

Metabolomic Approach to Identify Potential Biomarkers in KRAS-Mutant Pancreatic Cancer Cells

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cytotoxicity Assay
2.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.4. Sample Preparation for UHPLC/Q-TOF-MS
2.5. Liquid Chromatography and Mass Spectrometry
2.6. Data Processing and Analysis
2.7. Statistical Analysis
3. Results
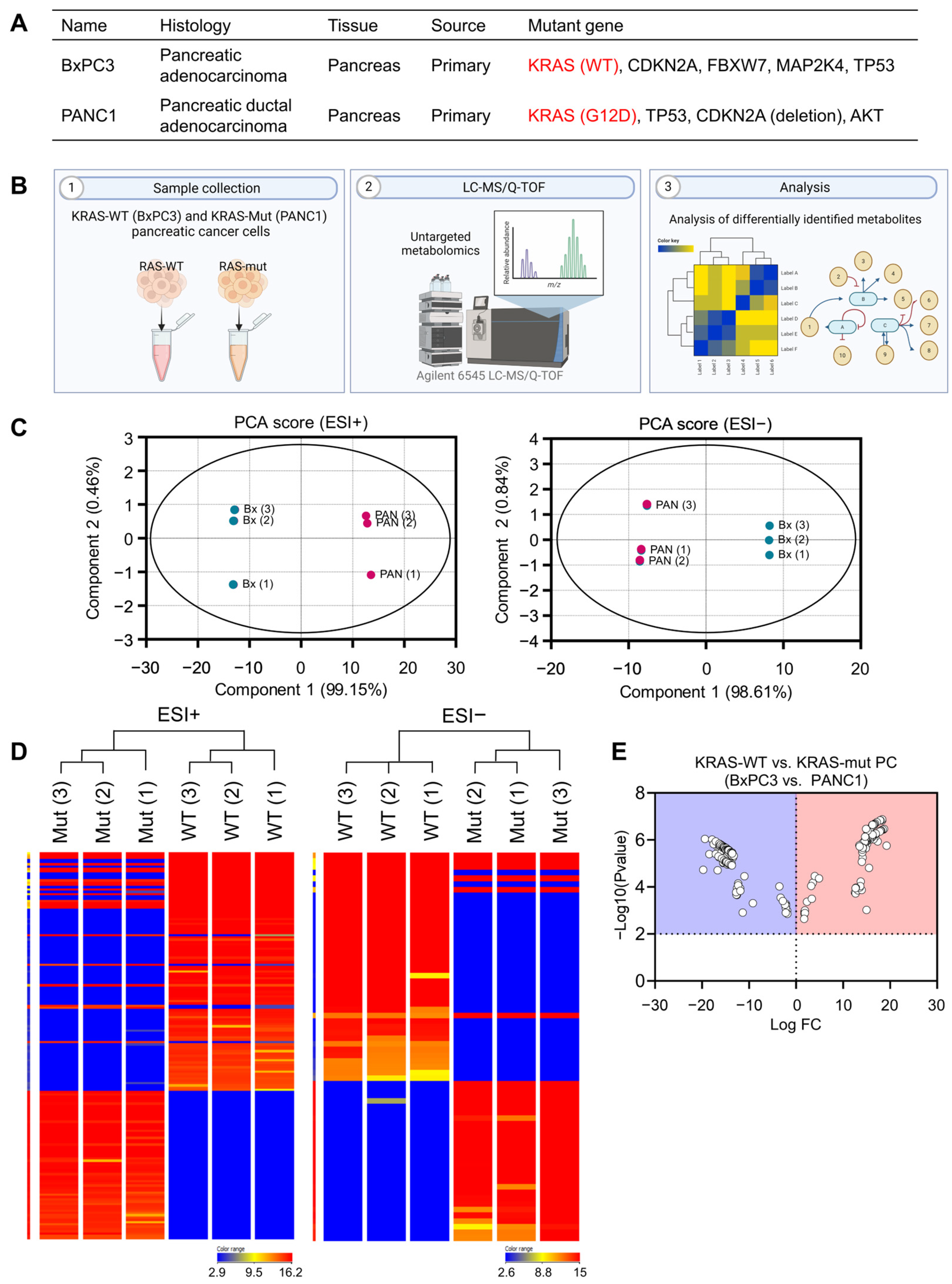
3.1. Different Metabolomic Profile between KRAS-Wildtype and KRAS-Mutant Pancreatic Cancer Cells
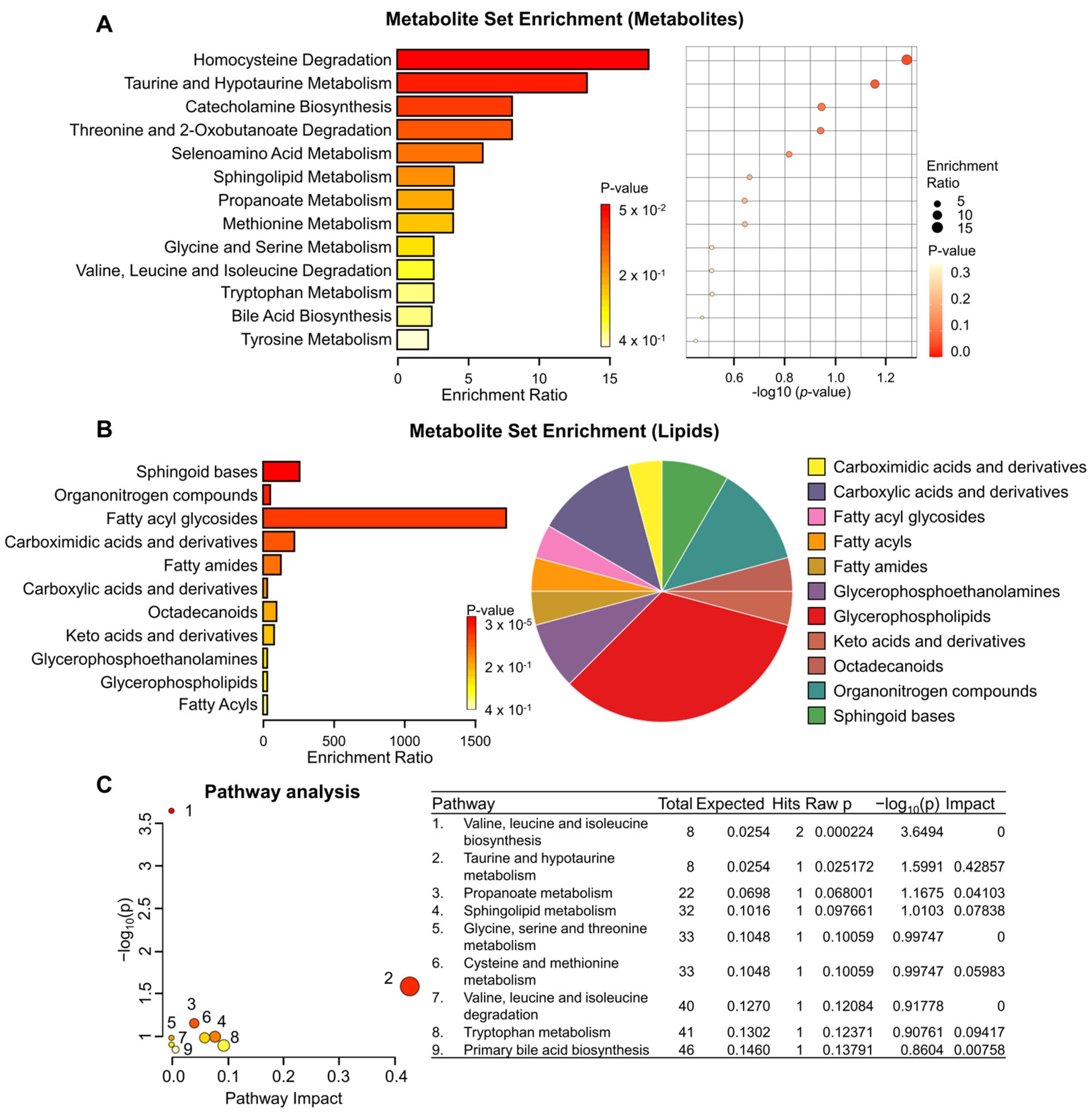
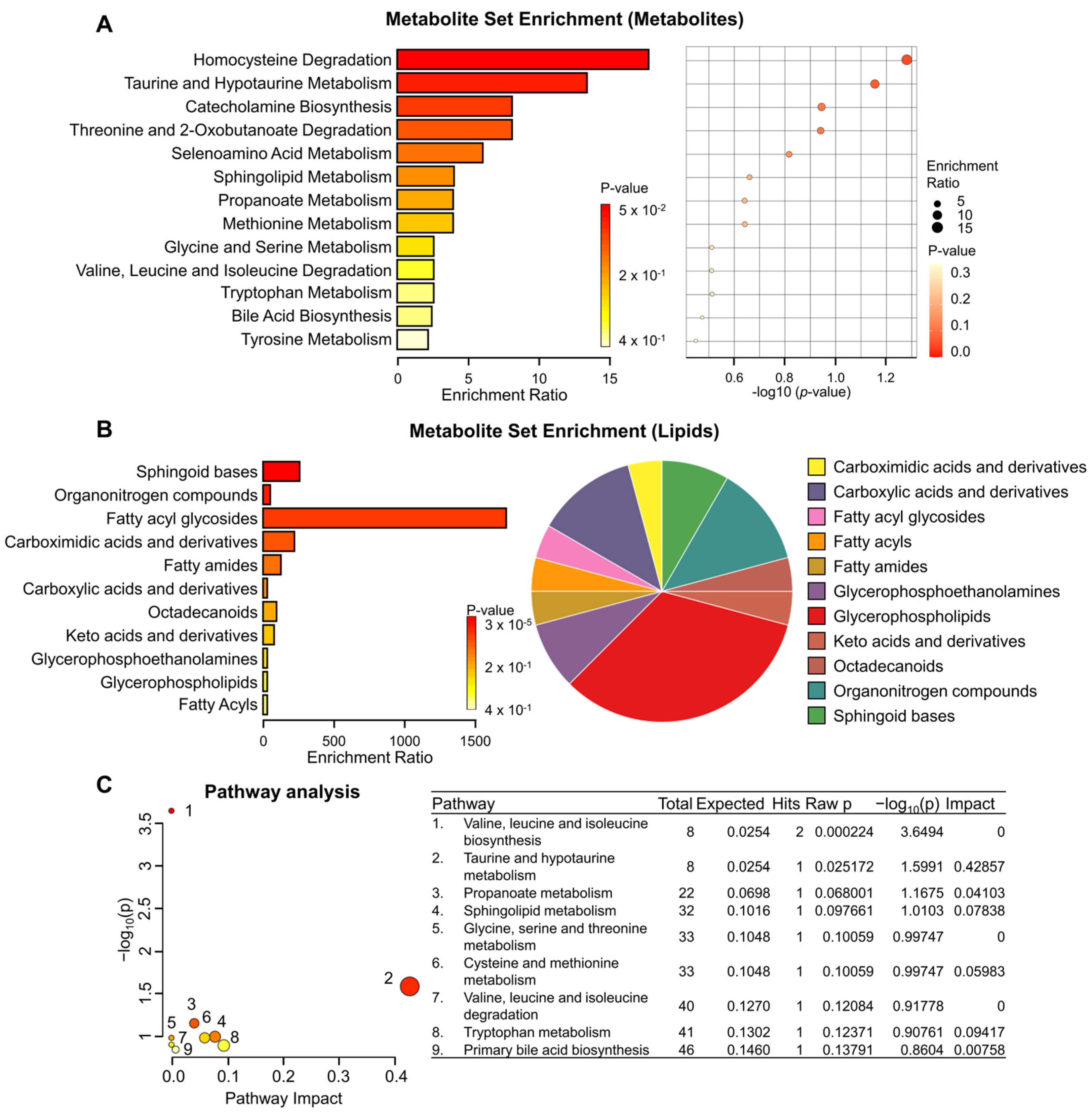
3.2. Metabolite Set Enrichment and Pathway Analysis of KRAS-Wildtype and KRAS-Mutant Pancreatic Cancer Cells
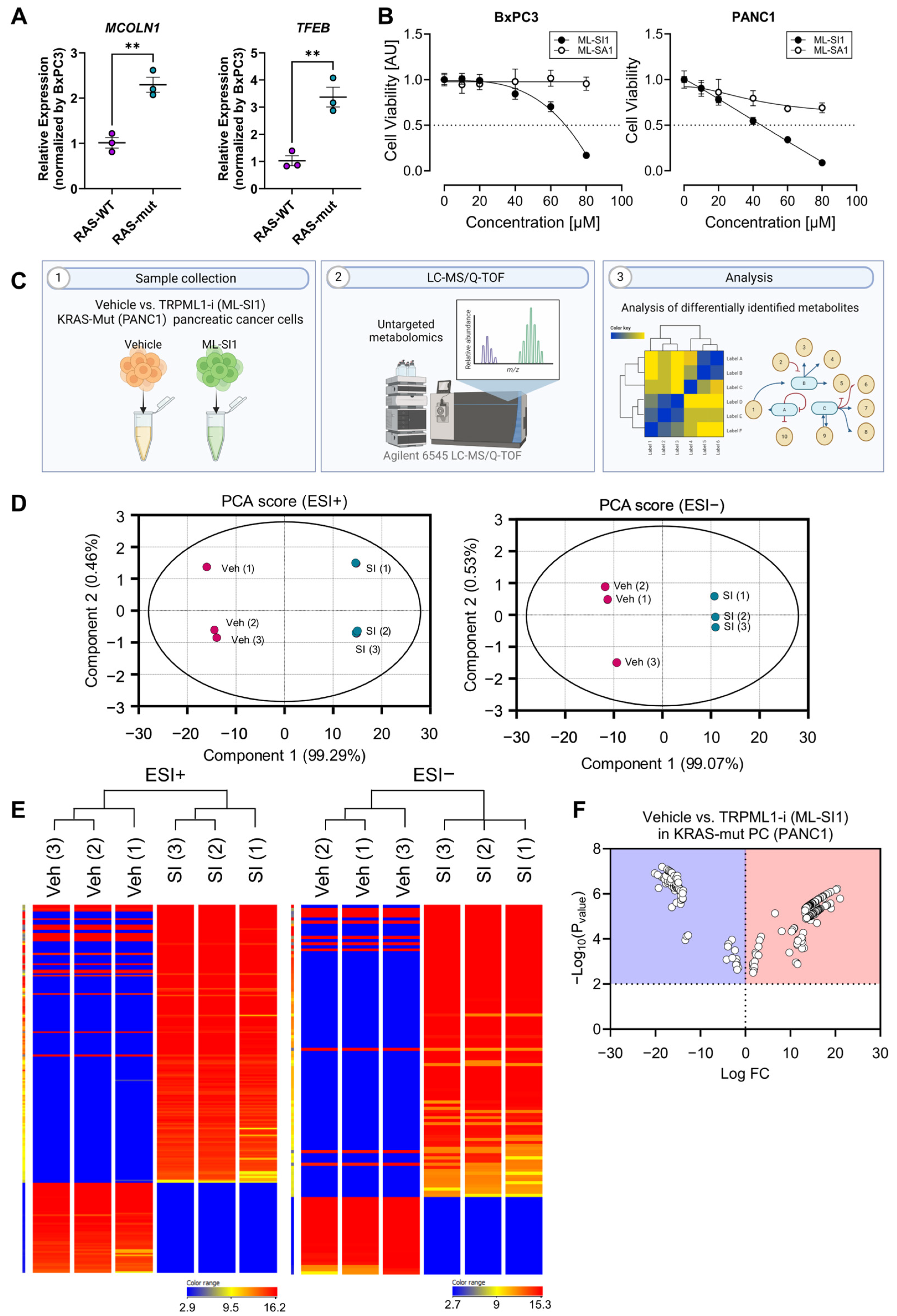
3.3. Alterations in Metabolomic Profile of KRAS-Mutant Pancreatic Cancer Cells by TRPML1 Inhibition
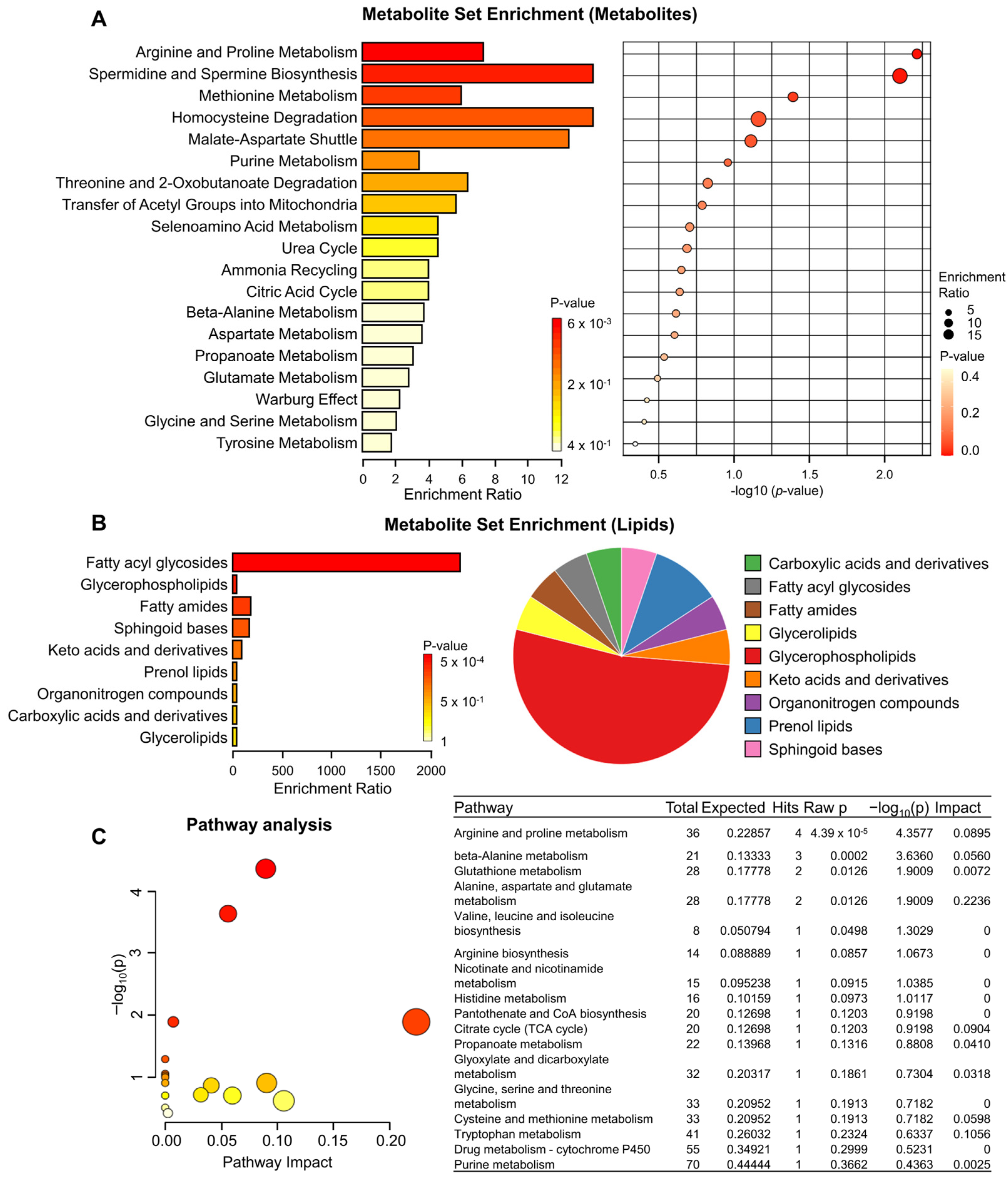
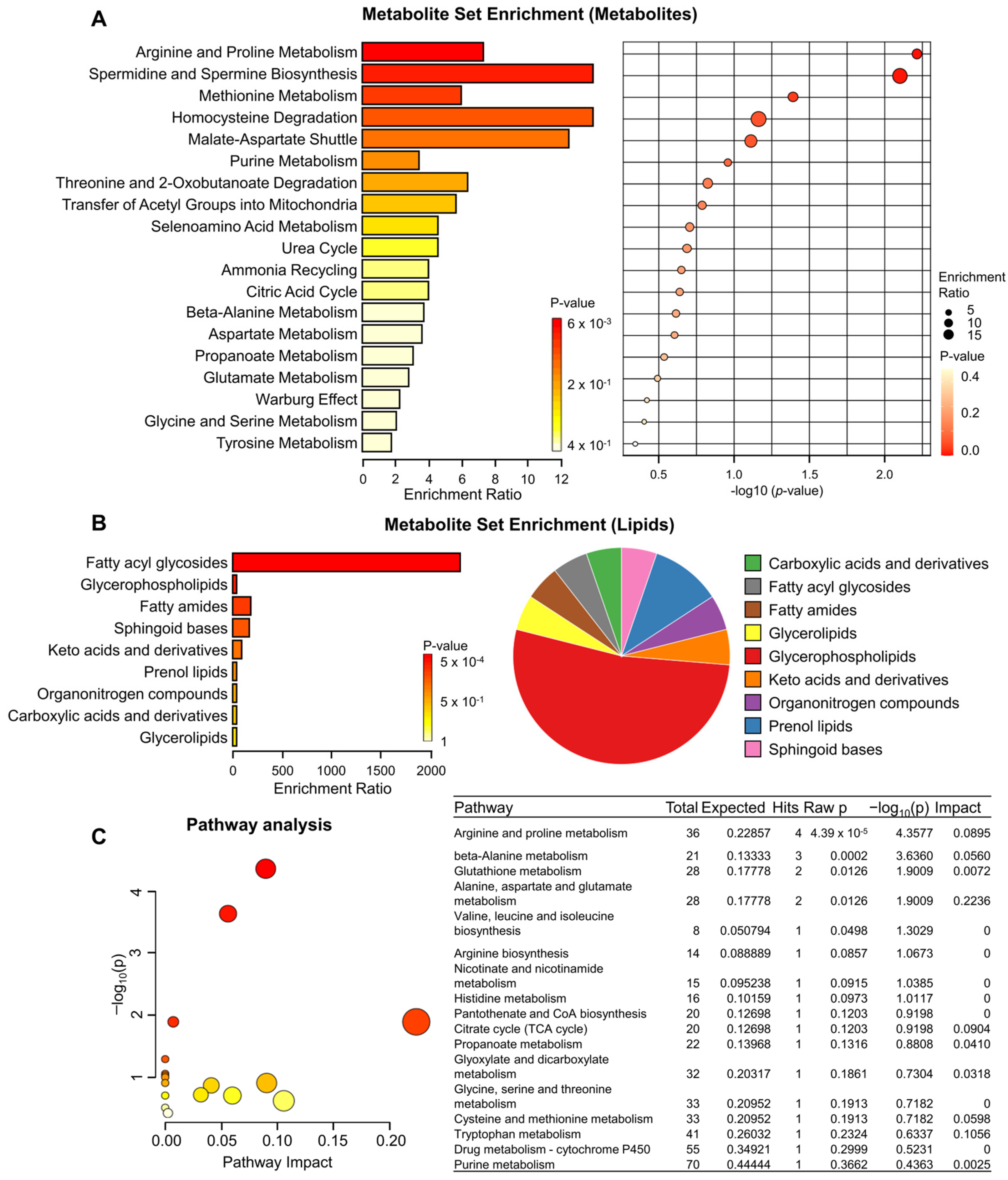
3.4. Metabolite Set Enrichment and Pathway Analysis of KRAS-mutant Pancreatic Cancer Cells by TRPML1 Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J Clin 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Gillen, S.; Schuster, T.; Meyer Zum Buschenfelde, C.; Friess, H.; Kleeff, J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Hasselluhn, M.C.; Neesse, A. A tangled tale of molecular subtypes in pancreatic cancer. Gut 2019, 68, 953–954. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.H.; Jang, K.T.; Lim, T.; Lee, J.; Choi, Y.L.; Jang, H.L.; Yi, J.H.; Baek, K.K.; Park, S.H.; et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol. Cancer Ther. 2011, 10, 1993–1999. [Google Scholar] [CrossRef]
