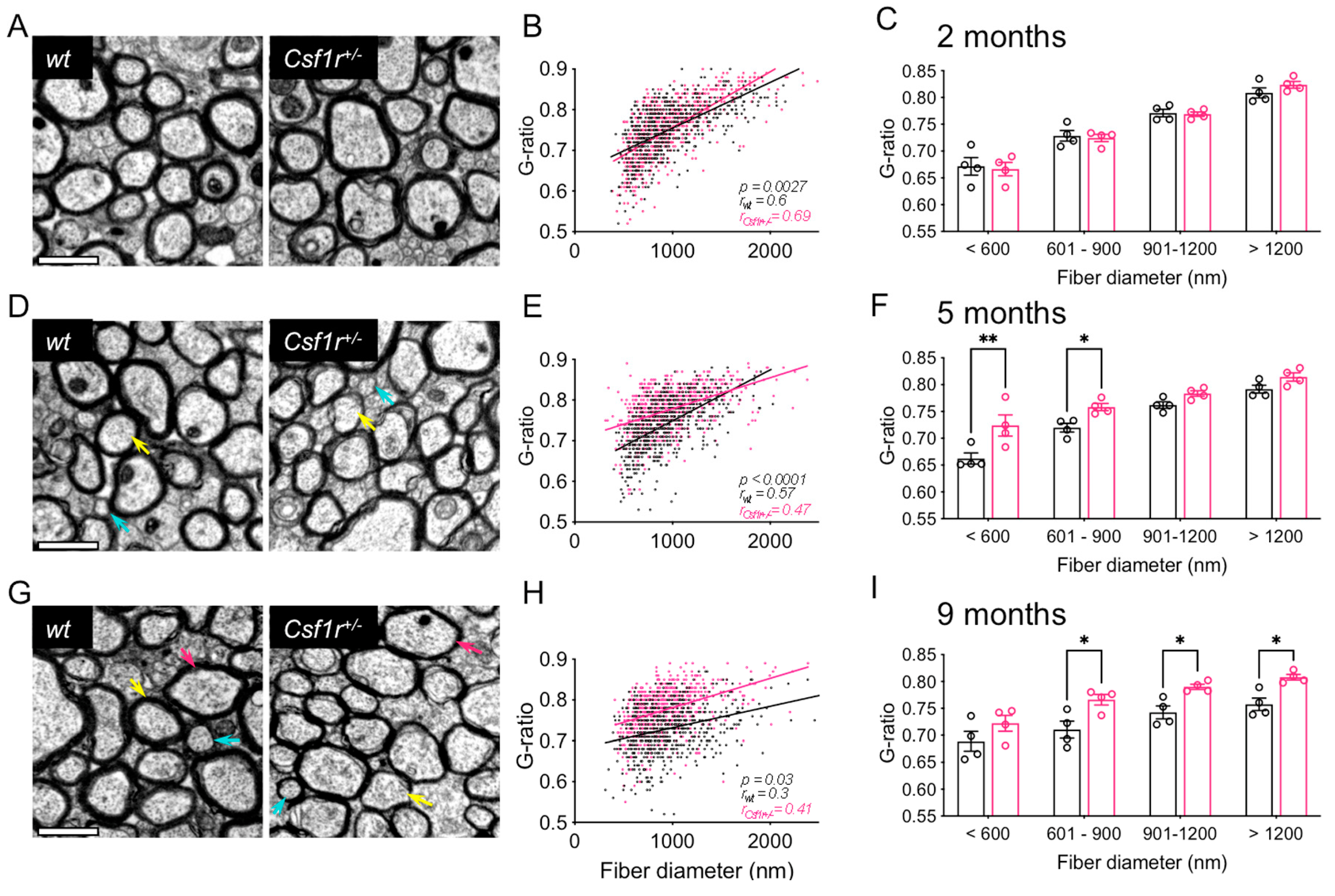
3.1. Callosal Demyelination is an Early Feature of Csf1r+/− Mouse Pathology
3.2. Cerebral Trem2 Expression Is Elevated in Csf1r+/− Brains by 4 Months of Age
3.3. Trem2 Deletion Attenuates Callosal Demyelination in 9-Month-Old Csf1r+/− Mice
3.4. Trem2 Deficiency Attenuates Callosal Axonal Degeneration and Loss of Layer V Neurons in 9-Month-Old Csf1r+/− Mice
3.5. Trem2 Ablation Prevents the Increase in Microglial Density and Alterations in Their Morphology in Csf1r+/− Mice
3.6. Trem2 Deficiency Does Not Cause the Upregulation of CSF-1R Expression in Macrophages
3.7. CSF-1R and TREM2 Have Opposite Effects on the Formation of Myelin-Derived Lipid Droplets in Macrophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akiyama, H.; Nishimura, T.; Kondo, H.; Ikeda, K.; Hayashi, Y.; McGeer, P.L. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res. 1994, 639, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Raivich, G.; Haas, S.; Werner, A.; Klein, M.A.; Kloss, C.; Kreutzberg, G.W. Regulation of MCSF receptors on microglia in the normal and injured mouse central nervous system: A quantitative immunofluorescence study using confocal laser microscopy. J. Comp. Neurol. 1998, 395, 342–358. [Google Scholar] [CrossRef]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef]
- Chitu, V.; Gokhan, S.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Baker, M.; Nicholson, A.M.; Rutherford, N.J.; Finch, N.; Soto-Ortolaza, A.; Lash, J.; Wider, C.; Wojtas, A.; DeJesus-Hernandez, M.; et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2011, 44, 200–205. [Google Scholar] [CrossRef]
- Konno, T.; Kasanuki, K.; Ikeuchi, T.; Dickson, D.W.; Wszolek, Z.K. CSF1R-related leukoencephalopathy: A major player in primary microgliopathies. Neurology 2018, 91, 1092–1104. [Google Scholar] [CrossRef]
- Konno, T.; Tada, M.; Tada, M.; Koyama, A.; Nozaki, H.; Harigaya, Y.; Nishimiya, J.; Matsunaga, A.; Yoshikura, N.; Ishihara, K.; et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 2013, 82, 139–148. [Google Scholar] [CrossRef]
- Kondo, Y.; Kinoshita, M.; Fukushima, K.; Yoshida, K.; Ikeda, S. Early involvement of the corpus callosum in a patient with hereditary diffuse leukoencephalopathy with spheroids carrying the de novo K793T mutation of CSF1R. Intern. Med. 2013, 52, 503–506. [Google Scholar] [CrossRef]
- Oyanagi, K.; Kinoshita, M.; Suzuki-Kouyama, E.; Inoue, T.; Nakahara, A.; Tokiwai, M.; Arai, N.; Satoh, J.I.; Aoki, N.; Jinnai, K.; et al. Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: Lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol. 2017, 27, 748–769. [Google Scholar] [CrossRef]
- Coleman, M. Axon degeneration mechanisms: Commonality amid diversity. Nat. Rev. Neurosci. 2005, 6, 889–898. [Google Scholar] [CrossRef]
