
Genetic Insights into Age-Related Macular Degeneration


Abstract
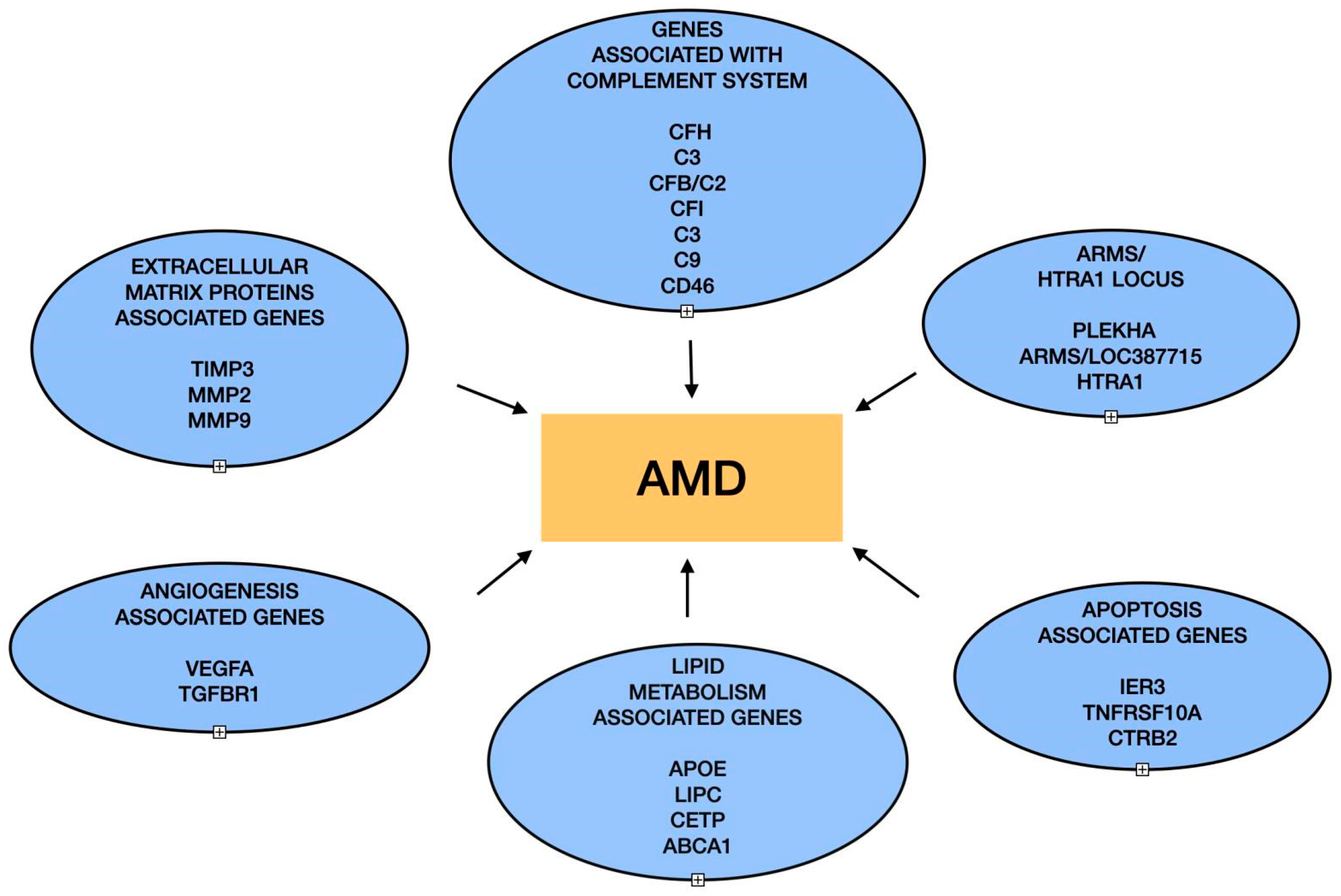
:1. Introduction
2. Role of Complement Proteins
2.1. CFH and AMD
2.2. C3 and AMD
2.3. CFB/C2, CFI, and AMD
2.4. CD46 and AMD
3. ARMS2/HTRA1 Locus
4. Lipid Metabolism Genes and AMD
4.1. Apolipoprotein E and AMD
4.2. LIPC, CETP, LPL, ABCA1, and AMD
5. Extracellular Matrix Proteins and AMD
Role of TIMP3 (Tissue Inhibitor of Metalloproteinase-3)
6. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Clark, A. Brief introduction on macula of retina. Ophthalmol. Clin. Ther. 2021, 5, 1–2. [Google Scholar]
- Abdelsalam, A.; Del Priore, L.; Zarbin, M.A. Drusen in Age-Related Macular Degeneration. Surv. Ophthalmol. 1999, 44, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Luthert, P.J.; Chong, V.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers Drusen as biomarkers of Immune-Mediated processes at the RPE-Bruch’s membrane interface in aging and Age-Related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732. [Google Scholar] [CrossRef] [PubMed]
- Bakri, S.J.; Bektas, M.; Sharp, D.; Luo, R.; Sarda, S.P.; Khan, S. Geographic atrophy: Mechanism of disease, pathophysiology, and role of the complement system. J. Manag. Care Spec. Pharm. 2023, 29, S2–S11. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, Z. Recent developments in the treatment of wet age-related macular degeneration. Curr. Med. Sci. 2020, 40, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Al-Zamil, W.M.; Yassin, S.A. Recent developments in age-related macular degeneration: A review. Clin. Interv. Aging 2017, 12, 1313–1330. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A.; Mousavi, M. Overview of Risk Factors for Age-Related Macular Degeneration (AMD). J. Stem Cells 2015, 10, 171–191. [Google Scholar] [PubMed]
- Guymer, R.H.; Chong, E. Modifiable risk factors for age-related macular degeneration. Med. J. Aust. 2006, 184, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bedell, M.; Zhang, K. Age-related macular degeneration: Genetic and environmental factors of disease. Mol. Interv. 2010, 10, 271–281. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-Related Macular Degeneration: Genetics and Biology coming together. Ann. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef]
- Schick, T.; Lorés-Motta, L.; Altay, L.; Fritsche, L.G.; Hollander, A.I.D.; Fauser, S. The effect of genetic variants associated with Age-Related macular degeneration varies with age. Investig. Ophthalmol. Vis. Sci. 2020, 61, 17. [Google Scholar] [CrossRef] [PubMed]
- Stradiotto, E.; Allegrini, D.; Fossati, G.; Raimondi, R.; Sorrentino, T.; Tripepi, D.; Barone, G.; Inforzato, A.; Romano, M.R. Genetic Aspects of Age-Related Macular Degeneration and their therapeutic potential. Int. J. Mol. Sci. 2022, 23, 13280. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.R.; Chew, E.Y.; Swaroop, A. Genetic studies of age-related macular degeneration. Ophthalmology 2012, 119, 2526–2536. [Google Scholar] [CrossRef] [PubMed]
- Shahid, H.; Khan, J.C.; Cipriani, V.; Sepp, T.; Matharu, B.K.; Bunce, C.; Harding, S.; Clayton, D.; Moore, A.T.; Yates, J.R. Age-related macular degeneration: The importance of family history as a risk factor. Br. J. Ophthalmol. 2011, 96, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J. Genetic association of Age-Related macular degeneration and polypoidal choroidal vasculopathy. Asia Pac. J. Ophthalmol. 2020, 9, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Graßmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Kronenberg, F.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2015, 48, 134–143. [Google Scholar] [CrossRef]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.J.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of Age-Related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef]
