
Unraveling the Cardiac Matrix: From Diabetes to Heart Failure, Exploring Pathways and Potential Medications

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
3. Collagen Synthesis
3.1. C-Terminal Propeptide of Procollagen Type I
3.2. Procollagen Type I N-Terminal Propeptide
3.3. Procollagen Type III Amino-Terminal Propeptide
4. Collagen Degradation
4.1. C-Terminal Telopeptide of Collagen Type I
4.2. Matrix Metalloproteinases
4.3. Tissue Inhibitors of Metalloproteinases
5. Collagen Metabolism
5.1. Transforming Growth Factor-Β and SMADS
5.2. Corin
6. Inflammatory Factors
6.1. Galectin-3
6.2. Tumor Necrosis Factor-α and Interleukins
6.3. MicroRNA
7. Insulin Resistance and Hyperinsulinemia
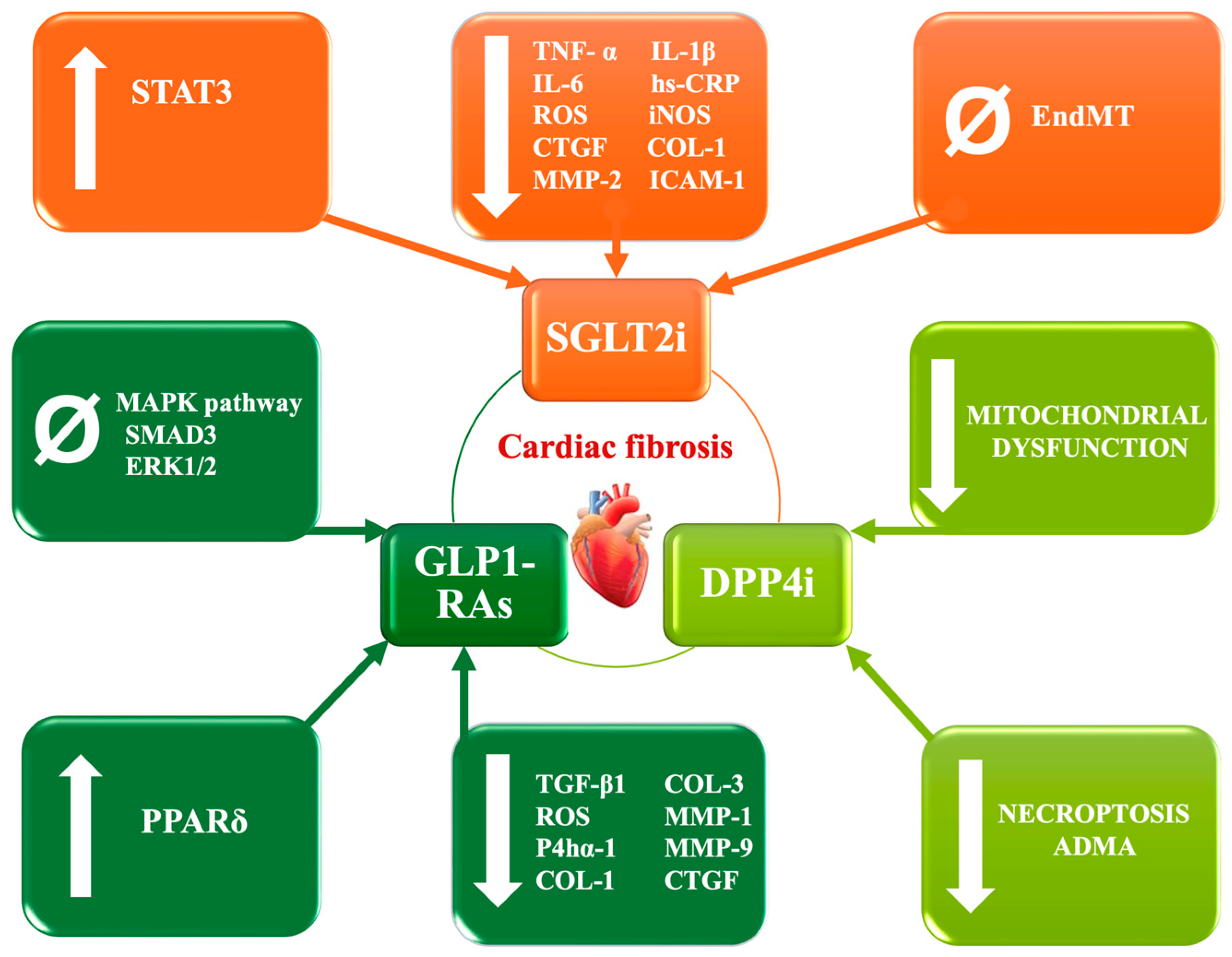
8. Novel Drugs and Their Anti-Fibrotic Role
8.1. SGLT2 Inhibitors
8.2. Glucagon-like Peptide-1 Receptor Agonists
8.3. Dipeptidyl Peptidase-4 Inhibitors
8.4. Neprilysin Inhibitor
8.5. Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase
9. Limitations and Future Directions
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Frantz, S.; Hundertmark, M.J.; Schulz-Menger, J.; Bengel, F.M.; Bauersachs, J. Left ventricular remodelling post-myocardial infarction: Pathophysiology, imaging, and novel therapies. Eur. Heart J. 2022, 43, 2549–2561. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akagi, S.; Saito, Y.; Ejiri, K.; Matsuo, N.; Ichikawa, K.; Iwasaki, K.; Naito, T.; et al. Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 3587. [Google Scholar] [CrossRef] [PubMed]
- Preis, S.R.; Pencina, M.J.; Hwang, S.J.; D’Agostino, R.B., Sr.; Savage, P.J.; Levy, D.; Fox, C.S. Trends in cardiovascular disease risk factors in individuals with and without diabetes mellitus in the Framingham Heart Study. Circulation 2009, 120, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Matheus, A.S.; Tannus, L.R.; Cobas, R.A.; Palma, C.C.; Negrato, C.A.; Gomes, M.B. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013, 2013, 653789. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of Hyperglycemia in Type 2 Diabetes, 2022. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2022, 45, 2753–2786. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, Y.; Zhang, W.; Jia, Q.; Wang, X.; Li, Y.; Lv, S.; Zhang, J. Roles of Biomarkers in Myocardial Fibrosis. Aging Dis. 2020, 11, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Ma, T.; Huang, X.; Zheng, H.; Huang, G.; Li, W.; Liu, X.; Liang, J.; Cao, Y.; Hu, Y.; Huang, Y. SFRP2 Improves Mitochondrial Dynamics and Mitochondrial Biogenesis, Oxidative Stress, and Apoptosis in Diabetic Cardiomyopathy. Oxidative Med. Cell Longev. 2021, 2021, 9265016. [Google Scholar] [CrossRef]
- Salib, M.; Girerd, N.; Simon, A.; Kearney-Schwartz, A.; Duarte, K.; Leroy, C.; Rossignol, P.; Benetos, A.; Frimat, L.; Girerd, S. Levels of Procollagen Type I C-Terminal Pro-Peptide and Galectin-3, Arterial Stiffness Measured By Pulse Wave Velocity, and Cardiovascular Morbidity and Mortality in 44 Patients 2 Years After Kidney Transplantation. Ann. Transplant. 2023, 28, e938137. [Google Scholar] [CrossRef]
- Ponikowska, B.; Iwanek, G.; Zdanowicz, A.; Urban, S.; Zymlinski, R.; Ponikowski, P.; Biegus, J. Biomarkers of Myocardial Injury and Remodeling in Heart Failure. J. Pers. Med. 2022, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.; Rossignol, P.; Pizard, A.; Machu, J.L.; Collier, T.; Girerd, N.; Huby, A.C.; Gonzalez, A.; Diez, J.; Lopez, B.; et al. Potential spironolactone effects on collagen metabolism biomarkers in patients with uncontrolled blood pressure. Heart 2019, 105, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; O’Meara, E.; Claggett, B.; Prescott, M.F.; Solomon, S.D.; Swedberg, K.; Packer, M.; McMurray, J.J.V.; Shi, V.; Lefkowitz, M.; et al. Effects of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFrEF. J. Am. Coll. Cardiol. 2019, 73, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, X.; Chen, C.; Jiang, W.; Lu, D.; Liu, Q.; Wang, K.; Yan, Y.; Jiang, Z.; Geng, J.; et al. Renal Denervation Effects on Myocardial Fibrosis and Ventricular Arrhythmias in Rats with Ischemic Cardiomyopathy. Cell Physiol. Biochem. 2018, 46, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Santanasto, A.J.; Cvejkus, R.K.; Wojczynski, M.K.; Marron, M.M.; Schupf, N.; Christensen, K.; Thyagarajan, B.; Zmuda, J.M. Circulating Procollagen Type III N-Terminal Peptide and Physical Function in Adults from the Long Life Family Study. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Osokina, A.; Karetnikova, V.; Polikutina, O.; Ivanova, A.; Gruzdeva, O.; Dyleva, Y.; Kokov, A.; Brel, N.; Pecherina, T.; Barbarash, O. Prognostic potential of cardiac structural and functional parameters and N-terminal propeptide of type III procollagen in predicting cardiac fibrosis one year after myocardial infarction with preserved left ventricular ejection fraction. Aging 2021, 13, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Jarolim, P.; Scirica, B.M.; Braunwald, E.; Park, J.G.; Das, S.; Sabatine, M.S.; Morrow, D.A. Biomarker of Collagen Turnover (C-Terminal Telopeptide) and Prognosis in Patients With Non- ST -Elevation Acute Coronary Syndromes. J. Am. Heart Assoc. 2019, 8, e011444. [Google Scholar] [CrossRef] [PubMed]
- Manhenke, C.; Orn, S.; Squire, I.; Radauceanu, A.; Alla, F.; Zannad, F.; Dickstein, K. The prognostic value of circulating markers of collagen turnover after acute myocardial infarction. Int. J. Cardiol. 2011, 150, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Owolabi, U.S.; Amraotkar, A.R.; Coulter, A.R.; Singam, N.S.V.; Aladili, B.N.; Singh, A.; Trainor, P.J.; Mitra, R.; DeFilippis, A.P. Change in matrix metalloproteinase 2, 3, and 9 levels at the time of and after acute atherothrombotic myocardial infarction. J. Thromb. Thrombolysis 2020, 49, 235–244. [Google Scholar] [CrossRef]
- Myasoedova, V.A.; Chistiakov, D.A.; Grechko, A.V.; Orekhov, A.N. Matrix metalloproteinases in pro-atherosclerotic arterial remodeling. J. Mol. Cell Cardiol. 2018, 123, 159–167. [Google Scholar] [CrossRef]
- Zhou, K.; Li, Y.; Xu, Y.; Guo, R. Circulating Matrix Metalloproteinase-28 Levels Are Related to GRACE Scores and Short-Term Outcomes in Patients with Acute Myocardial Infarction. Biomed. Res. Int. 2020, 2020, 9206703. [Google Scholar] [CrossRef] [PubMed]