- Beirowski, B.; Nogradi, A.; Babetto, E.; Garcia-Alias, G.; Coleman, M.P. Mechanisms of axonal spheroid formation in central nervous system Wallerian degeneration. J. Neuropathol. Exp. Neurol. 2010, 69, 455–472. [Google Scholar] [CrossRef]
- Alturkustani, M.; Keith, J.; Hazrati, L.N.; Rademakers, R.; Ang, L.C. Pathologic staging of white matter lesions in adult-onset leukoencephalopathy/leukodystrophy with axonal spheroids. J. Neuropathol. Exp. Neurol. 2015, 74, 233–240. [Google Scholar] [CrossRef]
- Zhan, F.X.; Zhu, Z.Y.; Liu, Q.; Zhou, H.Y.; Luan, X.H.; Huang, X.J.; Liu, X.L.; Tian, W.T.; Wang, S.G.; Song, X.X.; et al. Altered structural and functional connectivity in CSF1R-related leukoencephalopathy. Brain Imaging Behav. 2020, 15, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Gokhan, S.; Stanley, E.R. Modeling CSF-1 receptor deficiency diseases—How close are we? FEBS J. 2022, 289, 5049–5073. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Konno, T.; Tada, M.; Tezuka, T.; Miura, T.; Mezaki, N.; Okazaki, K.I.; Arakawa, M.; Itoh, K.; Yamamoto, T.; et al. Characteristic microglial features in patients with hereditary diffuse leukoencephalopathy with spheroids. Ann. Neurol. 2016, 80, 554–565. [Google Scholar] [CrossRef]
- Kempthorne, L.; Yoon, H.; Madore, C.; Smith, S.; Wszolek, Z.K.; Rademakers, R.; Kim, J.; Butovsky, O.; Dickson, D.W. Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy. Acta Neuropathol. Commun. 2020, 8, 72. [Google Scholar] [CrossRef]
- Chitu, V.; Gokhan, S.; Gulinello, M.; Branch, C.A.; Patil, M.; Basu, R.; Stoddart, C.; Mehler, M.F.; Stanley, E.R. Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP). Neurobiol. Dis. 2015, 74, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Biundo, F.; Shlager, G.G.L.; Park, E.S.; Wang, P.; Gulinello, M.E.; Gokhan, S.; Ketchum, H.C.; Saha, K.; DeTure, M.A.; et al. Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling. Cell Rep. 2020, 30, 3004–3019. [Google Scholar] [CrossRef] [PubMed]
- Biundo, F.; Chitu, V.; Shlager, G.G.L.; Park, E.S.; Gulinello, M.E.; Saha, K.; Ketchum, H.C.; Fernandes, C.; Gokhan, S.; Mehler, M.F.; et al. Microglial reduction of colony stimulating factor-1 receptor expression is sufficient to confer adult onset leukodystrophy. Glia 2021, 69, 779–791. [Google Scholar] [CrossRef]
- Kober, D.L.; Alexander-Brett, J.M.; Karch, C.M.; Cruchaga, C.; Colonna, M.; Holtzman, M.J.; Brett, T.J. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 2016, 5, e20391. [Google Scholar] [CrossRef]
- Sudom, A.; Talreja, S.; Danao, J.; Bragg, E.; Kegel, R.; Min, X.; Richardson, J.; Zhang, Z.; Sharkov, N.; Marcora, E.; et al. Molecular basis for the loss-of-function effects of the Alzheimer’s disease-associated R47H variant of the immune receptor TREM2. J. Biol. Chem. 2018, 293, 12634–12646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Grabowski, G.A. Lysosomal acid lipase and atherosclerosis. Curr. Opin. Lipidol. 2004, 15, 539–544. [Google Scholar] [CrossRef]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854. [Google Scholar] [CrossRef] [PubMed]