- Klein, R.J.; Zeiss, C.J.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.; Mayne, S.T.; et al. Complement factor H Polymorphism in Age-Related Macular Degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Zareparsi, S.; Branham, K.; Li, M.; Shah, S.; Klein, R.J.; Ott, J.; Hoh, J.; Abecasis, G.R.; Swaroop, A. Strong Association of the Y402H Variant in Complement Factor H at 1q32 with Susceptibility to Age-Related Macular Degeneration. Am. J. Hum. Genet. 2005, 77, 149–153. [Google Scholar] [CrossRef]
- Dunkelberger, J.; Song, W. Complement and its role in innate and adaptive immune responses. Cell Res. 2009, 20, 34–50. [Google Scholar] [CrossRef]
- Nesargikar, P.; Spiller, O.B.; Chávez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef]
- Khandhadia, S.; Cipriani, V.; Yates, J.R.; Lotery, A. Age-related macular degeneration and the complement system. Immunobiology 2012, 217, 127–146. [Google Scholar] [CrossRef]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef]
- Kumar-Singh, R. The role of complement membrane attack complex in dry and wet AMD—From hypothesis to clinical trials. Exp. Eye Res. 2019, 184, 266–277. [Google Scholar] [CrossRef]
- Xie, C.; Jane-wit, D.; Pober, J.S. Complement Membrane Attack Complex. Am. J. Pathol. 2020, 190, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Bora, P.S.; Sohn, J.H.; Cruz, J.M.; Jha, P.; Nishihori, H.; Wang, Y.; Kaliappan, S.; Kaplan, H.J.; Bora, N.S. Role of complement and complement membrane attack complex in Laser-Induced Choroidal Neovascularization. J. Immunol. 2005, 174, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Bora, N.S.; Jha, P.; Bora, P.S. The role of complement in ocular pathology. Semin. Immunopathol. 2008, 30, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O.; Ritter, R.; Abel, K.; Manning, A.K.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and Age-Related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef]
- Harboe, M.; Mollnes, T.E. The alternative complement pathway revisited. J. Cell. Mol. Med. 2008, 12, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Stampoulis, D.; Gunter, C.E.; Greenwood, J.; Adamson, P.; Moss, S.E. Regulation of C3 activation by the alternative complement pathway in the mouse retina. PLoS ONE 2016, 11, e0161898. [Google Scholar] [CrossRef] [PubMed]
- Toomey, C.B.; Johnson, L.V.; Rickman, C.B. Complement factor H in AMD: Bridging genetic associations and pathobiology. Prog. Retin. Eye Res. 2018, 62, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.P.; Pangburn, M.K.; Cortés, C. Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Licht, C.; Pluthero, F.G.; Li, L.; Christensen, H.; Habbig, S.; Höppe, B.; Geary, D.F.; Zipfel, P.F.; Kahr, W.H.A. Platelet-associated complement factor H in healthy persons and patients with atypical HUS. Blood 2009, 114, 4538–4545. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Li, Q.; Bu, H.; Lin, F. Mesenchymal stem cells inhibit complement activation by secreting factor H. Stem Cells Dev. 2010, 19, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Mandal, N.A.; Ayyagari, R. Complement factor H: Spatial and temporal expression and localization in the eye. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4091. [Google Scholar] [CrossRef] [PubMed]
- Sivapathasuntharam, C.; Hayes, M.J.; Shinhmar, H.; Kam, J.H.; Sivaprasad, S.; Jeffery, G. Complement factor H regulates retinal development and its absence may establish a footprint for age related macular degeneration. Sci. Rep. 2019, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- De Córdoba, S.R.; Lublin, D.M.; Rubinstein, P.; Atkinson, J.P. Human genes for three complement components that regulate the activation of C3 are tightly linked. J. Exp. Med. 1985, 161, 1189–1195. [Google Scholar] [CrossRef]