- Basia, D.; Gupta, M.D.; Kunal, S.; Muheeb, G.; Girish, M.P.; Bansal, A.; Batra, V.; Yusuf, J.; Mukhopadhyay, S.; Tyagi, S.; et al. Matrix metalloproteinases and their gene polymorphism in young ST-segment elevation myocardial infarction. Indian Heart J. 2022, 74, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Somuncu, M.U.; Pusuroglu, H.; Karakurt, H.; Bolat, I.; Karakurt, S.T.; Demir, A.R.; Isiksacan, N.; Akgul, O.; Surgit, O. The prognostic value of elevated matrix metalloproteinase-9 in patients undergoing primary percutaneous coronary intervention for ST-elevation myocardial infarction: A two-year prospective study. Rev. Port. Cardiol. 2020, 39, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Spiers, J.P.; Waterstone, M.; Russell-Hallinan, A.; Gallagher, J.; McDonald, K.; Ryan, C.; Gilmer, J.; Ledwidge, M. Investigation of association of genetic variant rs3918242 of matrix metalloproteinase-9 with hypertension, myocardial infarction and progression of ventricular dysfunction in Irish Caucasian patients with diabetes: A report from the STOP-HF follow-up programme. BMC Cardiovasc. Disord. 2021, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.T.; Dang, Y.; Zhang, F.F.; Zhang, Q.H.; Wu, H.B.; Liu, G. Combination of serum TIMP-3, CA125, and NT-proBNP in predicting ventricular remodeling in patients with heart failure following acute myocardial infarction. Cardiovasc. Diagn. Ther. 2020, 10, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.F.; Gao, Y.; Liu, J.M.; Lu, J.P.; Wang, Y.P.; Wang, S.M.; Hou, L.B.; Tian, A.X.; Gao, Y. Prognostic value of tissue inhibitor of metalloproteinase-matrix metalloproteinase biomarkers at 30 days in patients with acute myocardial infarction without reperfusion therapy. Chin. Med. J. 2020, 134, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.G.; Wei, Y.; Jiang, J.; Wang, L.; Liang, H.Y.; Lei, C.B. Effect of TGF-beta1 on myocardial cell apoptosis in rats with acute myocardial infarction via MAPK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, J.; Zhang, Q.; Shao, J.; Du, K.; Xu, X.; Kong, Y. Prognostic Value of Plasma Soluble Corin in Patients With Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 67, 2008–2014. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.C.; Liu, Y.; Yang, H.; Du, K.; Kong, Y.; Xu, X.H. Plasma Corin as a Predictor of Cardiovascular Events in Patients With Chronic Heart Failure. JACC Heart Fail. 2016, 4, 664–669. [Google Scholar] [CrossRef]
- Hu, W.; Chen, S.; Song, Y.; Zhu, F.; Shi, J.; Han, X.; Zhou, D.; Zhi, Z.; Zhang, F.; Shen, Y.; et al. Serum Soluble Corin Deficiency Predicts Major Disability within 3 Months after Acute Stroke. PLoS ONE 2016, 11, e0163731. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.F.; Chou, R.H.; Lin, S.J.; Li, S.Y.; Huang, P.H. Serum PCSK6 and corin levels are not associated with cardiovascular outcomes in patients undergoing coronary angiography. PLoS ONE 2019, 14, e0226129. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, J.; Li, P.; Han, P.; Kang, Y.J.; Zhang, W. Gene expression patterns and related pathways in the hearts of rhesus monkeys subjected to prolonged myocardial ischemia. Exp. Biol. Med. 2023, 248, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Vucic, R.M.; Andrejic, O.M.; Stokanovic, D.; Stoimenov, T.J.; McClements, L.; Nikolic, V.N.; Sreckovic, M.; Veselinovic, M.; Aleksandric, S.; Popadic, V.; et al. Galectin-3 as a Prognostic Biomarker in Patients with First Acute Myocardial Infarction without Heart Failure. Diagnostics 2023, 13, 3348. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, K.; Huang, X.; Feng, L.; Yuan, Y.; Li, J.; Lao, Y.; Guo, Z. Association Between Serum Galectin-3 Levels and Coronary Stenosis Severity in Patients With Coronary Artery Disease. Front. Cardiovasc. Med. 2022, 9, 818162. [Google Scholar] [CrossRef] [PubMed]
- Kokturk, U.; Pusuroglu, H.; Somuncu, M.U.; Akgul, O.; Uygur, B.; Ozyilmaz, S.; Isiksacan, N.; Surgit, O.; Yildirim, A. Short and Long-Term Prognostic Significance of Galectin-3 in Patients with ST-Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. Angiology 2023, 74, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huai, W.; Ye, X.; Pan, Y.; Yang, X.; Chen, M.; Ma, Q.B.; Gao, Y.; Zhang, Y. Circulating plasma galectin-3 predicts new-onset atrial fibrillation in patients after acute myocardial infarction during hospitalization. BMC Cardiovasc. Disord. 2022, 22, 392. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, O.; Karaayvaz, E.; Erdogan, T.; Panc, C.; Sarikaya, R.; Oncul, A.; Bilge, A.K. A new biomarker that predicts ventricular arrhythmia in patients with ischemic dilated cardiomyopathy: Galectin-3. Rev. Port. Cardiol. 2021, 40, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Obeid, S.; Yousif, N.; Davies, A.; Loretz, R.; Saleh, L.; Niederseer, D.; Noor, H.A.; Amin, H.; Mach, F.; Gencer, B.; et al. Prognostic role of plasma galectin-3 levels in acute coronary syndrome. Eur. Heart J. Acute Cardiovasc. Care 2020, 9, 869–878. [Google Scholar] [CrossRef]
- Grandin, E.W.; Jarolim, P.; Murphy, S.A.; Ritterova, L.; Cannon, C.P.; Braunwald, E.; Morrow, D.A. Galectin-3 and the development of heart failure after acute coronary syndrome: Pilot experience from PROVE IT-TIMI 22. Clin. Chem. 2012, 58, 267–273. [Google Scholar] [CrossRef]
- Cassaglia, P.; Penas, F.; Betazza, C.; Fontana Estevez, F.; Miksztowicz, V.; Martinez Naya, N.; Llamosas, M.C.; Noli Truant, S.; Wilensky, L.; Volberg, V.; et al. Genetic Deletion of Galectin-3 Alters the Temporal Evolution of Macrophage Infiltration and Healing Affecting the Cardiac Remodeling and Function after Myocardial Infarction in Mice. Am. J. Pathol. 2020, 190, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Targeting galectin-3 in myocardial infarction: A unique opportunity for biomarker-guided therapy. Cardiovasc. Res. 2023, 119, 2495–2496. [Google Scholar] [CrossRef] [PubMed]
- Daseke, M.J., 2nd; Valerio, F.M.; Kalusche, W.J.; Ma, Y.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res. Cardiol. 2019, 114, 37. [Google Scholar] [CrossRef] [PubMed]
- Daseke, M.J., 2nd; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell Signal 2021, 77, 109816. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10, 1676. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jin, Q.; Zhang, L.; He, S.; Song, Y.; Xu, L.; Deng, C.; Wang, L.; Qin, X.; Xie, M. Ultrasonic Microbubble Cavitation Deliver Gal-3 shRNA to Inhibit Myocardial Fibrosis after Myocardial Infarction. Pharmaceutics 2023, 15, 729. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Frangogiannis, N.G. Macrophages in myocardial infarction. Am. J. Physiol. Cell Physiol. 2022, 323, C1304–C1324. [Google Scholar] [CrossRef] [PubMed]
- Mo, D.; Tian, W.; Zhang, H.N.; Feng, Y.D.; Sun, Y.; Quan, W.; Hao, X.W.; Wang, X.Y.; Liu, X.X.; Li, C.; et al. Cardioprotective effects of galectin-3 inhibition against ischemia/reperfusion injury. Eur. J. Pharmacol. 2019, 863, 172701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cheng, K.; Chen, H.; Tu, J.; Shen, Y.; Pang, L.; Wu, W. Galectin-3 knock down inhibits cardiac ischemia-reperfusion injury through interacting with bcl-2 and modulating cell apoptosis. Arch. Biochem. Biophys. 2020, 694, 108602. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Kang, P.; Wang, J.; Fang, D.; Fang, T.; Yu, Y.; Zhang, W.; Shen, L.; Li, Z.; Wang, H.; Ye, H.; et al. Activation of ALDH2 attenuates high glucose induced rat cardiomyocyte fibrosis and necroptosis. Free Radic. Biol. Med. 2020, 146, 198–210. [Google Scholar] [CrossRef]
- Fang, L.; Ellims, A.H.; Beale, A.L.; Taylor, A.J.; Murphy, A.; Dart, A.M. Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Am. J. Transl. Res. 2017, 9, 5063–5073. [Google Scholar]
- Bhandary, B.; Meng, Q.; James, J.; Osinska, H.; Gulick, J.; Valiente-Alandi, I.; Sargent, M.A.; Bhuiyan, M.S.; Blaxall, B.C.; Molkentin, J.D.; et al. Cardiac Fibrosis in Proteotoxic Cardiac Disease is Dependent Upon Myofibroblast TGF -beta Signaling. J. Am. Heart Assoc. 2018, 7, e010013. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, C.; Song, Q.; Li, Y.; Da, X.; Yu, Y.; Li, H.; Clark, I.M.; Chen, Q.; Wang, Q.K. ADAMTS16 activates latent TGF-beta, accentuating fibrosis and dysfunction of the pressure-overloaded heart. Cardiovasc. Res. 2020, 116, 956–969. [Google Scholar] [CrossRef] [PubMed]