- Schultz, N.A.; Roslind, A.; Christensen, I.J.; Horn, T.; Hogdall, E.; Pedersen, L.N.; Kruhoffer, M.; Burcharth, F.; Wojdemann, M.; Johansen, J.S. Frequencies and prognostic role of KRAS and BRAF mutations in patients with localized pancreatic and ampullary adenocarcinomas. Pancreas 2012, 41, 759–766. [Google Scholar] [CrossRef]
- Shin, S.H.; Kim, S.C.; Hong, S.M.; Kim, Y.H.; Song, K.B.; Park, K.M.; Lee, Y.J. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas 2013, 42, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef] [PubMed]
- Harsha, H.C.; Kandasamy, K.; Ranganathan, P.; Rani, S.; Ramabadran, S.; Gollapudi, S.; Balakrishnan, L.; Dwivedi, S.B.; Telikicherla, D.; Selvan, L.D.; et al. A compendium of potential biomarkers of pancreatic cancer. PLoS Med. 2009, 6, e1000046. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Pfeiffer, P.; Vilgrain, V.; Lamarca, A.; Seufferlein, T.; O’Reilly, E.M.; Hackert, T.; Golan, T.; Prager, G.; Haustermans, K.; et al. Pancreatic cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Diamandis, E.P.; Blasutig, I.M. Strategies for discovering novel pancreatic cancer biomarkers. J. Proteomics 2013, 81, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kulasingam, V.; Diamandis, E.P. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat. Clin. Pract. Oncol. 2008, 5, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef]