- Ripoche, J.; Day, A.J.; Harris, T.; Sim, R.B. The complete amino acid sequence of human complement factor H. Biochem. J. 1988, 249, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, E.; Jokiranta, T.S.; Male, D.A.; Ranganathan, S.; Ormsby, R.J.; Fischetti, V.A.; Mold, C.; Gordon, D.L. A common site within factor H SCR 7 responsible for binding heparin, C-reactive protein and streptococcal M protein. Eur. J. Immunol. 2003, 33, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Magnússon, K.P.; Duan, S.; Sigurðsson, H.; Pétursson, H.; Yang, Z.; Zhao, Y.; Bernstein, P.S.; Ge, J.; Jónasson, F.; Stefánsson, E.; et al. CFH Y402H confers similar risk of soft Drusen and both forms of advanced AMD. PLOS Med. 2005, 3, e5. [Google Scholar] [CrossRef]
- Sepp, T.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.; Moore, A.T.; Bird, A.C.; Yates, J.R. Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Investig. Ophthalmol. Vis. Sci. 2006, 47, 536. [Google Scholar] [CrossRef]
- Nazari Khanamiri, H.; Ghasemi Falavarjani, K.; Sanati, M.H.; Aryan, H.; Irani, A.; Hashemi, M.; Modarres, M.; Parvaresh, M.M.; Nikeghbali, A. Complement Factor H Y402H and LOC387715 A69S Polymorphisms in Association with Age-Related Macular Degeneration in Iran. J. Ophthalmic Vis. Res. 2014, 9, 181–187. [Google Scholar] [PubMed]
- Landowski, M.; Kelly, U.; Klingeborn, M.; Groelle, M.; Ding, J.; Grigsby, D.; Rickman, C.B. Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 3703–3711. [Google Scholar] [CrossRef]
- Maugeri, A.; Barchitta, M.; Agodi, A. The association between complement factor H rs1061170 polymorphism and age-related macular degeneration: A comprehensive meta-analysis stratified by stage of disease and ethnicity. Acta Ophthalmol. 2018, 97, e8–e21. [Google Scholar] [CrossRef]
- Supanji, S.; Ilham Perdamaian, A.B.; Khalifa Farzana, I.A.; Sasongko, M.B.; Agni, A.N.; Wardhana, F.S.; Widayanti, T.W.; Prayogo, M.E. Strong Linkage Disequilibrium and Haplotype Association of Neovascular Age-Related Macular Degeneration in Indonesian Patients. Malays. J. Med. Health Sci. 2023, 19, 138–144. [Google Scholar] [CrossRef]
- Nahla, A.F.; Marianne, S.I.; Mohamed, A.Z.; Riham, A. Association of Complement Factor H (CFH) Y402H Gene Polymorphism and Serum CFH with Risk of Age-Related Macular Degeneration in Egyptian Patients. Med. J. Cairo Univ. 2019, 87, 497. [Google Scholar] [CrossRef]
- Gotoh, N.; Yamada, R.; Hiratani, H.; Renault, V.; Kuroiwa, S.; Monet, M.; Toyoda, S.; Chida, S.; Mandai, M.; Otani, A.; et al. No association between complement factor H gene polymorphism and exudative age-related macular degeneration in Japanese. Hum. Genet. 2006, 120, 139–143. [Google Scholar] [CrossRef]
- Xu, Y.; Guan, N.; Xu, J.; Yang, X.; Ma, K.; Zhou, H.; Zhang, F.; Snellingen, T.; Jiao, Y.; Liu, X.; et al. Association of CFH, LOC387715, and HTRA1 Polymorphisms with Exudative Age-Related Macular Degeneration in a Northern Chinese Population. Mol. Vis. 2008, 14, 1373–1381. [Google Scholar]
- Nielsen, M.K.; Subhi, Y.; Molbech, C.R.; Grønskov, K.; Sørensen, T.L. Distribution of Risk Alleles in Patients with Age-Related Macular Degeneration. Dan. Med. J. 2020, 67, A05190295. [Google Scholar]
- Kondo, N.; Honda, S.; Kuno, S.; Negi, A. Coding Variant I62V in the Complement Factor H Gene Is Strongly Associated with Polypoidal Choroidal Vasculopathy. Ophthalmology 2009, 116, 304–310. [Google Scholar] [CrossRef]
- Wu, M.; Guo, Y.; Ma, Y.; Zhang, Z.; Wang, Q.; Zhou, X. Association of Two Polymorphisms, rs1061170 and rs1410996, in Complement Factor H with Age-Related Macular Degeneration in an Asian Population: A Meta-Analysis. Ophthalmic Res. 2016, 55, 135–144. [Google Scholar] [CrossRef]