- Dzialo, E.; Czepiel, M.; Tkacz, K.; Siedlar, M.; Kania, G.; Blyszczuk, P. WNT/beta-Catenin Signaling Promotes TGF-beta-Mediated Activation of Human Cardiac Fibroblasts by Enhancing IL-11 Production. Int. J. Mol. Sci. 2021, 22, 10072. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Hung, C.S.; Liao, C.W.; Wei, L.H.; Chen, C.W.; Shun, C.T.; Wen, W.F.; Wan, C.H.; Wu, X.M.; Chang, Y.Y.; et al. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc. Res. 2018, 114, 690–702. [Google Scholar] [CrossRef]
- Tanaka, H.; Sun, T.; Kinashi, H.; Kamiya, K.; Yamaguchi, M.; Nobata, H.; Sakata, F.; Kim, H.; Mizuno, M.; Kunoki, S.; et al. Interleukin-6 blockade reduces salt-induced cardiac inflammation and fibrosis in subtotal nephrectomized mice. Am. J. Physiol. Renal Physiol. 2022, 323, F654–F665. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.C.S.; Huibers, M.M.H.; van Kuik, J.; de Weger, R.A.; Vink, A.; de Jonge, N. The Interleukin-33/ST2 Pathway Is Expressed in the Failing Human Heart and Associated with Pro-fibrotic Remodeling of the Myocardium. J. Cardiovasc. Transl. Res. 2018, 11, 15–21. [Google Scholar] [CrossRef]
- Zhong, C.; Min, K.; Zhao, Z.; Zhang, C.; Gao, E.; Huang, Y.; Zhang, X.; Baldini, M.; Roy, R.; Yang, X.; et al. MAP Kinase Phosphatase-5 Deficiency Protects Against Pressure Overload-Induced Cardiac Fibrosis. Front. Immunol. 2021, 12, 790511. [Google Scholar] [CrossRef]
- Mia, M.M.; Cibi, D.M.; Ghani, S.; Singh, A.; Tee, N.; Sivakumar, V.; Bogireddi, H.; Cook, S.A.; Mao, J.; Singh, M.K. Loss of Yap/Taz in cardiac fibroblasts attenuates adverse remodelling and improves cardiac function. Cardiovasc. Res. 2022, 118, 1785–1804. [Google Scholar] [CrossRef]
- Pordzik, J.; Jakubik, D.; Jarosz-Popek, J.; Wicik, Z.; Eyileten, C.; De Rosa, S.; Indolfi, C.; Siller-Matula, J.M.; Czajka, P.; Postula, M. Significance of circulating microRNAs in diabetes mellitus type 2 and platelet reactivity: Bioinformatic analysis and review. Cardiovasc. Diabetol. 2019, 18, 113. [Google Scholar] [CrossRef]
- Zhao, Y.; Du, D.; Chen, S.; Chen, Z.; Zhao, J. New Insights into the Functions of MicroRNAs in Cardiac Fibrosis: From Mechanisms to Therapeutic Strategies. Genes 2022, 13, 1390. [Google Scholar] [CrossRef]
- Guo, R.; Nair, S. Role of microRNA in diabetic cardiomyopathy: From mechanism to intervention. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2070–2077. [Google Scholar] [CrossRef]
- Jin, Z.Q. MicroRNA targets and biomarker validation for diabetes-associated cardiac fibrosis. Pharmacol. Res. 2021, 174, 105941. [Google Scholar] [CrossRef]
- Rawal, S.; Munasinghe, P.E.; Nagesh, P.T.; Lew, J.K.S.; Jones, G.T.; Williams, M.J.A.; Davis, P.; Bunton, D.; Galvin, I.F.; Manning, P.; et al. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the Type 2 diabetic human and mouse heart. Clin. Sci. 2017, 131, 847–863. [Google Scholar] [CrossRef]
- Geng, H.; Guan, J. MiR-18a-5p inhibits endothelial-mesenchymal transition and cardiac fibrosis through the Notch2 pathway. Biochem. Biophys. Res. Commun. 2017, 491, 329–336. [Google Scholar] [CrossRef]
- Liu, X.; Guo, B.; Zhang, W.; Ma, B.; Li, Y. MiR-20a-5p overexpression prevented diabetic cardiomyopathy via inhibition of cardiomyocyte apoptosis, hypertrophy, fibrosis and JNK/NF-kappaB signalling pathway. J. Biochem. 2021, 170, 349–362. [Google Scholar] [CrossRef]
- Tang, C.M.; Zhang, M.; Huang, L.; Hu, Z.Q.; Zhu, J.N.; Xiao, Z.; Zhang, Z.; Lin, Q.X.; Zheng, X.L.; Yang, M.; et al. CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Sci. Rep. 2017, 7, 40342. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.H.; Zhang, Y.Y.; Wang, Y.Z.; Wang, J.; Zhao, Y.; Jin, X.X.; Xue, G.L.; Li, P.H.; Sun, Y.L.; et al. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFbeta1 and miR-29 pathways. Sci. Rep. 2016, 6, 23010. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Puthanveetil, P.; Feng, B.; Matkovich, S.J.; Dorn, G.W., 2nd; Chakrabarti, S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell Mol. Med. 2014, 18, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Wang, Y.; Li, H.; Li, Y.; Sahil, A.; Lv, J.; Liu, Y.; Yang, Z.; Dong, R.; Xue, H.; et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-beta1/Smads signaling in diabetic cardiomyopathy. FASEB J. 2020, 34, 5282–5298. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chen, S.; Gordon, A.D.; Chakrabarti, S. miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J. Mol. Cell Cardiol. 2017, 105, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Cao, Y.; Chen, S.; Chu, X.; Chu, Y.; Chakrabarti, S. miR-200b Mediates Endothelial-to-Mesenchymal Transition in Diabetic Cardiomyopathy. Diabetes 2016, 65, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, X.; Lin, Q.; Xu, Q. Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA. Gene 2019, 715, 143995. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Gao, L.; Xiao, L.; Yao, R.; Du, B.; Li, Y.; Wu, L.; Liang, C.; Huang, Z.; et al. miR-222 inhibits cardiac fibrosis in diabetic mice heart via regulating Wnt/beta-catenin-mediated endothelium to mesenchymal transition. J. Cell Physiol. 2020, 235, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xu, W.; Zhang, W.; Wang, W.; Liu, T.; Zhou, X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics 2019, 9, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yao, Y.; Shi, S.; Zhou, M.; Zhou, Y.; Wang, M.; Chiu, J.J.; Huang, Z.; Zhang, W.; Liu, M.; et al. Inhibition of miR-21 alleviated cardiac perivascular fibrosis via repressing EndMT in T1DM. J. Cell Mol. Med. 2020, 24, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, W.; Xu, M.; Huang, H.; Wang, J.; Chen, X. Micro-RNA 21Targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can. J. Cardiol. 2014, 30, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Meng, C.; Han, F.; Yang, J.; Wang, J.; Zhu, Y.; Cui, X.; Zuo, M.; Xu, J.; Chang, B. Vildagliptin Attenuates Myocardial Dysfunction and Restores Autophagy via miR-21/SPRY1/ERK in Diabetic Mice Heart. Front. Pharmacol. 2021, 12, 634365. [Google Scholar] [CrossRef]
- Shi, P.; Zhao, X.D.; Shi, K.H.; Ding, X.S.; Tao, H. MiR-21-3p triggers cardiac fibroblasts pyroptosis in diabetic cardiac fibrosis via inhibiting androgen receptor. Exp. Cell Res. 2021, 399, 112464. [Google Scholar] [CrossRef]
