
FDA-Approved Trifluoromethyl Group-Containing Drugs: A Review of 20 Years

 , , and
, , and
Abstract
:1. Introduction
2. FDA-Approved Drugs Containing TFM
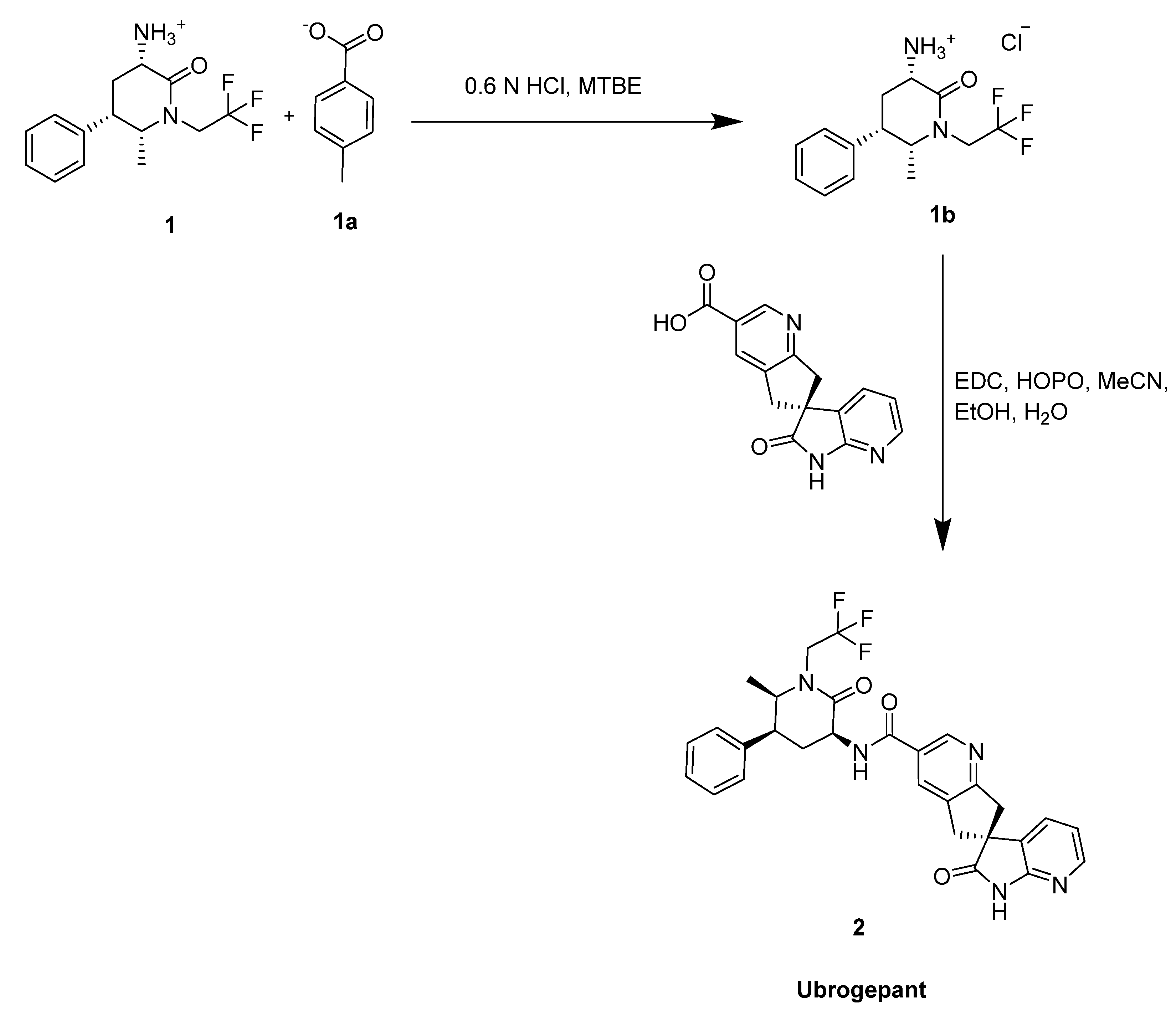