- Cruz-González, F.; Cieza-Borrella, C.; Valverde, G.L.; Lorenzo-Pérez, R.; Hernández-Galilea, E.; González-Sarmiento, R. CFH (rs1410996), HTRA1 (rs112000638) and ARMS2 (rs10490923) Gene Polymorphisms are Associated with AMD Risk in Spanish Patients. Ophthalmic Genet. 2013, 35, 68–73. [Google Scholar] [CrossRef]
- Salman, A.; Song, W.; Young, S.; McClements, M.E.; MacLaren, R.E. CRISPR Approach for Allele-Specific Targeting of SNPs Associated with Age-Related Macular Degeneration in ARPE19 Cells. Hum. Genet. 2023, 64, 756. [Google Scholar]
- Sahu, A.; Lambris, J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001, 180, 35–48. [Google Scholar] [CrossRef]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P. Complement component C3—The “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar] [CrossRef]
- Delanghe, J.; Speeckaert, R.; Speeckaert, M.M. Complement C3 and its polymorphism: Biological and clinical consequences. Pathology 2014, 46, 1–10. [Google Scholar] [CrossRef]
- Bora, P.S.; Hu, Z.; Tezel, T.H.; Sohn, J.H.; Kang, S.S.; Cruz, J.M.; Bora, N.S.; Garen, A.; Kaplan, H.J. Immunotherapy for choroidal neovascularization in a laser-induced mouse model simulating exudative (wet) macular degeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 2679–2684. [Google Scholar] [CrossRef]
- Kopplin, L.J.; Wang, Y.; Ajudua, S.; Igo Jr, R.P.; Chew, E.Y.; Clemons, T.E.; Henning, A.K.; Francis, P.J.; Klein, M.L.; Iyengar, S.K. C3 Variant Is Associated with Neovascular AMD & Geographic Atrophy in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2669. [Google Scholar]
- Yates, J.R.W.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement C3 Variant and the Risk of Age-Related Macular Degeneration. N. Engl. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Pauer, G.J.; Nerone, P.; Bamba, S.; Hagstrom, S.A.; Cleveland AMD Study Group. A Strong Association Between the Complement C3 Arg80Gly Polymorphism and Advanced AMD. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1707. [Google Scholar]
- Yanagisawa, S.; Kondo, N.; Miki, A.; Matsumiya, W.; Kusuhara, S.; Tsukahara, Y.; Honda, S.; Negi, A. A Common Complement C3 Variant Is Associated with Protection against Wet Age-Related Macular Degeneration in a Japanese Population. PLoS ONE 2011, 6, e28847. [Google Scholar] [CrossRef]
- Duvvari, M.R.; Păun, C.; Buitendijk, G.H.S.; Saksens, N.T.M.; Volokhina, E.; Ristau, T.; Schoenmaker-Koller, F.E.; Van De Ven, J.P.H.; Groenewoud, J.M.M.; Van Den Heuvel, L.P.W.J.; et al. Analysis of Rare Variants in the C3 Gene in Patients with Age-Related Macular Degeneration. PLoS ONE 2014, 9, e94165. [Google Scholar] [CrossRef]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.M.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Zhao, M.; Li, X. CFB/C2Gene Polymorphisms and Risk of Age-Related Macular Degeneration: A Systematic Review and Meta-Analysis. Curr. Eye Res. 2012, 37, 259–271. [Google Scholar] [CrossRef]
- Nilsson, S.C.; Sim, R.B.; Lea, S.M.; Frémeaux-Bacchi, V.; Blom, A.M. Complement factor I in health and disease. Mol. Immunol. 2011, 48, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, P.J. The story of complement factor I. Immunobiology 2019, 224, 511–517. [Google Scholar] [CrossRef]
- Fagerness, J.; Maller, J.; Neale, B.M.; Reynolds, R.; Daly, M.J.; Seddon, J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2008, 17, 100–104. [Google Scholar] [CrossRef]
- Hollander, A.I.D.; Van De Ven, J.P.; Nilsson, S.C.; Tan, P.L.; Buitendijk, G.H.S.; Ristau, T.; Katsanis, N.; Klaver, C.C.W.; Blom, A.M.; Hoyng, C.C.B. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat. Genet. 2013, 45, 813–817. [Google Scholar] [CrossRef]
- Saksens, N.T.M.; Geerlings, M.J.; Bakker, B.; Schick, T.; Daha, M.R.; Fauser, S.; Boon, C.J.F.; De Jong, E.K.; Hoyng, C.B.; Hollander, A.I.D. Rare genetic variants associated with development of Age-Related macular degeneration. JAMA Ophthalmol. 2016, 134, 287. [Google Scholar] [CrossRef]