- Che, H.; Wang, Y.; Li, Y.; Lv, J.; Li, H.; Liu, Y.; Dong, R.; Sun, Y.; Xu, X.; Zhao, J.; et al. Inhibition of microRNA-150-5p alleviates cardiac inflammation and fibrosis via targeting Smad7 in high glucose-treated cardiac fibroblasts. J. Cell Physiol. 2020, 235, 7769–7779. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, W.; Xu, X.; Guo, L.; Zhang, Y.; Han, S.; Shen, D. The mechanism of TGF-beta/miR-155/c-Ski regulates endothelial-mesenchymal transition in human coronary artery endothelial cells. Biosci. Rep. 2017, 37, BSR20160603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Cui, Y.; Li, B.; Luo, X.; Li, B.; Tang, Y. miR-155 regulates high glucose-induced cardiac fibrosis via the TGF-beta signaling pathway. Mol. Biosyst. 2016, 13, 215–224. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, M. GAS5 regulates diabetic cardiomyopathy via miR-221-3p/p27 axis-associated autophagy. Mol. Med. Rep. 2021, 23, 135. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, X.; Chen, X.; Yang, S.; Chen, H. Inhibition of miR-223 attenuates the NLRP3 inflammasome activation, fibrosis, and apoptosis in diabetic cardiomyopathy. Life Sci. 2020, 256, 117980. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, Y.; Deng, Y.; Li, H. MicroRNA-223 Regulates Cardiac Fibrosis After Myocardial Infarction by Targeting RASA1. Cell Physiol. Biochem. 2018, 46, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Gao, L.; Liu, Y.; Liu, Y.; Yao, R.; Li, Y.; Xiao, L.; Wu, L.; Du, B.; Huang, Z.; et al. MiR-451 antagonist protects against cardiac fibrosis in streptozotocin-induced diabetic mouse heart. Life Sci. 2019, 224, 12–22. [Google Scholar] [CrossRef]
- Valkov, N.; King, M.E.; Moeller, J.; Liu, H.; Li, X.; Zhang, P. MicroRNA-1-Mediated Inhibition of Cardiac Fibroblast Proliferation Through Targeting Cyclin D2 and CDK6. Front. Cardiovasc. Med. 2019, 6, 65. [Google Scholar] [CrossRef]
- Zhong, C.; Wang, K.; Liu, Y.; Lv, D.; Zheng, B.; Zhou, Q.; Sun, Q.; Chen, P.; Ding, S.; Xu, Y.; et al. miR-19b controls cardiac fibroblast proliferation and migration. J. Cell Mol. Med. 2016, 20, 1191–1197. [Google Scholar] [CrossRef]
- Yuan, X.; Pan, J.; Wen, L.; Gong, B.; Li, J.; Gao, H.; Tan, W.; Liang, S.; Zhang, H.; Wang, X. MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J. Cell Mol. Med. 2020, 24, 227–237. [Google Scholar] [CrossRef]
- Huang, Y.; Qi, Y.; Du, J.Q.; Zhang, D.F. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert. Opin. Ther. Targets 2014, 18, 1355–1365. [Google Scholar] [CrossRef]
- Wei, Y.; Yan, X.; Yan, L.; Hu, F.; Ma, W.; Wang, Y.; Lu, S.; Zeng, Q.; Wang, Z. Inhibition of microRNA-155 ameliorates cardiac fibrosis in the process of angiotensin II-induced cardiac remodeling. Mol. Med. Rep. 2017, 16, 7287–7296. [Google Scholar] [CrossRef]
- Yuan, X.; Pan, J.; Wen, L.; Gong, B.; Li, J.; Gao, H.; Tan, W.; Liang, S.; Zhang, H.; Wang, X. MiR-144-3p Enhances Cardiac Fibrosis After Myocardial Infarction by Targeting PTEN. Front. Cell Dev. Biol. 2019, 7, 249. [Google Scholar] [CrossRef]
- Zhou, X.L.; Xu, H.; Liu, Z.B.; Wu, Q.C.; Zhu, R.R.; Liu, J.C. miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1. J. Cell Mol. Med. 2018, 22, 3816–3824. [Google Scholar] [CrossRef]
- Surina, S.; Fontanella, R.A.; Scisciola, L.; Marfella, R.; Paolisso, G.; Barbieri, M. miR-21 in Human Cardiomyopathies. Front. Cardiovasc. Med. 2021, 8, 767064. [Google Scholar] [CrossRef]
- Wang, X.; Morelli, M.B.; Matarese, A.; Sardu, C.; Santulli, G. Cardiomyocyte-derived exosomal microRNA-92a mediates post-ischemic myofibroblast activation both in vitro and ex vivo. ESC Heart Fail. 2020, 7, 284–288. [Google Scholar] [CrossRef]
- Morelli, M.B.; Shu, J.; Sardu, C.; Matarese, A.; Santulli, G. Cardiosomal microRNAs Are Essential in Post-Infarction Myofibroblast Phenoconversion. Int. J. Mol. Sci. 2019, 21, 201. [Google Scholar] [CrossRef]
- Ranjan, P.; Kumari, R.; Goswami, S.K.; Li, J.; Pal, H.; Suleiman, Z.; Cheng, Z.; Krishnamurthy, P.; Kishore, R.; Verma, S.K. Myofibroblast-Derived Exosome Induce Cardiac Endothelial Cell Dysfunction. Front. Cardiovasc. Med. 2021, 8, 676267. [Google Scholar] [CrossRef]
- Jazbutyte, V.; Fiedler, J.; Kneitz, S.; Galuppo, P.; Just, A.; Holzmann, A.; Bauersachs, J.; Thum, T. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age 2013, 35, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Li, H.; Cao, H.; Dong, Y.; Gao, L.; Liu, Z.; Ge, J.; Zhu, H. Therapeutic silencing miR-146b-5p improves cardiac remodeling in a porcine model of myocardial infarction by modulating the wound reparative phenotype. Protein Cell 2021, 12, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, O.A.; Alghamdi, M.; Alfaifi, J.; Alamri, M.M.S.; Al-Shahrani, A.M.; Alharthi, M.H.; Alshahrani, A.M.; Alhalafi, A.H.; Adam, M.I.E.; Bahashwan, E.; et al. The emerging role of miRNAs in myocardial infarction: From molecular signatures to therapeutic targets. Pathol. Res. Pract. 2024, 253, 155087. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Dasgupta, C.; Mulder, C.; Zhang, L. MicroRNA-210 Controls Mitochondrial Metabolism and Protects Heart Function in Myocardial Infarction. Circulation 2022, 145, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, S.; Xu, G.; Chen, F.; Ding, F. MicroRNA-15a inhibition protects against hypoxia/reoxygenation-induced apoptosis of cardiomyocytes by targeting mothers against decapentaplegic homolog 7. Mol. Med. Rep. 2017, 15, 3699–3705. [Google Scholar] [CrossRef] [PubMed]
- Taubel, J.; Hauke, W.; Rump, S.; Viereck, J.; Batkai, S.; Poetzsch, J.; Rode, L.; Weigt, H.; Genschel, C.; Lorch, U.; et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: Results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur. Heart J. 2021, 42, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, Y.; Yuan, J.; Gao, W.; Zhong, X.; Yao, K.; Lin, L.; Ge, J. Dendritic cell-derived exosomal miR-494-3p promotes angiogenesis following myocardial infarction. Int. J. Mol. Med. 2021, 47, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Seferovic, P.M.; Petrie, M.C.; Filippatos, G.S.; Anker, S.D.; Rosano, G.; Bauersachs, J.; Paulus, W.J.; Komajda, M.; Cosentino, F.; de Boer, R.A.; et al. Type 2 diabetes mellitus and heart failure: A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 853–872. [Google Scholar] [CrossRef]
- Lee, M.M.Y.; McMurray, J.J.V.; Lorenzo-Almoros, A.; Kristensen, S.L.; Sattar, N.; Jhund, P.S.; Petrie, M.C. Diabetic cardiomyopathy. Heart 2019, 105, 337–345. [Google Scholar] [CrossRef]
- Zamora, M.; Villena, J.A. Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2833. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Lawrence, S.P.; Lizunov, V.A.; Cushman, S.W.; Holman, G.D. A common trafficking route for GLUT4 in cardiomyocytes in response to insulin, contraction and energy-status signalling. J. Cell Sci. 2009, 122, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.L.; Feng, Q.; Pan, S.; Fu, W.J.; Liu, Z.; Liu, Z. Diabetic cardiomyopathy: Early diagnostic biomarkers, pathogenetic mechanisms, and therapeutic interventions. Cell Death Discov. 2023, 9, 256. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Dong, X.; Li, S.; Jiang, F.; Chen, J.; Yu, S.; Dong, B.; Su, Q. Effects of (Pro)renin Receptor on Diabetic Cardiomyopathy Pathological Processes in Rats via the PRR-AMPK-YAP Pathway. Front. Physiol. 2021, 12, 657378. [Google Scholar] [CrossRef] [PubMed]
- Bodiga, V.L.; Eda, S.R.; Bodiga, S. Advanced glycation end products: Role in pathology of diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Cavalera, M.; Wang, J.; Russo, I.; Shinde, A.; Kong, P.; Gonzalez-Quesada, C.; Rai, V.; Dobaczewski, M.; Lee, D.W.; et al. Smad3 Signaling Promotes Fibrosis While Preserving Cardiac and Aortic Geometry in Obese Diabetic Mice. Circ. Heart Fail. 2015, 8, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Mou, Y.L.; Xie, Y.Y. Research progress in relations between renin angiotensin system and diabetic cardiomyopathy. Sheng Li Ke Xue Jin Zhan 2011, 42, 269–275. [Google Scholar] [PubMed]