- Perez-Rambla, C.; Puchades-Carrasco, L.; Garcia-Flores, M.; Rubio-Briones, J.; Lopez-Guerrero, J.A.; Pineda-Lucena, A. Non-invasive urinary metabolomic profiling discriminates prostate cancer from benign prostatic hyperplasia. Metabolomics 2017, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Locasale, J.W. Metabolomics: A Primer. Trends Biochem. Sci. 2017, 42, 274–284. [Google Scholar] [CrossRef]
- Jung, J.; Venkatachalam, K. TRPML1 and RAS-driven cancers—Exploring a link with great therapeutic potential. Channels 2019, 13, 374–381. [Google Scholar] [CrossRef]
- Jung, J.; Cho, K.J.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20, e46685. [Google Scholar] [CrossRef]
- Qi, J.; Xing, Y.; Liu, Y.; Wang, M.M.; Wei, X.; Sui, Z.; Ding, L.; Zhang, Y.; Lu, C.; Fei, Y.H.; et al. MCOLN1/TRPML1 finely controls oncogenic autophagy in cancer by mediating zinc influx. Autophagy 2021, 17, 4401–4422. [Google Scholar] [CrossRef]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef]
- Santoni, G.; Santoni, M.; Maggi, F.; Marinelli, O.; Morelli, M.B. Emerging Role of Mucolipins TRPML Channels in Cancer. Front. Oncol. 2020, 10, 659. [Google Scholar] [CrossRef]
- Kim, B.; Kim, G.; Kim, H.; Song, Y.S.; Jung, J. Modulation of Cisplatin Sensitivity through TRPML1-Mediated Lysosomal Exocytosis in Ovarian Cancer Cells: A Comprehensive Metabolomic Approach. Cells 2024, 13, 115. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef]
- Hu, Z.D.; Yan, J.; Cao, K.Y.; Yin, Z.Q.; Xin, W.W.; Zhang, M.F. MCOLN1 Promotes Proliferation and Predicts Poor Survival of Patients with Pancreatic Ductal Adenocarcinoma. Dis. Markers 2019, 2019, 9436047. [Google Scholar] [CrossRef]