- Bonyadi, M.; Norouzi, N.; Babaei, E.; Bonyadi, M.R.; Javadzadeh, A.; Yaseri, M.; Soheilian, M. Association of polymorphisms of complement factor I rs141853578 (G119R) with age-related macular degeneration in Iranian population. Int. Ophthalmol. 2019, 39, 551. [Google Scholar] [CrossRef]
- Yu, Q.; Zhu, J.; Yao, Y.; Sun, C. Complement family member CFI polymorphisms and AMD susceptibility from a comprehensive analysis. Biosci. Rep. 2020, 40, BSR20200406. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, M.K.; Post, T.W.; Atkinson, J.P. Membrane cofactor protein (MCP or CD46): Newest member of the regulators of complement activation gene cluster. Ann. Rev. Immunol. 1991, 9, 431–455. [Google Scholar] [CrossRef]
- Bora, N.S.; Lublin, D.M.; Kumar, B.V.; Hockett, R.D.; Holers, V.M.; Atkinson, J.P. Structural gene for human membrane cofactor protein (MCP) of complement maps to within 100 kb of the 3’ end of the C3b/C4b receptor gene. J. Exp. Med. 1989, 169, 597–602. [Google Scholar] [CrossRef]
- Seya, T.; Turner, J.R.; Atkinson, J.P. Purification and characterization of a membrane protein (gp45-70) that is a cofactor for cleavage of C3b and C4b. J. Exp. Med. 1986, 163, 837–855. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Leung, M.K.; Cui, W.; Subramanian, V.; Parkinson, J.; Barlow, P.N.; Manchester, M.; Atkinson, J.P. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46). J. Biol. Chem. 2000, 275, 37692–37701. [Google Scholar] [CrossRef] [PubMed]
- Lyzogubov, V.V.; Wu, X.; Jha, P.; Tytarenko, R.G.; Triebwasser, M.; Kolar, G.R.; Bertram, P.; Bora, P.S.; Atkinson, J.P.; Bora, N.S. Complement Regulatory Protein CD46 Protects against Choroidal Neovascularization in Mice. Am. J. Path. 2014, 184, 2537–2548. [Google Scholar] [CrossRef]
- Lyzogubov, V.; Wu, X.; Kolar, G.; Bora, P.; Atkinson, J.; Bora, N. CD46-/- Mouse as a Model of Dry-Type Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4591. [Google Scholar]
- Lyzogubov, V.V.; Bora, P.S.; Wu, X.; Horn, L.E.; De Roque, R.; Rudolf, X.V.; Atkinson, J.P.; Bora, N.S. The complement regulatory protein CD46 deficient mouse spontaneously develops Dry-Type Age-Related macular Degeneration–Like phenotype. Am. J. Pathol. 2016, 186, 2088–2104. [Google Scholar] [CrossRef]
- Fierz, W. Age-Related Macular Degeneration: A Connection between Human Herpes Virus-6A-Induced CD46 Downregulation and Complement Activation? Front. Immunol. 2017, 8, 1314. [Google Scholar] [CrossRef]
- Wang, G. Chromosome 10q26 locus and age-related macular degeneration: A progress update. Exp. Eye Res. 2014, 119, 1–7. [Google Scholar] [CrossRef]
- Jakobsdóttir, J.; Conley, Y.P.; Weeks, D.E.; Mah, T.S.; Ferrell, R.E.; Gorin, M.B. Susceptibility Genes for Age-Related Maculopathy on Chromosome 10q26. Am. J. Hum. Genet. 2005, 77, 389–407. [Google Scholar] [CrossRef]
- Rivera, A.; Fisher, S.; Fritsche, L.G.; Keilhauer, C.N.; Lichtner, P.; Meitinger, T.; Weber, B.H.F. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum. Mol. Genet. 2005, 14, 3227–3236. [Google Scholar] [CrossRef]
- Grassmann, F.; Heid, I.M.; Weber, B.H.F. Recombinant haplotypes narrow the ARMS2/HTRA1 association signal for Age-Related macular degeneration. Genetics 2017, 205, 919–924. [Google Scholar] [CrossRef]
- DeWan, A.T.; Liu, M.; Hartman, S.E.; Zhang, S.S.M.; Liu, D.T.L.; Zhao, C.; Tam, P.O.S.; Chan, W.M.; Lam, D.S.C.; Snyder, M.; et al. HTRA1 promoter polymorphism in wet Age-Related macular degeneration. Science 2006, 314, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Loenhardt, T.; Janßen, A.; Fisher, S.; Rivera, A.; Keilhauer, C.N.; Weber, B.H.F. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat. Genet. 2008, 40, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Myers, C.E.; Meuer, S.M.; Gangnon, R.E.; Sivakumaran, T.A.; Iyengar, S.K.; Lee, K.E.; Klein, B.E.K. Risk alleles in CFH and ARMS2 and the long-term natural history of Age-Related Macular Degeneration. JAMA Ophthalmol. 2013, 131, 383. [Google Scholar] [CrossRef] [PubMed]