- Fuentes-Antras, J.; Ioan, A.M.; Tunon, J.; Egido, J.; Lorenzo, O. Activation of toll-like receptors and inflammasome complexes in the diabetic cardiomyopathy-associated inflammation. Int. J. Endocrinol. 2014, 2014, 847827. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Pal, P.B.; Sonowal, H.; Shukla, K.; Srivastava, S.K.; Ramana, K.V. Aldose Reductase Mediates NLRP3 Inflammasome-Initiated Innate Immune Response in Hyperglycemia-Induced Thp1 Monocytes and Male Mice. Endocrinology 2017, 158, 3661–3675. [Google Scholar] [CrossRef]
- Grubic Rotkvic, P.; Cigrovski Berkovic, M.; Bulj, N.; Rotkvic, L.; Celap, I. Sodium-glucose cotransporter 2 inhibitors’ mechanisms of action in heart failure. World J. Diabetes 2020, 11, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Garla, V.V.; Butler, J.; Lien, L.F. SGLT-2 Inhibitors in Heart Failure: Guide for Prescribing and Future Perspectives. Curr. Cardiol. Rep. 2021, 23, 59. [Google Scholar] [CrossRef] [PubMed]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef]
- Ghezzi, C.; Hirayama, B.A.; Gorraitz, E.; Loo, D.D.; Liang, Y.; Wright, E.M. SGLT2 inhibitors act from the extracellular surface of the cell membrane. Physiol. Rep. 2014, 2, e12058. [Google Scholar] [CrossRef] [PubMed]
- Al Jobori, H.; Daniele, G.; Adams, J.; Cersosimo, E.; Solis-Herrera, C.; Triplitt, C.; DeFronzo, R.A.; Abdul-Ghani, M. Empagliflozin Treatment Is Associated With Improved beta-Cell Function in Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2018, 103, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Merovci, A.; Mari, A.; Solis-Herrera, C.; Xiong, J.; Daniele, G.; Chavez-Velazquez, A.; Tripathy, D.; Urban McCarthy, S.; Abdul-Ghani, M.; DeFronzo, R.A. Dapagliflozin lowers plasma glucose concentration and improves beta-cell function. J. Clin. Endocrinol. Metab. 2015, 100, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Luna-Marco, C.; Iannantuoni, F.; Hermo-Argibay, A.; Devos, D.; Salazar, J.D.; Victor, V.M.; Rovira-Llopis, S. Cardiovascular benefits of SGLT2 inhibitors and GLP-1 receptor agonists through effects on mitochondrial function and oxidative stress. Free Radic. Biol. Med. 2024, 213, 19–35. [Google Scholar] [CrossRef]
- Wang, C.; Qin, Y.; Zhang, X.; Yang, Y.; Wu, X.; Liu, J.; Qin, S.; Chen, K.; Xiao, W. Effect of Dapagliflozin on Indicators of Myocardial Fibrosis and Levels of Inflammatory Factors in Heart Failure Patients. Dis. Markers 2022, 2022, 5834218. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKalpha/TGF-beta/Smad signalling in type 2 diabetic rats. J. Cell Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef] [PubMed]
- Katsurada, K.; Nandi, S.S.; Sharma, N.M.; Patel, K.P. Enhanced Expression and Function of Renal SGLT2 (Sodium-Glucose Cotransporter 2) in Heart Failure: Role of Renal Nerves. Circ. Heart Fail. 2021, 14, e008365. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; DeMets, D.L.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Langkilde, A.M.; Martinez, F.A.; Bengtsson, O.; Ponikowski, P.; Sabatine, M.S.; et al. A trial to evaluate the effect of the sodium-glucose co-transporter 2 inhibitor dapagliflozin on morbidity and mortality in patients with heart failure and reduced left ventricular ejection fraction (DAPA-HF). Eur. J. Heart Fail. 2019, 21, 665–675. [Google Scholar] [CrossRef]
- Gager, G.M.; von Lewinski, D.; Sourij, H.; Jilma, B.; Eyileten, C.; Filipiak, K.; Hulsmann, M.; Kubica, J.; Postula, M.; Siller-Matula, J.M. Effects of SGLT2 Inhibitors on Ion Homeostasis and Oxidative Stress associated Mechanisms in Heart Failure. Biomed. Pharmacother. 2021, 143, 112169. [Google Scholar] [CrossRef] [PubMed]
- Faridvand, Y.; Kazemzadeh, H.; Vahedian, V.; Mirzajanzadeh, P.; Nejabati, H.R.; Safaie, N.; Maroufi, N.F.; Pezeshkian, M.; Nouri, M.; Jodati, A. Dapagliflozin attenuates high glucose-induced endothelial cell apoptosis and inflammation through AMPK/SIRT1 activation. Clin. Exp. Pharmacol. Physiol. 2022, 49, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Li, F.F.; Gao, G.; Li, Q.; Zhu, H.H.; Su, X.F.; Wu, J.D.; Ye, L.; Ma, J.H. Influence of Dapagliflozin on Glycemic Variations in Patients with Newly Diagnosed Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016, 5347262. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.W.; Que, J.Q.; Liu, S.; Huang, K.Y.; Qian, L.; Weng, Y.B.; Rong, F.N.; Wang, L.; Zhou, Y.Y.; Xue, Y.J.; et al. Sodium-Glucose Co-transporter-2 Inhibitor of Dapagliflozin Attenuates Myocardial Ischemia/Reperfusion Injury by Limiting NLRP3 Inflammasome Activation and Modulating Autophagy. Front. Cardiovasc. Med. 2021, 8, 768214. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Efentakis, P.; Balafas, E.; Togliatto, G.; Davos, C.H.; Varela, A.; Dimitriou, C.A.; Nikolaou, P.E.; Maratou, E.; Lambadiari, V.; et al. Empagliflozin Limits Myocardial Infarction in Vivo and Cell Death in Vitro: Role of STAT3, Mitochondria, and Redox Aspects. Front. Physiol. 2017, 8, 1077. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Yurista, S.R.; Sillje, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.M.; Pavez Giani, M.G.; Hillebrands, J.L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef]
- Daud, E.; Ertracht, O.; Bandel, N.; Moady, G.; Shehadeh, M.; Reuveni, T.; Atar, S. The impact of empagliflozin on cardiac physiology and fibrosis early after myocardial infarction in non-diabetic rats. Cardiovasc. Diabetol. 2021, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.; Wang, C.Y.; Su, M.M.; Lin, L.Y.; Yang, W.S. Effect of Empagliflozin on Cardiac Function, Adiposity, and Diffuse Fibrosis in Patients with Type 2 Diabetes Mellitus. Sci. Rep. 2019, 9, 15348. [Google Scholar] [CrossRef] [PubMed]
- Adel, S.M.H.; Jorfi, F.; Mombeini, H.; Rashidi, H.; Fazeli, S. Effect of a low dose of empagliflozin on short-term outcomes in type 2 diabetics with acute coronary syndrome after percutaneous coronary intervention. Saudi Med. J. 2022, 43, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.A.; Zhang, Y.; Desjardins, J.F.; Nghiem, L.; Visram, A.; Batchu, S.N.; Yerra, V.G.; Kabir, G.; Thai, K.; Advani, A.; et al. Load-independent effects of empagliflozin contribute to improved cardiac function in experimental heart failure with reduced ejection fraction. Cardiovasc. Diabetol. 2020, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, X.; Liu, H.; Chen, Y.; Li, P.; Liu, L.; Li, J.; Ren, Y.; Huang, J.; Xiong, E.; et al. Empagliflozin Ameliorates Diabetic Cardiomyopathy via Attenuating Oxidative Stress and Improving Mitochondrial Function. Oxidative Med. Cell Longev. 2022, 2022, 1122494. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Hodrea, J.; Saeed, A.; Molnar, A.; Fintha, A.; Barczi, A.; Wagner, L.J.; Szabo, A.J.; Fekete, A.; Balogh, D.B. SGLT2 inhibitor dapagliflozin prevents atherosclerotic and cardiac complications in experimental type 1 diabetes. PLoS ONE 2022, 17, e0263285. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Li, X.; Kerindongo, R.P.; Preckel, B.; Kalina, J.O.; Hollmann, M.W.; Zuurbier, C.J.; Weber, N.C. Canagliflozin inhibits inflammasome activation in diabetic endothelial cells—Revealing a novel calcium-dependent anti-inflammatory effect of canagliflozin on human diabetic endothelial cells. Biomed. Pharmacother. 