- Xu, M.; Almasi, S.; Yang, Y.; Yan, C.; Sterea, A.M.; Rizvi Syeda, A.K.; Shen, B.; Richard Derek, C.; Huang, P.; Gujar, S.; et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium 2019, 79, 80–88. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, H.; Liu, X.; Zhang, H.; Zhang, Y.; Bai, X.; Wang, L.; Li, H.; Li, X.; Zhang, S.; et al. Downregulated MCOLN1 Attenuates The Progression Of Non-Small-Cell Lung Cancer By Inhibiting Lysosome-Autophagy. Cancer Manag. Res. 2019, 11, 8607–8617. [Google Scholar] [CrossRef]
- Jung, J.; Liao, H.; Coker, S.A.; Liang, H.; Hancock, J.F.; Denicourt, C.; Venkatachalam, K. p53 mitigates the effects of oncogenic HRAS in urothelial cells via the repression of MCOLN1. iScience 2021, 24, 102701. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes. Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Landman, A. 2016 ASCO Gastrointestinal Cancers Symposium. Lancet Oncol 2016, 17, 282. [Google Scholar] [CrossRef]
- Makrilia, N.; Syrigos, K.N.; Saif, M.W. Treatment for refractory pancreatic cancer. Highlights from the “2011 ASCO Gastrointestinal Cancers Symposium”. San Francisco, CA, USA. January 20-22, 2011. JOP 2011, 12, 110–113. [Google Scholar]
- Huanwen, W.; Zhiyong, L.; Xiaohua, S.; Xinyu, R.; Kai, W.; Tonghua, L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 2009, 8, 125. [Google Scholar] [CrossRef]
- Rathos, M.J.; Joshi, K.; Khanwalkar, H.; Manohar, S.M.; Joshi, K.S. Molecular evidence for increased antitumor activity of gemcitabine in combination with a cyclin-dependent kinase inhibitor, P276-00 in pancreatic cancers. J. Transl. Med. 2012, 10, 161. [Google Scholar] [CrossRef]