- Gouna, G.; Klose, C.; Bosch-Queralt, M.; Liu, L.; Gokce, O.; Schifferer, M.; Cantuti-Castelvetri, L.; Simons, M. TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J. Exp. Med. 2021, 218, e20210227. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef]
- Otero, K.; Turnbull, I.R.; Poliani, P.L.; Vermi, W.; Cerutti, E.; Aoshi, T.; Tassi, I.; Takai, T.; Stanley, S.L.; Miller, M.; et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat. Immunol. 2009, 10, 734–743. [Google Scholar] [CrossRef]
- Cheng, Q.; Danao, J.; Talreja, S.; Wen, P.; Yin, J.; Sun, N.; Li, C.M.; Chui, D.; Tran, D.; Koirala, S.; et al. TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer’s disease variant Trem2(R47H) on murine myeloid cell function. J. Biol. Chem. 2018, 293, 12620–12633. [Google Scholar] [CrossRef]
- Bianchin, M.M.; Martin, K.C.; de Souza, A.C.; de Oliveira, M.A.; Rieder, C.R. Nasu-Hakola disease and primary microglial dysfunction. Nat. Rev. Neurol. 2010, 6, 523. [Google Scholar] [CrossRef]
- Paloneva, J.; Kestila, M.; Wu, J.; Salminen, A.; Bohling, T.; Ruotsalainen, V.; Hakola, P.; Bakker, A.B.; Phillips, J.H.; Pekkarinen, P.; et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 2000, 25, 357–361. [Google Scholar] [CrossRef]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef]
- Cheng, B.; Li, X.; Dai, K.; Duan, S.; Rong, Z.; Chen, Y.; Lu, L.; Liu, Z.; Huang, X.; Xu, H.; et al. Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) Interacts with Colony-Stimulating Factor 1 Receptor (CSF1R) but Is Not Necessary for CSF1/CSF1R-Mediated Microglial Survival. Front. Immunol. 2021, 12, 633796. [Google Scholar] [CrossRef]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell. Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Linnartz-Gerlach, B.; Bodea, L.G.; Klaus, C.; Ginolhac, A.; Halder, R.; Sinkkonen, L.; Walter, J.; Colonna, M.; Neumann, H. TREM2 triggers microglial density and age-related neuronal loss. Glia 2019, 67, 539–550. [Google Scholar] [CrossRef]
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Kurti, A.; Baker, K.E.; Liu, C.C.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M.; Bu, G.; Fryer, J.D. Behavioral and transcriptomic analysis of Trem2-null mice: Not all knockout mice are created equal. Hum. Mol. Genet. 2018, 27, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Gu, L.Z.; Zhang, Y.D.; Tan, L. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol. Aging 2014, 35, 1243–1251. [Google Scholar] [CrossRef]
- Peters, A.; Folger Sethares, C. The Fine Structure of the Aging Brain. Available online: http://www.bu.edu/agingbrain (accessed on 5 December 2017).