- Micklisch, S.; Lin, Y.; Jacob, S.; Karlstetter, M.; Dannhausen, K.; Dasari, P.; Von Der Heide, M.; Dahse, H.M.; Schmölz, L.; Graßmann, F.; et al. Age-related macular degeneration associated polymorphism rs10490924 in ARMS2 results in deficiency of a complement activator. J. Neuroinflamm. 2017, 14, 4. [Google Scholar] [CrossRef]
- Körtvely, E.; Hauck, S.M.; Duetsch, G.; Gloeckner, C.J.; Kremmer, E.; Alge-Priglinger, C.S.; Deeg, C.A.; Ueffing, M. ARMS2 Is a Constituent of the Extracellular Matrix Providing a Link between Familial and Sporadic Age-Related Macular Degenerations. Investig. Ophthalmol. Vis. Sci. 2010, 51, 79. [Google Scholar] [CrossRef]
- Gursky, O. Apolipoprotein structure and dynamics. Curr. Opin. Lipidol. 2005, 16, 287–294. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Hu, M.L.; Quinn, J.; Xue, K. Interactions between Apolipoprotein E Metabolism and Retinal Inflammation in Age-Related Macular Degeneration. Life 2021, 11, 635. [Google Scholar] [CrossRef]
- Klaver, C.C.W.; Kliffen, M.; Van Duijn, C.M.; Hofman, A.; Cruts, M.; Grobbee, D.E.; Van Broeckhoven, C.; De Jong, P.T.V.M. Genetic Association of Apolipoprotein E with Age-Related Macular Degeneration. Am. J. Hum. Genet. 1998, 63, 200–206. [Google Scholar] [CrossRef]
- Mao, X.; Wu, W.B.; Fang, W.; Liu, Q. Association of Apolipoprotein E Polymorphisms with Age-related Macular Degeneration Subtypes: An Updated Systematic Review and Meta-analysis. Arch. Med. Res. 2017, 48, 370–377. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef]
- Pirim, D.; Bunker, C.H.; Hokanson, J.E.; Hamman, R.F.; Demirci, F.; Kamboh, M.I. Hepatic lipase (LIPC) sequencing in individuals with extremely high and low high-density lipoprotein cholesterol levels. PLoS ONE 2020, 15, e0243919. [Google Scholar] [CrossRef]
- Yu, Y.; Reynolds, R.; Fagerness, J.; Rosner, B.; Daly, M.J.; Seddon, J.M. Association of Variants in theLIPCandABCA1Genes with Intermediate and Large Drusen and Advanced Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4663. [Google Scholar] [CrossRef]
- Lee, J.; Zeng, J.; Hughes, G.; Chen, Y.; Grob, S.; Zhao, L.; Lee, C.; Krupa, M.; Quach, J.; Luo, J.; et al. Association of LIPC and advanced age-related macular degeneration. Eye 2013, 27, 265–271. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, J.; Hou, X.; Nguyen, D.H.; Cao, G.; Li, G.; Qiu, G.; Zhang, K.; Zhang, M.; Su, Z. CETP Gene may be Associated with Advanced Age-Related Macular Degeneration in the Chinese Population. Ophthalmic Genet. 2014, 36, 303–308. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Han, Y.; Zhang, R.; Li, Q.; Wang, M.; Ma, L. CETP/LPL/LIPC gene polymorphisms and susceptibility to age-related macular degeneration. Sci. Rep. 2015, 5, 15711. [Google Scholar] [CrossRef]
- Curcio, C.A.; Johnson, M.; Rudolf, M.; Huang, J. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011, 95, 1638–1645. [Google Scholar] [CrossRef]
- Jacobo-Albavera, L.; Domínguez-Pérez, M.; Medina-Leyte, D.J.; González-Garrido, A.; Villarreal-Molina, T. The role of the ATP-Binding Cassette A1 (ABCA1) in human disease. Int. J. Mol. Sci. 2021, 22, 1593. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, Q.; Wang, L.; Wang, Y.; Wang, D.; Ding, H. Role of ABCA1 in cardiovascular disease. J. Pers. Med. 2022, 12, 1010. [Google Scholar] [CrossRef]
- Peters, F.; Ebner, L.J.A.; Atac, D.; Maggi, J.; Berger, W.; Hollander, A.I.D.; Grimm, C. Regulation of ABCA1 by AMD-Associated Genetic Variants and hypoxia in IPSC-RPE. Int. J. Mol. Sci. 2022, 23, 3194. [Google Scholar] [CrossRef]