- Kim, Y.; Han, D.; Min, H.; Jin, J.; Yi, E.C.; Kim, Y. Comparative proteomic profiling of pancreatic ductal adenocarcinoma cell lines. Mol. Cells 2014, 37, 888–898. [Google Scholar] [CrossRef]
- Liu, Q.; Lan, J.; Martinez-Jarquin, S.; Ge, W.; Zenobi, R. Screening Metabolic Biomarkers in KRAS Mutated Mouse Acinar and Human Pancreatic Cancer Cells via Single-Cell Mass Spectrometry. Anal. Chem. 2024, 96, 4918–4924. [Google Scholar] [CrossRef]
- Weber, N.; Hatsch, A.; Labagnere, L.; Heider, H. Production of (S)-2-aminobutyric acid and (S)-2-aminobutanol in Saccharomyces cerevisiae. Microb. Cell Fact. 2017, 16, 51. [Google Scholar] [CrossRef]
- Danchin, A.; Dondon, L. Serine sensitivity of Escherichia coli K 12: Partial characterization of a serine resistnat mutant that is extremely sensitive to 2-ketobutyrate. Mol. Gen. Genet. 1980, 178, 155–164. [Google Scholar] [CrossRef]
- Shigematsu, M.; Nakagawa, R.; Tomonaga, S.; Funaba, M.; Matsui, T. Fluctuations in metabolite content in the liver of magnesium-deficient rats. Br. J. Nutr. 2016, 116, 1694–1699. [Google Scholar] [CrossRef]
- Montalbano, S.; Raboni, S.; Sidoli, S.; Mozzarelli, A.; Bettati, S.; Buschini, A. Post-Translational Modifications of Histone Variants in the Absence and Presence of a Methionine-Depleting Enzyme in Normal and Cancer Cells. Cancers 2023, 15, 527. [Google Scholar] [CrossRef]
- Poirson-Bichat, F.; Goncalves, R.A.; Miccoli, L.; Dutrillaux, B.; Poupon, M.F. Methionine depletion enhances the antitumoral efficacy of cytotoxic agents in drug-resistant human tumor xenografts. Clin. Cancer Res. 2000, 6, 643–653. [Google Scholar]