- Young, K.; Morrison, H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018, 136, e57648. [Google Scholar] [CrossRef]
- Rolfe, A.J.; Bosco, D.B.; Broussard, E.N.; Ren, Y. In Vitro Phagocytosis of Myelin Debris by Bone Marrow-Derived Macrophages. J. Vis. Exp. 2017, 130, e56322. [Google Scholar] [CrossRef]
- Larocca, J.N.; Norton, W.T. Isolation of myelin. Curr. Protoc. Cell Biol. 2007, Supplement 33, 3–25. [Google Scholar] [CrossRef]
- Sun, X.; Wang, X.; Chen, T.; Li, T.; Cao, K.; Lu, A.; Chen, Y.; Sun, D.; Luo, J.; Fan, J.; et al. Myelin activates FAK/Akt/NF-kappaB pathways and provokes CR3-dependent inflammatory response in murine system. PLoS ONE 2010, 5, e9380. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Ma, J.; Tanaka, K.F.; Shimizu, T.; Bernard, C.C.; Kakita, A.; Takahashi, H.; Pfeiffer, S.E.; Ikenaka, K. Microglial cystatin F expression is a sensitive indicator for ongoing demyelination with concurrent remyelination. J. Neurosci. Res. 2011, 89, 639–649. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Brioschi, S. Neuroinflammation and neurodegeneration in human brain at single-cell resolution. Nat. Rev. Immunol. 2020, 20, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Peters, A. Structural changes that occur during normal aging of primate cerebral hemispheres. Neurosci. Biobehav. Rev. 2002, 26, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.D.; Pieropan, F.; Chacon-De-La-Rocha, I.; Lecca, D.; Abbracchio, M.P.; Azim, K.; Butt, A.M. Functional genomic analyses highlight a shift in Gpr17-regulated cellular processes in oligodendrocyte progenitor cells and underlying myelin dysregulation in the aged mouse cerebrum. Aging Cell 2021, 20, e13335. [Google Scholar] [CrossRef]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Hill, R.A.; Li, A.M.; Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 2018, 21, 683–695. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biundo, F.; Chitu, V.; Gökhan, Ş.; Chen, E.; Oppong-Asare, J.; Stanley, E.R. Trem2 Enhances Demyelination in the Csf1r+/− Mouse Model of Leukoencephalopathy. Biomedicines 2023, 11, 2094. https://doi.org/10.3390/biomedicines11082094
Biundo F, Chitu V, Gökhan Ş, Chen E, Oppong-Asare J, Stanley ER. Trem2 Enhances Demyelination in the Csf1r+/− Mouse Model of Leukoencephalopathy. Biomedicines. 2023; 11(8):2094. https://doi.org/10.3390/biomedicines11082094
Chicago/Turabian StyleBiundo, Fabrizio, Violeta Chitu, Şölen Gökhan, Edward Chen, Jude Oppong-Asare, and E. Richard Stanley. 2023. "Trem2 Enhances Demyelination in the Csf1r+/− Mouse Model of Leukoencephalopathy" Biomedicines 11, no. 8: 2094. https://doi.org/10.3390/biomedicines11082094