2023, 159, 114228. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.J.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2020, 36, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Mazer, C.D.; Yan, A.T.; Mason, T.; Garg, V.; Teoh, H.; Zuo, F.; Quan, A.; Farkouh, M.E.; Fitchett, D.H.; et al. Effect of Empagliflozin on Left Ventricular Mass in Patients With Type 2 Diabetes Mellitus and Coronary Artery Disease: The EMPA-HEART CardioLink-6 Randomized Clinical Trial. Circulation 2019, 140, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.M.; Gandy, S.; McCrimmon, R.; Houston, J.G.; Struthers, A.D.; Lang, C.C. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: The DAPA-LVH trial. Eur. Heart J. 2020, 41, 3421–3432. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S. Anti-inflammatory effects of empagliflozin in patients with type 2 diabetes and insulin resistance. Diabetol. Metab. Syndr. 2018, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Sezai, A.; Sekino, H.; Unosawa, S.; Taoka, M.; Osaka, S.; Tanaka, M. Canagliflozin for Japanese patients with chronic heart failure and type II diabetes. Cardiovasc. Diabetol. 2019, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Effect of Empagliflozin on Endothelial Function in Patients With Type 2 Diabetes and Cardiovascular Disease: Results from the Multicenter, Randomized, Placebo-Controlled, Double-Blind EMBLEM Trial. Diabetes Care 2019, 42, e159–e161. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jun, H.S. Anti-diabetic actions of glucagon-like peptide-1 on pancreatic beta-cells. Metabolism 2014, 63, 9–19. [Google Scholar] [CrossRef]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Orskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef]
- Sharma, D.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements & challenges. Biomed. Pharmacother. 2018, 108, 952–962. [Google Scholar] [CrossRef]
- Ussher, J.R.; Drucker, D.J. Glucagon-like peptide 1 receptor agonists: Cardiovascular benefits and mechanisms of action. Nat. Rev. Cardiol. 2023, 20, 463–474. [Google Scholar] [CrossRef]
- Tan, Q.; Akindehin, S.E.; Orsso, C.E.; Waldner, R.C.; DiMarchi, R.D.; Muller, T.D.; Haqq, A.M. Recent Advances in Incretin-Based Pharmacotherapies for the Treatment of Obesity and Diabetes. Front. Endocrinol. 2022, 13, 838410. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.P.; Pratley, R.E. GLP-1 Analogs and DPP-4 Inhibitors in Type 2 Diabetes Therapy: Review of Head-to-Head Clinical Trials. Front. Endocrinol. 2020, 11, 178. [Google Scholar] [CrossRef]
- Uccellatore, A.; Genovese, S.; Dicembrini, I.; Mannucci, E.; Ceriello, A. Comparison Review of Short-Acting and Long-Acting Glucagon-like Peptide-1 Receptor Agonists. Diabetes Ther. 2015, 6, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Lin, H.; Tian, S.; Liu, S.; Li, J.; Lv, X.; Chen, S.; Zhao, L.; Pu, F.; Chen, X.; et al. Glucagon-like peptide-1 receptor activation maintains extracellular matrix integrity by inhibiting the activity of mitogen-activated protein kinases and activator protein-1. Free Radic. Biol. Med. 2021, 177, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.K.; Ma, D.X.; Wang, Z.M.; Hu, X.F.; Li, S.L.; Tian, H.Z.; Wang, M.J.; Shu, Y.W.; Yang, J. The glucagon-like peptide-1 (GLP-1) analog liraglutide attenuates renal fibrosis. Pharmacol. Res. 2018, 131, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lin, T.; Shi, M.; Chen, X.; Wu, P. Liraglutide suppresses production of extracellular matrix proteins and ameliorates renal injury of diabetic nephropathy by enhancing Wnt/beta-catenin signaling. Am. J. Physiol. Renal Physiol. 2020, 319, F458–F468. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, L.; Feng, B.; He, N.; Zhang, Y.; Ye, H. Protective effects of glucagon-like peptide-1 on cardiac remodeling by inhibiting oxidative stress through mammalian target of rapamycin complex 1/p70 ribosomal protein S6 kinase pathway in diabetes mellitus. J. Diabetes Investig. 2020, 11, 39–51. [Google Scholar] [CrossRef]
- Picatoste, B.; Ramirez, E.; Caro-Vadillo, A.; Iborra, C.; Ares-Carrasco, S.; Egido, J.; Tunon, J.; Lorenzo, O. Sitagliptin reduces cardiac apoptosis, hypertrophy and fibrosis primarily by insulin-dependent mechanisms in experimental type-II diabetes. Potential roles of GLP-1 isoforms. PLoS ONE 2013, 8, e78330. [Google Scholar] [CrossRef]
- Zhu, Q.; Luo, Y.; Wen, Y.; Wang, D.; Li, J.; Fan, Z. Semaglutide inhibits ischemia/reperfusion-induced cardiomyocyte apoptosis through activating PKG/PKCepsilon/ERK1/2 pathway. Biochem. Biophys. Res. Commun. 2023, 647, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bin Dayel, A.F.; Alonazi, A.S.; Alrasheed, N.M.; Alamin, M.A.; Sarawi, W.S.; Alharbi, A.O.; Alabbad, N.A.; Albuaijan, D.A.; Alassiri, D.N.; Aljarbua, A.F.; et al. Role of the integrin-linked kinase/TGF-beta/SMAD pathway in sitagliptin-mediated cardioprotective effects in a rat model of diabetic cardiomyopathy. J. Pharm. Pharmacol. 2024, 76, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Chen, H.; Xu, F.; Wang, J.; Liu, Y.; Xing, X.; Guo, L.; Zhang, M.; Lu, Q. Liraglutide alleviates cardiac fibrosis through inhibiting P4halpha-1 expression in STZ-induced diabetic cardiomyopathy. Acta Biochim. Biophys. Sin. 2019, 51, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, X.; Yang, L.; Yang, L.; Ma, H. Liraglutide ameliorates myocardial damage in experimental diabetic rats by inhibiting pyroptosis via Sirt1/AMPK signaling. Iran. J. Basic Med. Sci. 2021, 24, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Noyan-Ashraf, M.H.; Momen, M.A.; Ban, K.; Sadi, A.M.; Zhou, Y.Q.; Riazi, A.M.; Baggio, L.L.; Henkelman, R.M.; Husain, M.; Drucker, D.J. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 2009, 58, 975–983. [Google Scholar] [CrossRef]
- Wu, L.; Wang, K.; Wang, W.; Wen, Z.; Wang, P.; Liu, L.; Wang, D.W. Glucagon-like peptide-1 ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via the PPARalpha pathway. Aging Cell 2018, 17, e12763. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Kobayashi, T.; Saheki, T.; Japar, S.; Iwama, H.; Matsumura, Y.; Ozaki, M.; et al. Exendin-4 Increases Scavenger Receptor Class BI Expression via Activation of AMPK/FoxO1 in Human Vascular Endothelial Cells. Curr. Issues Mol. Biol. 2022, 44, 5474–5484. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Tong, G.; Fan, H.; Zhen, C.; Zeng, L.; Xue, L.; Chen, J.; Sun, Z.; He, P. Exendin-4 alleviates myocardial ischemia reperfusion injury by enhancing autophagy through promoting nuclear translocation of TFEB. Exp. Cell Res. 2023, 423, 113469. [Google Scholar] [CrossRef] [PubMed]
- Nuamnaichati, N.; Mangmool, S.; Chattipakorn, N.; Parichatikanond, W. Stimulation of GLP-1 Receptor Inhibits Methylglyoxal-Induced Mitochondrial Dysfunctions in H9c2 Cardiomyoblasts: Potential Role of Epac/PI3K/Akt Pathway. Front. Pharmacol. 2020, 11, 805. [Google Scholar] [CrossRef]
- Wei, H.; Bu, R.; Yang, Q.; Jia, J.; Li, T.; Wang, Q.; Chen, Y. Exendin-4 Protects against Hyperglycemia-Induced Cardiomyocyte Pyroptosis via the AMPK-TXNIP Pathway. J. Diabetes Res. 2019, 2019, 8905917. [Google Scholar] [CrossRef]