- Gao, X.; Locasale, J.W.; Reid, M.A. Serine and Methionine Metabolism: Vulnerabilities in Lethal Prostate Cancer. Cancer Cell 2019, 35, 339–341. [Google Scholar] [CrossRef]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P., Jr.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Sun, Y.; Fowke, J.H.; Liang, X.; Mozhui, K.; Sen, S.; Bao, W.; Liu, B.; Snetselaar, L.G.; Wallace, R.B.; Shadyab, A.H.; et al. Changes in Dietary Intake of Methionine, Folate/Folic Acid and Vitamin B12 and Survival in Postmenopausal Women with Breast Cancer: A Prospective Cohort Study. Nutrients 2022, 14, 4747. [Google Scholar] [CrossRef]
- Perez-Miguelsanz, J.; Vallecillo, N.; Garrido, F.; Reytor, E.; Perez-Sala, D.; Pajares, M.A. Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1165–1182. [Google Scholar] [CrossRef]
- Pajares, M.A.; Perez-Sala, D. Betaine homocysteine S-methyltransferase: Just a regulator of homocysteine metabolism? Cell Mol. Life Sci. 2006, 63, 2792–2803. [Google Scholar] [CrossRef]
- Sakura, T.; Hayakawa, F.; Sugiura, I.; Murayama, T.; Imai, K.; Usui, N.; Fujisawa, S.; Yamauchi, T.; Yujiri, T.; Kakihana, K.; et al. High-dose methotrexate therapy significantly improved survival of adult acute lymphoblastic leukemia: A phase III study by JALSG. Leukemia 2018, 32, 626–632. [Google Scholar] [CrossRef]
- Baldwin, C.M.; Perry, C.M. Pemetrexed: A review of its use in the management of advanced non-squamous non-small cell lung cancer. Drugs 2009, 69, 2279–2302. [Google Scholar] [CrossRef]