2.1. Ubrogepant
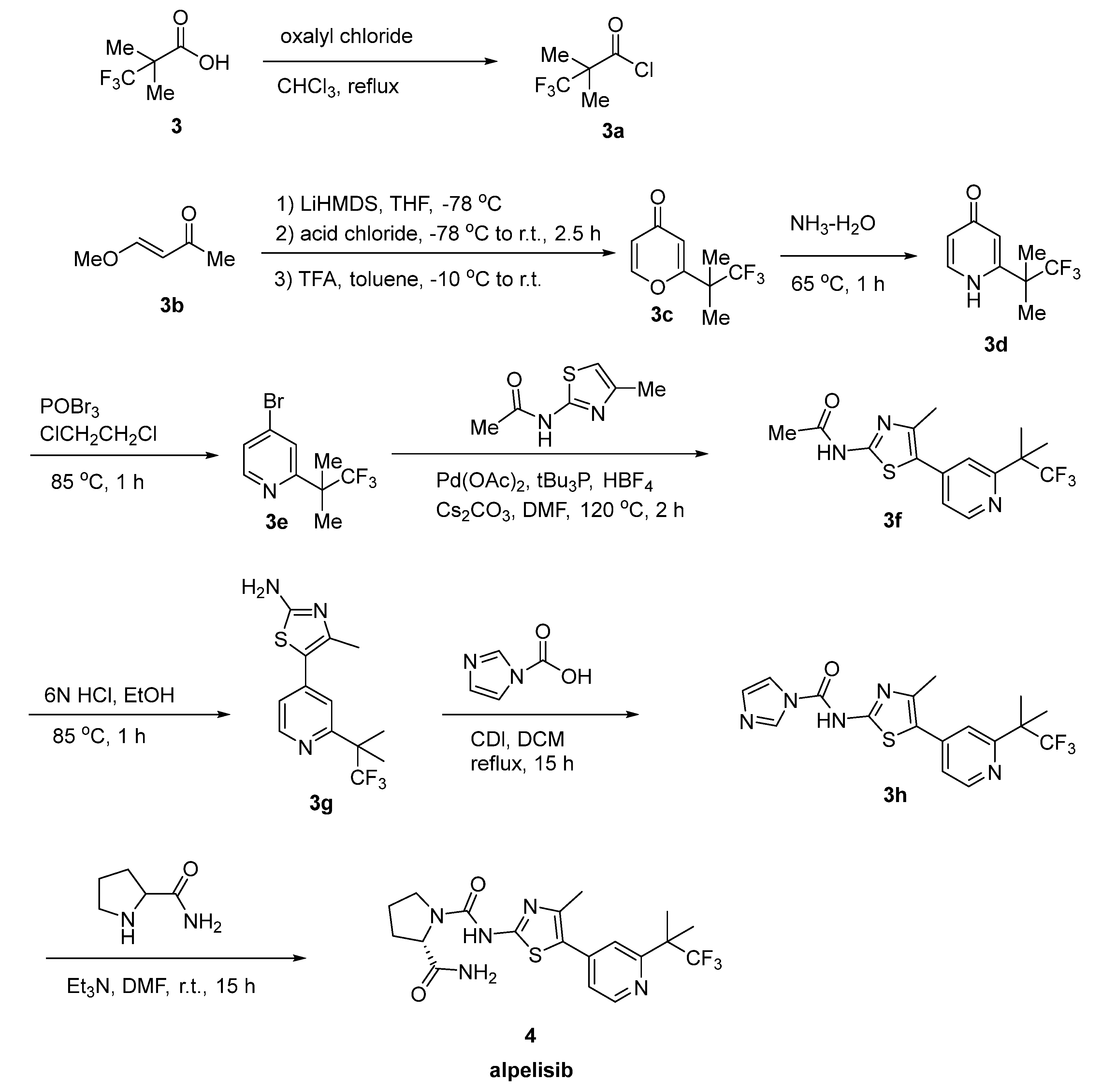
2.2. Alpelisib