- Wassef, M.A.E.; Tork, O.M.; Rashed, L.A.; Ibrahim, W.; Morsi, H.; Rabie, D.M.M. Mitochondrial Dysfunction in Diabetic Cardiomyopathy: Effect of Mesenchymal Stem Cell with PPAR-gamma Agonist or Exendin-4. Exp. Clin. Endocrinol. Diabetes 2018, 126, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.A.; Khalil, M.A.; Alkhateeb, M.A.; Eleawa, S.M.; Zaki, M.S.A.; El-Kott, A.F.; Al-Shraim, M.; El-Sayed, F.; Eldeen, M.A.; Bin-Meferij, M.M.; et al. Exendin-4 Attenuates Remodeling in the Remote Myocardium of Rats After an Acute Myocardial Infarction by Activating beta-Arrestin-2, Protein Phosphatase 2A, and Glycogen Synthase Kinase-3 and Inhibiting beta-Catenin. Cardiovasc. Drugs Ther. 2021, 35, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, S.; Wang, L.; Zhou, W.; Li, P.; Deng, N.; Tang, Q.; Li, Y.; Wu, L.; Chen, J.; et al. Exendin-4 inhibits atrial arrhythmogenesis in a model of myocardial infarction-induced heart failure via the GLP-1 receptor signaling pathway. Exp. Ther. Med. 2020, 20, 3669–3678. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, D.; Wang, F.; Shi, S.; Chen, Y.; Yang, B.; Tang, Y.; Huang, C. Exendin-4 inhibits structural remodeling and improves Ca(2+) homeostasis in rats with heart failure via the GLP-1 receptor through the eNOS/cGMP/PKG pathway. Peptides 2017, 90, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.J.; Hodson, N.W.; Sherratt, M.J.; Kassem, M.; Lewis, A.L.; Wallrapp, C.; Malik, N.; Holt, C.M. Combined MSC and GLP-1 Therapy Modulates Collagen Remodeling and Apoptosis following Myocardial Infarction. Stem Cells Int. 2016, 2016, 7357096. [Google Scholar] [CrossRef] [PubMed]
- Withaar, C.; Meems, L.M.G.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Sillje, H.H.W.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.S.P.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.D.; Huang, H.F.; Yang, Q.; Chen, X.Q. Liraglutide improves myocardial fibrosis after myocardial infarction through inhibition of CTGF by activating cAMP in mice. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4648–4656. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, T.; Hibi, K.; Konishi, M.; Maejima, N.; Iwahashi, N.; Tsukahara, K.; Kosuge, M.; Ebina, T.; Umemura, S.; Kimura, K. Plasma Glucagon-Like Peptide-1 and Tissue Characteristics of Coronary Plaque in Non-Diabetic Acute Coronary Syndrome Patients. Circ. J. 2016, 80, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, M.; Fu, E.L.; Szummer, K.; Norhammar, A.; Lundman, P.; Wanner, C.; Sjolander, A.; Jernberg, T.; Carrero, J.J. Glucagon-like peptide-1 receptor agonists and the risk of cardiovascular events in diabetes patients surviving an acute myocardial infarction. Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, 104–111. [Google Scholar] [CrossRef]
- Trombara, F.; Cosentino, N.; Bonomi, A.; Ludergnani, M.; Poggio, P.; Gionti, L.; Baviera, M.; Colacioppo, P.; Roncaglioni, M.C.; Leoni, O.; et al. Impact of chronic GLP-1 RA and SGLT-2I therapy on in-hospital outcome of diabetic patients with acute myocardial infarction. Cardiovasc. Diabetol. 2023, 22, 26. [Google Scholar] [CrossRef]
- Marfella, R.; Prattichizzo, F.; Sardu, C.; Rambaldi, P.F.; Fumagalli, C.; Marfella, L.V.; La Grotta, R.; Frige, C.; Pellegrini, V.; D’Andrea, D.; et al. GLP-1 receptor agonists-SGLT-2 inhibitors combination therapy and cardiovascular events after acute myocardial infarction: An observational study in patients with type 2 diabetes. Cardiovasc. Diabetol. 2024, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Paiman, E.H.M.; van Eyk, H.J.; van Aalst, M.M.A.; Bizino, M.B.; van der Geest, R.J.; Westenberg, J.J.M.; Geelhoed-Duijvestijn, P.H.; Kharagjitsingh, A.V.; Rensen, P.C.N.; Smit, J.W.A.; et al. Effect of Liraglutide on Cardiovascular Function and Myocardial Tissue Characteristics in Type 2 Diabetes Patients of South Asian Descent Living in the Netherlands: A Double-Blind, Randomized, Placebo-Controlled Trial. J. Magn. Reson. Imaging 2020, 51, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, R.S.; Hobbs, T.M.; Wells, B.J.; Kong, S.X.; Kattan, M.W.; Bouchard, J.; Chagin, K.M.; Yu, C.; Sakurada, B.; Milinovich, A.; et al. Association of glucagon-like peptide-1 receptor agonist use and rates of acute myocardial infarction, stroke and overall mortality in patients with type 2 diabetes mellitus in a large integrated health system. Diabetes Obes. Metab. 2017, 19, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.J.; Huo, J.L.; Mao, Z.H.; Pan, S.K.; Liu, D.W.; Liu, Z.S.; Wu, P.; Gao, Z.X. Emerging role of antidiabetic drugs in cardiorenal protection. Front. Pharmacol. 2024, 15, 1349069. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Hayashino, Y.; Akai, Y.; Yabuta, M.; Tsujii, S. Dipeptidyl peptidase-4 inhibitors as preferable oral hypoglycemic agents in terms of treatment satisfaction: Results from a multicenter, 12-week, open label, randomized controlled study in Japan (PREFERENCE 4 study). J. Diabetes Investig. 2018, 9, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meininger, G.; Sheng, D.; Terranella, L.; Stein, P.P.; Sitagliptin Study, G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: A randomized, double-blind, non-inferiority trial. Diabetes Obes. Metab. 2007, 9, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Jermendy, G. Incretin-based antidiabetic treatment and diseases of the pancreas (pancreatitis, pancreas carcinoma). Orv. Hetil. 2016, 157, 523–528. [Google Scholar] [CrossRef]
- Nicholson, G.; Hall, G.M. Diabetes mellitus: New drugs for a new epidemic. Br. J. Anaesth. 2011, 107, 65–73. [Google Scholar] [CrossRef]
- Xie, D.; Wang, Q.; Huang, W.; Zhao, L. Dipeptidyl-peptidase-4 inhibitors have anti-inflammatory effects in patients with type 2 diabetes. Eur. J. Clin. Pharmacol. 2023, 79, 1291–1301. [Google Scholar] [CrossRef]
- Pham, T.K.; Nguyen, T.H.T.; Yi, J.M.; Kim, G.S.; Yun, H.R.; Kim, H.K.; Won, J.C. Evogliptin, a DPP-4 inhibitor, prevents diabetic cardiomyopathy by alleviating cardiac lipotoxicity in db/db mice. Exp. Mol. Med. 2023, 55, 767–778. [Google Scholar] [CrossRef]
- Moon, J.Y.; Woo, J.S.; Seo, J.W.; Lee, A.; Kim, D.J.; Kim, Y.G.; Kim, S.Y.; Lee, K.H.; Lim, S.J.; Cheng, X.W.; et al. The Dose-Dependent Organ-Specific Effects of a Dipeptidyl Peptidase-4 Inhibitor on Cardiovascular Complications in a Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0150745. [Google Scholar] [CrossRef]
- Arruda-Junior, D.F.; Salles, T.A.; Martins, F.L.; Antonio, E.L.; Tucci, P.J.F.; Gowdak, L.H.W.; Tavares, C.A.M.; Girardi, A.C. Unraveling the interplay between dipeptidyl peptidase 4 and the renin-angiotensin system in heart failure. Life Sci. 2022, 305, 120757. [Google Scholar] [CrossRef]
- Wojcicka, G.; Pradiuch, A.; Fornal, E.; Stachniuk, A.; Korolczuk, A.; Marzec-Kotarska, B.; Nikolaichuk, H.; Czechowska, G.; Kozub, A.; Trzpil, A.; et al. The effect of exenatide (a GLP-1 analogue) and sitagliptin (a DPP-4 inhibitor) on asymmetric dimethylarginine (ADMA) metabolism and selected biomarkers of cardiac fibrosis in rats with fructose-induced metabolic syndrome. Biochem. Pharmacol. 2023, 214, 115637. [Google Scholar] [CrossRef]