- Pokrovsky, V.S.; Abo Qoura, L.; Morozova, E.; Bunik, V.I. Predictive markers for efficiency of the amino-acid deprivation therapies in cancer. Front. Med. (Lausanne) 2022, 9, 1035356. [Google Scholar] [CrossRef]
- Lien, E.C.; Ghisolfi, L.; Geck, R.C.; Asara, J.M.; Toker, A. Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci. Signal 2017, 10, 510. [Google Scholar] [CrossRef]
- Mecham, J.O.; Rowitch, D.; Wallace, C.D.; Stern, P.H.; Hoffman, R.M. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef]
- Halpern, B.C.; Clark, B.R.; Hardy, D.N.; Halpern, R.M.; Smith, R.A. The effect of replacement of methionine by homocystine on survival of malignant and normal adult mammalian cells in culture. Proc. Natl. Acad. Sci. USA 1974, 71, 1133–1136. [Google Scholar] [CrossRef]
- He, D.; Feng, H.; Sundberg, B.; Yang, J.; Powers, J.; Christian, A.H.; Wilkinson, J.E.; Monnin, C.; Avizonis, D.; Thomas, C.J.; et al. Methionine oxidation activates pyruvate kinase M2 to promote pancreatic cancer metastasis. Mol. Cell 2022, 82, 3045–3060.E11. [Google Scholar] [CrossRef]
- Fan, K.; Zhang, S.; Ni, X.; Shen, S.; Wang, J.; Sun, W.; Suo, T.; Liu, H.; Ni, X.; Liu, H. KRAS G12D mutation eliminates reactive oxygen species through the Nrf2/CSE/H (2)S axis and contributes to pancreatic cancer growth. Acta Biochim. Biophys. Sin. 2022, 54, 1731–1739. [Google Scholar] [CrossRef]
- Medina, C.B.; Mehrotra, P.; Arandjelovic, S.; Perry, J.S.A.; Guo, Y.; Morioka, S.; Barron, B.; Walk, S.F.; Ghesquiere, B.; Krupnick, A.S.; et al. Metabolites released from apoptotic cells act as tissue messengers. Nature 2020, 580, 130–135. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BxPC3 vs. PANC1 | Veh vs. ML-SI1 in PANC1 | ||