- Liutkevičienė, R.; Vilkevičiūtė, A.; Streleckienė, G.; Kriaučiūnienė, L.; Chaleckis, R.; Deltuva, V.P. Associations of cholesteryl ester transfer protein (CETP) gene variants with predisposition to age-related macular degeneration. Gene 2017, 636, 30–35. [Google Scholar] [CrossRef]
- Liutkevičienė, R.; Vilkevičiūtė, A.; Kriaučiūnienė, L.; Banevičius, M.; Būdienė, B.; Stanislovaitienė, D.; Žemaitienė, R.; Deltuva, V.P. Association of genetic variants at CETP, AGER, and CYP4F2 locus with the risk of atrophic age-related macular degeneration. Mol. Genet. Genom. Med. 2020, 8, e1357. [Google Scholar] [CrossRef]
- Cheng, C.; Yamashiro, K.; Chen, L.J.; Ahn, J.; Huang, L.; Huang, L.; Cheung, C.M.G.; Miyake, M.; Cackett, P.; Yeo, I.; et al. New loci and coding variants confer risk for age-related macular degeneration in East Asians. Nat. Commun. 2015, 6, 6063. [Google Scholar] [CrossRef]
- Klagsbrun, M.; Folkman, J. Angiogenesis. In Handbook of Experimental Pharmacology; Sporn, M.B., Roberts, A.B., Eds.; Springer: Berlin/Heidelberg, Germany, 2024; pp. 549–586. [Google Scholar]
- Kamei, M.; Hollyfield, J.G. TIMP-3 in Bruch’s Membrane: Changes During Aging and in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2367–2375. [Google Scholar]
- Warwick, A.; Gibson, J.; Sood, R.; Lotery, A. A rare penetrant TIMP3 mutation confers relatively late onset choroidal neovascularisation which can mimic age-related macular degeneration. Eye 2015, 30, 488–491. [Google Scholar] [CrossRef]
- Chen, W.; Stambolian, D.; Edwards, A.O.; Branham, K.; Othman, M.; Jakobsdóttir, J.; Tosakulwong, N.; Pericak-Vance, M.A.; Campochiaro, P.A.; Klein, M.L.; et al. Genetic variants near TIMP3 and high-density lipoprotein–associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401–7406. [Google Scholar] [CrossRef]
- Qin, S.; Dong, N.; Yang, M.; Wang, J.; Feng, X.; Wang, Y. Complement Inhibitors in Age-Related Macular Degeneration: A Potential Therapeutic Option. J. Immunol. Res. 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Shughoury, A.; Sevgi, D.D.; Ciulla, T.A. The Complement System: A Novel Therapeutic Target for Age-Related Macular Degeneration. Expert Opin. Pharmacother. 2023, 24, 1887–1899. [Google Scholar] [CrossRef]
- Xu, H.; Yi, C.; Chen, M. The Complement Pathway as a Therapeutic Target for Neovascular Age-Related Macular Degeneration-Mediated Subretinal Fibrosis. Curr. Opin. Pharmacol. 2024, 76, 102448. [Google Scholar] [CrossRef]
- Cruz-Pimentel, M.; Wu, L. Complement Inhibitors for Advanced Dry Age-Related Macular Degeneration (Geographic Atrophy): Some Light at the End of the Tunnel? J. Clin. Med. 2023, 12, 5131. [Google Scholar] [CrossRef]
- Tan, C.S.; Ngo, W.K.; Chay, I.W.; Ting, D.S.; Sadda, S.R. Neovascular Age-Related Macular Degeneration (nAMD): A Review of Emerging Treatment Options. Clin. Ophthalmol. 2022, 16, 917–933. [Google Scholar] [CrossRef]
- Khanani, A.M.; Thomas, M.J.; Aziz, A.A.; Weng, C.Y.; Danzig, C.J.; Yiu, G.; Kiss, S.; Waheed, N.K.; Kaiser, P.K. Review of Gene Therapies for Age-Related Macular Degeneration. Eye 2022, 36, 303–311. [Google Scholar] [CrossRef]
- Choi, E.H.; Suh, S.; Sears, A.E.; Hołubowicz, R.; Kedhar, S.R.; Browne, A.W.; Palczewski, K. Genome Editing in the Treatment of Ocular Diseases. Exp. Mol. Med. 2023, 55, 1678–1690. [Google Scholar] [CrossRef]