- Shao, S.; Xu, Q.; Yu, X.; Pan, R.; Chen, Y. Dipeptidyl peptidase 4 inhibitors and their potential immune modulatory functions. Pharmacol. Ther. 2020, 209, 107503. [Google Scholar] [CrossRef]
- Adhikari, J.; Hirai, T.; Kawakita, E.; Iwai, K.; Koya, D.; Kanasaki, K. Linagliptin ameliorated cardiac fibrosis and restored cardiomyocyte structure in diabetic mice associated with the suppression of necroptosis. J. Diabetes Investig. 2023, 14, 844–855. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Tran, D.; Bajaj, M.; Ye, Y. DPP-4 inhibition by linagliptin prevents cardiac dysfunction and inflammation by targeting the Nlrp3/ASC inflammasome. Basic Res. Cardiol. 2019, 114, 35. [Google Scholar] [CrossRef]
- Beraldo, J.I.; Benetti, A.; Borges-Junior, F.A.; Arruda-Junior, D.F.; Martins, F.L.; Jensen, L.; Dariolli, R.; Shimizu, M.H.; Seguro, A.C.; Luchi, W.M.; et al. Cardioprotection Conferred by Sitagliptin Is Associated with Reduced Cardiac Angiotensin II/Angiotensin-(1-7) Balance in Experimental Chronic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1940. [Google Scholar] [CrossRef]
- Hong, S.K.; Choo, E.H.; Ihm, S.H.; Chang, K.; Seung, K.B. Dipeptidyl peptidase 4 inhibitor attenuates obesity-induced myocardial fibrosis by inhibiting transforming growth factor-betal and Smad2/3 pathways in high-fat diet-induced obesity rat model. Metabolism 2017, 76, 42–55. [Google Scholar] [CrossRef]
- Chen, Y.F.; Zhang, L.J.; Liu, Q.H.; Li, X.W. Effects of Sitagliptin on myocardial remodeling and autophagy in diabetic mice and its mechanism. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2021, 37, 534–537. [Google Scholar] [CrossRef]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Hasan, I.H.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mahmoud, A.M. Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats. Drug Des. Dev. Ther. 2016, 10, 2095–2107. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Yang, Y.; Suo, Y.; Liu, R.; Qiu, J.; Zhao, Y.; Jiang, N.; Liu, C.; Tse, G.; et al. Alogliptin prevents diastolic dysfunction and preserves left ventricular mitochondrial function in diabetic rabbits. Cardiovasc. Diabetol. 2018, 17, 160. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Zhao, Y.; Jiang, N.; Qiu, J.; Yang, Y.; Li, J.; Liang, X.; Wang, X.; Tse, G.; et al. Alogliptin, a Dipeptidyl Peptidase-4 Inhibitor, Alleviates Atrial Remodeling and Improves Mitochondrial Function and Biogenesis in Diabetic Rabbits. J. Am. Heart Assoc. 2017, 6, e005945. [Google Scholar] [CrossRef]
- Esser, N.; Zraika, S. Neprilysin inhibition: A new therapeutic option for type 2 diabetes? Diabetologia 2019, 62, 1113–1122. [Google Scholar] [CrossRef]
- Potter, L.R. Natriuretic peptide metabolism, clearance and degradation. FEBS J. 2011, 278, 1808–1817. [Google Scholar] [CrossRef]
- McMurray, J.J. Neprilysin inhibition to treat heart failure: A tale of science, serendipity, and second chances. Eur. J. Heart Fail. 2015, 17, 242–247. [Google Scholar] [CrossRef]
- Jordan, J.; Stinkens, R.; Jax, T.; Engeli, S.; Blaak, E.E.; May, M.; Havekes, B.; Schindler, C.; Albrecht, D.; Pal, P.; et al. Improved Insulin Sensitivity With Angiotensin Receptor Neprilysin Inhibition in Individuals With Obesity and Hypertension. Clin. Pharmacol. Ther. 2017, 101, 254–263. [Google Scholar] [CrossRef]
- Nougue, H.; Pezel, T.; Picard, F.; Sadoune, M.; Arrigo, M.; Beauvais, F.; Launay, J.M.; Cohen-Solal, A.; Vodovar, N.; Logeart, D. Effects of sacubitril/valsartan on neprilysin targets and the metabolism of natriuretic peptides in chronic heart failure: A mechanistic clinical study. Eur. J. Heart Fail. 2019, 21, 598–605. [Google Scholar] [CrossRef]
- Suematsu, Y.; Miura, S.; Goto, M.; Matsuo, Y.; Arimura, T.; Kuwano, T.; Imaizumi, S.; Iwata, A.; Yahiro, E.; Saku, K. LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur. J. Heart Fail. 2016, 18, 386–393. [Google Scholar] [CrossRef]
- Ge, Q.; Zhao, L.; Ren, X.M.; Ye, P.; Hu, Z.Y. LCZ696, an angiotensin receptor-neprilysin inhibitor, ameliorates diabetic cardiomyopathy by inhibiting inflammation, oxidative stress and apoptosis. Exp. Biol. Med. 2019, 244, 1028–1039. [Google Scholar] [CrossRef]
- Gronholm, T.; Cheng, Z.J.; Palojoki, E.; Eriksson, A.; Backlund, T.; Vuolteenaho, O.; Finckenberg, P.; Laine, M.; Mervaala, E.; Tikkanen, I. Vasopeptidase inhibition has beneficial cardiac effects in spontaneously diabetic Goto-Kakizaki rats. Eur. J. Pharmacol. 2005, 519, 267–276. [Google Scholar] [CrossRef]
- Malek, V.; Sharma, N.; Gaikwad, A.B. Simultaneous inhibition of neprilysin and activation of ACE2 prevented diabetic cardiomyopathy. Pharmacol. Rep. 2019, 71, 958–967. [Google Scholar] [CrossRef]
- Torre, E.; Arici, M.; Lodrini, A.M.; Ferrandi, M.; Barassi, P.; Hsu, S.C.; Chang, G.J.; Boz, E.; Sala, E.; Vagni, S.; et al. SERCA2a stimulation by istaroxime improves intracellular Ca2+ handling and diastolic dysfunction in a model of diabetic cardiomyopathy. Cardiovasc. Res. 2022, 118, 1020–1032. [Google Scholar] [CrossRef]
- Lou, S.; Zhu, W.; Yu, T.; Zhang, Q.; Wang, M.; Jin, L.; Xiong, Y.; Xu, J.; Wang, Q.; Chen, G.; et al. Compound SJ-12 attenuates streptozocin-induced diabetic cardiomyopathy by stabilizing SERCA2a. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167140. [Google Scholar] [CrossRef]
- Xiong, X.; Zhang, X.; Zhang, Y.; Xie, J.; Bian, Y.; Yin, Q.; Tong, R.; Yu, D.; Pan, L. Sarco/endoplasmic reticulum Ca(2+) ATPase (SERCA)-mediated ER stress crosstalk with autophagy is involved in tris(2-chloroethyl) phosphate stress-induced cardiac fibrosis. J. Inorg. Biochem. 2022, 236, 111972. [Google Scholar] [CrossRef]
- Katz, M.G.; Brandon-Warner, E.; Fargnoli, A.S.; Williams, R.D.; Kendle, A.P.; Hajjar, R.J.; Schrum, L.W.; Bridges, C.R. Mitigation of myocardial fibrosis by molecular cardiac surgery-mediated gene overexpression. J. Thorac. Cardiovasc. Surg. 2016, 151, 1191–1200.e3. [Google Scholar] [CrossRef]
- Banks, T.E.; Rajapaksha, M.; Zhang, L.H.; Bai, F.; Wang, N.P.; Zhao, Z.Q. Suppression of angiotensin II-activated NOX4/NADPH oxidase and mitochondrial dysfunction by preserving glucagon-like peptide-1 attenuates myocardial fibrosis and hypertension. Eur. J. Pharmacol. 2022, 927, 175048. [Google Scholar] [CrossRef]

{kind=link}
| Novel Medication | Antifibrotic Effect |
|---|---|
| SGLT2i |
|
| GLP-1RAs |
|
| DPP4i |
|
| ARNi |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tudurachi, B.-S.; Anghel, L.; Tudurachi, A.; Sascău, R.A.; Zanfirescu, R.-L.; Stătescu, C. Unraveling the Cardiac Matrix: From Diabetes to Heart Failure, Exploring Pathways and Potential Medications. Biomedicines 2024, 12, 1314. https://doi.org/10.3390/biomedicines12061314
Tudurachi B-S, Anghel L, Tudurachi A, Sascău RA, Zanfirescu R-L, Stătescu C. Unraveling the Cardiac Matrix: From Diabetes to Heart Failure, Exploring Pathways and Potential Medications. Biomedicines. 2024; 12(6):1314. https://doi.org/10.3390/biomedicines12061314
Chicago/Turabian StyleTudurachi, Bogdan-Sorin, Larisa Anghel, Andreea Tudurachi, Radu Andy Sascău, Răzvan-Liviu Zanfirescu, and Cristian Stătescu. 2024. "Unraveling the Cardiac Matrix: From Diabetes to Heart Failure, Exploring Pathways and Potential Medications" Biomedicines 12, no. 6: 1314. https://doi.org/10.3390/biomedicines12061314