|---|---|---|---|---|
| Log FC | P (corr) | Log FC | P (corr) | |
| 2-Ketobutyric acid | 17.191 | 3.20 × 10−5 | −17.387 | 1.72 × 10−5 |
| Eicosanoyl-EA | 14.908 | 1.38 × 10−4 | −15.064 | 9.04 × 10−5 |
| PE(19:1(9Z)/0:0) | −14.896 | 1.46 × 10−4 | 2.085 | 2.53 × 10−2 |
| PI(20:4(5Z,8Z,11Z,14Z)/0:0) | −14.625 | 3.64 × 10−4 | 11.528 | 3.03 × 10−2 |
| PS(21:0/20:5(5Z,8Z,11Z,14Z,17Z)) | −14.342 | 3.64 × 10−4 | 12.680 | 2.22 × 10−3 |
| (4E,8E,10E-d18:3)sphingosine | −14.531 | 1.44 × 10−4 | 14.428 | 1.25 × 10−4 |
| 4,4′-Biphenyldithiol | −17.092 | 1.71 × 10−4 | 15.919 | 5.51 × 10−4 |
| LysoPE(0:0/20:0) | −13.412 | 2.20 × 10−4 | 16.017 | 1.24 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.; Jung, J. Metabolomic Approach to Identify Potential Biomarkers in KRAS-Mutant Pancreatic Cancer Cells. Biomedicines 2024, 12, 865. https://doi.org/10.3390/biomedicines12040865
Kim B, Jung J. Metabolomic Approach to Identify Potential Biomarkers in KRAS-Mutant Pancreatic Cancer Cells. Biomedicines. 2024; 12(4):865. https://doi.org/10.3390/biomedicines12040865
Chicago/Turabian StyleKim, Boyun, and Jewon Jung. 2024. "Metabolomic Approach to Identify Potential Biomarkers in KRAS-Mutant Pancreatic Cancer Cells" Biomedicines 12, no. 4: 865. https://doi.org/10.3390/biomedicines12040865
APA StyleKim, B., & Jung, J. (2024). Metabolomic Approach to Identify Potential Biomarkers in KRAS-Mutant Pancreatic Cancer Cells. Biomedicines, 12(4), 865. https://doi.org/10.3390/biomedicines12040865