- Gelfman, C.M.; Grishanin, R.; Bender, K.O.; Nguyen, A.; Greengard, J.; Sharma, P.; Nieves, J.; Kiss, S.; Gasmi, M. Comprehensive Preclinical Assessment of ADVM-022, an Intravitreal Anti-VEGF Gene Therapy for the Treatment of Neovascular AMD and Diabetic Macular Edema. J. Ocul. Pharmacol. Ther. 2021, 37, 181–190. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Ciupińska, J.; Derwich, M.; Szczepanska, J.; Kaarniranta, K. A New Generation of Gene Therapies as the Future of Wet AMD Treatment. Int. J. Mol. Sci. 2024, 25, 2386. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yang, Q.; Zhang, R.H.; Wei, W.B. Artificial Intelligence for the Detection of Age-Related Macular Degeneration in Color Fundus Photographs: A Systematic Review and Meta-Analysis. EClinicalMedicine 2021, 35, 100875. [Google Scholar] [CrossRef]
- Crincoli, E.; Sacconi, R.; Querques, L.; Querques, G. Artificial Intelligence in Age-Related Macular Degeneration: State of the Art and Recent Updates. BMC Ophthalmol. 2024, 24, 121. [Google Scholar] [CrossRef]
- Yan, Q.; Jiang, Y.; Huang, H.; Swaroop, A.; Chew, E.Y.; Weeks, D.E.; Chen, W.; Ding, Y. Genome-Wide Association Studies-Based Machine Learning for Prediction of Age-Related Macular Degeneration Risk. Transl. Vis. Sci. Technol. 2021, 10, 29. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Genes | Role | Mechanism of Action in the Pathogenesis of AMD |
|---|---|---|
| CFH | CFH is a crucial regulator of complement syste. Variants result in the reduced protective activity of CFH. | Misregulation leads to excessive inflammation, oxidative stress, damage to retinal cells, and drusen formation. |
| C3 | Cleavage products C3a and C3b promote inflammation and opsonization, leading to the formation of the membrane attack complex which is crucial for the immune defense. | Variants may alter the ability to inhibit the formation of the membrane attack complex, leading to chronic inflammation and cell damage in the retina. |
| CD46 | Acts as a cofactor for the cleavage for the inactivation of C3b and C4b, thereby moderating the activity of the complement system. | Variants lead to the misregulation of the complement pathway, leading to choroidal neovascularization and drusen formation in wet and dry AMD, respectively. |
| ARMS2 | ARMS2 is linked to AMD, particularly influencing the extracellular matrix and mitochondrial function in retinal cells. | Its involvement may be through the modulation of local inflammation and oxidative stress, impacting retinal cell integrity. |
| HTRA1 | Serine protease involved in extracellular matrix remodeling. | Overexpression can lead to the increased breakdown of the extracellular matrix and promote neovascular AMD. |
| TIMP3 | TIMP3 regulates extracellular matrix remodeling by inhibiting matrix metalloproteinases; mutations can lead to Sorsby’s fundus dystrophy and AMD. | Dysregulation contributes to drusen formation and choroidal neovascularization. |
| ABCA1 | ABCA1 gene impacts cholesterol efflux from cells, affecting lipid deposits in the retina. | Impaired function can lead to abnormal lipid accumulation and increased oxidative stress, promoting AMD development. |
| APOE | APOE is involved in lipid transport and clearance. The ε4 allele has been associated with a lower AMD risk, while ε2 has been linked to a higher risk. | Its role in lipid processing affects drusen composition and size, crucial in AMD pathogenesis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhumika; Bora, N.S.; Bora, P.S. Genetic Insights into Age-Related Macular Degeneration. Biomedicines 2024, 12, 1479. https://doi.org/10.3390/biomedicines12071479
Bhumika, Bora NS, Bora PS. Genetic Insights into Age-Related Macular Degeneration. Biomedicines. 2024; 12(7):1479. https://doi.org/10.3390/biomedicines12071479
Chicago/Turabian StyleBhumika, Nalini S. Bora, and Puran S. Bora. 2024. "Genetic Insights into Age-Related Macular Degeneration" Biomedicines 12, no. 7: 1479. https://doi.org/10.3390/biomedicines12071479
APA StyleBhumika, Bora, N. S., & Bora, P. S. (2024). Genetic Insights into Age-Related Macular Degeneration. Biomedicines, 12(7), 1479. https://doi.org/10.3390/biomedicines12071479