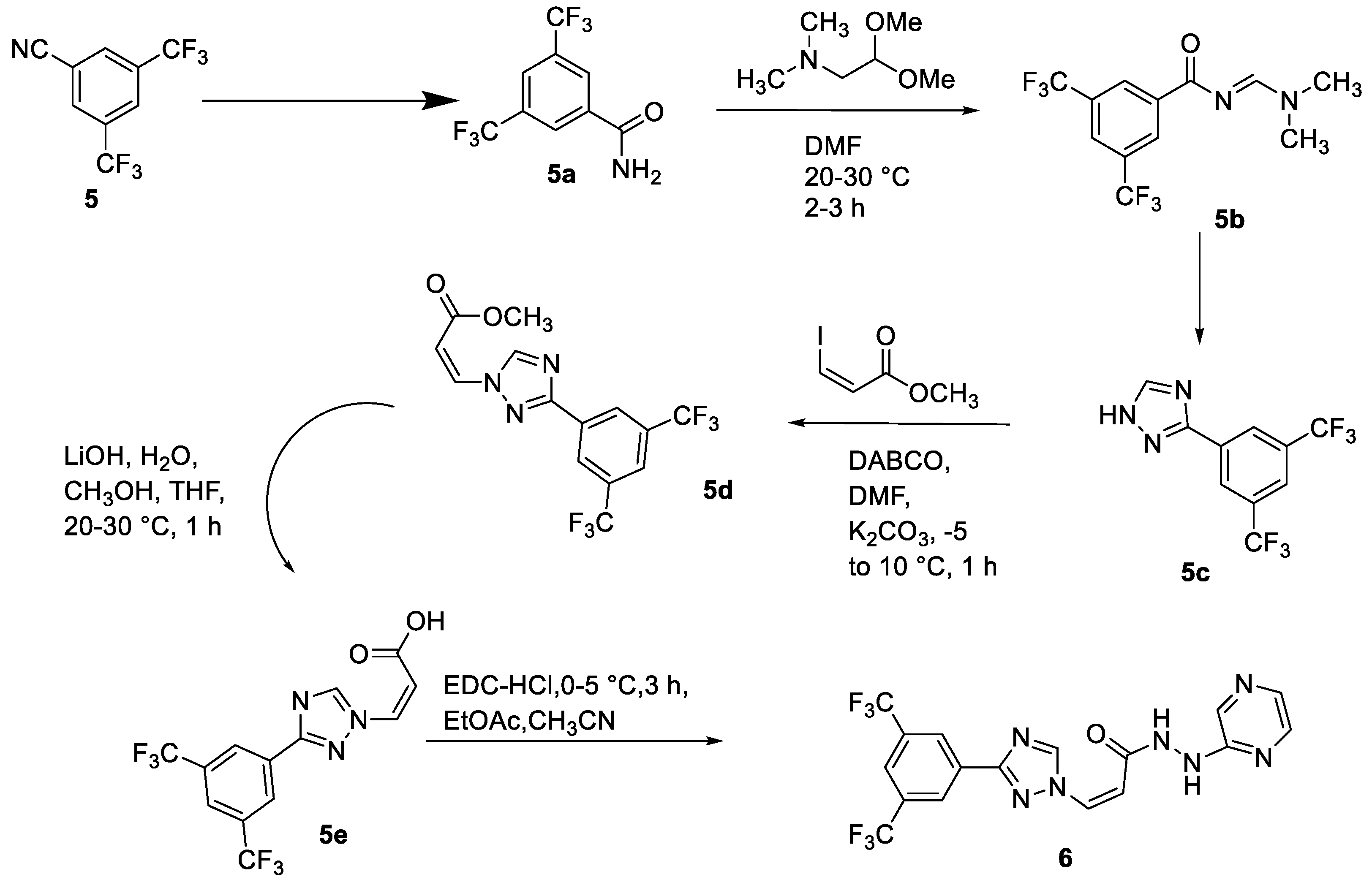
2.3. Selinexor
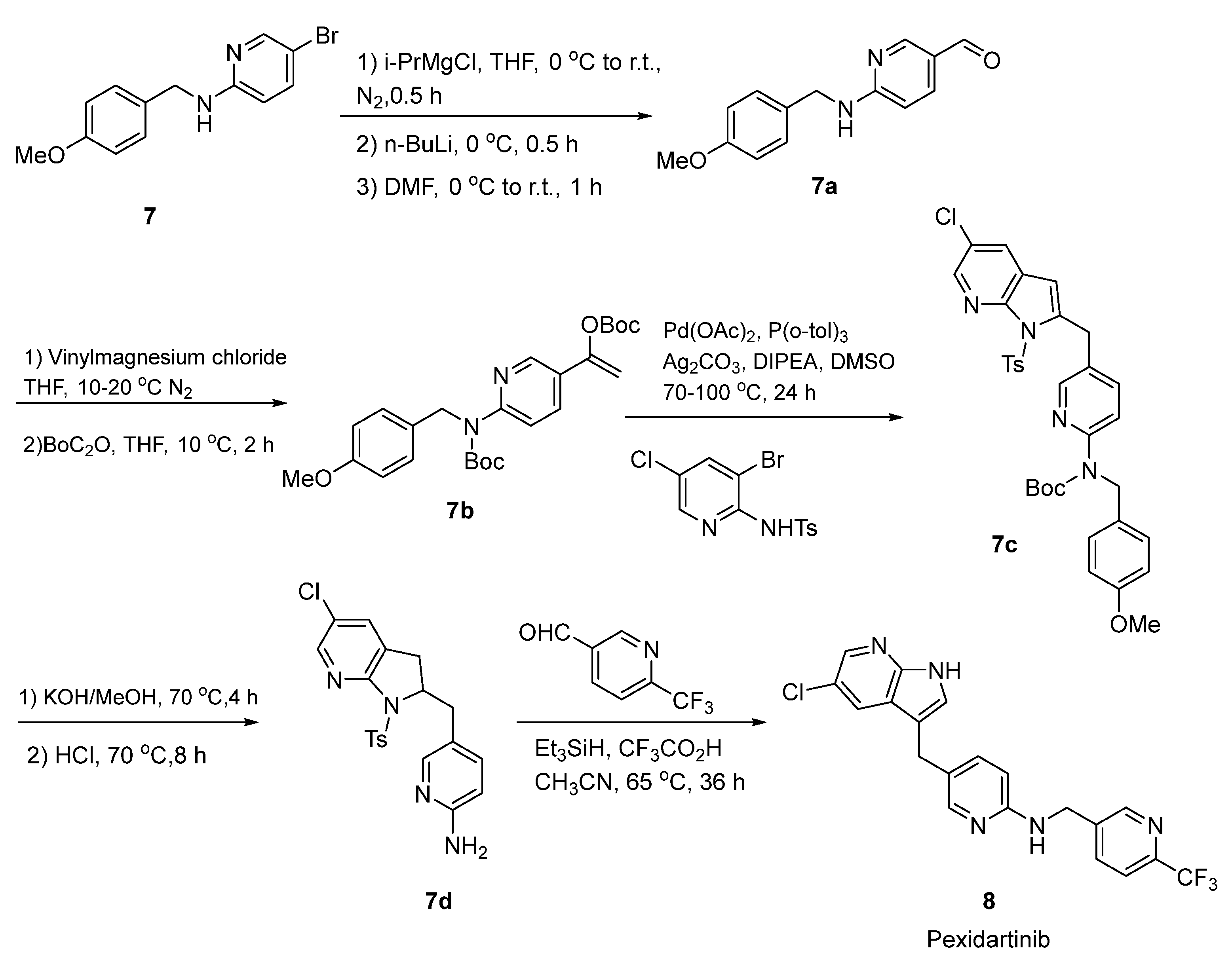
2.4. Pexidartinib
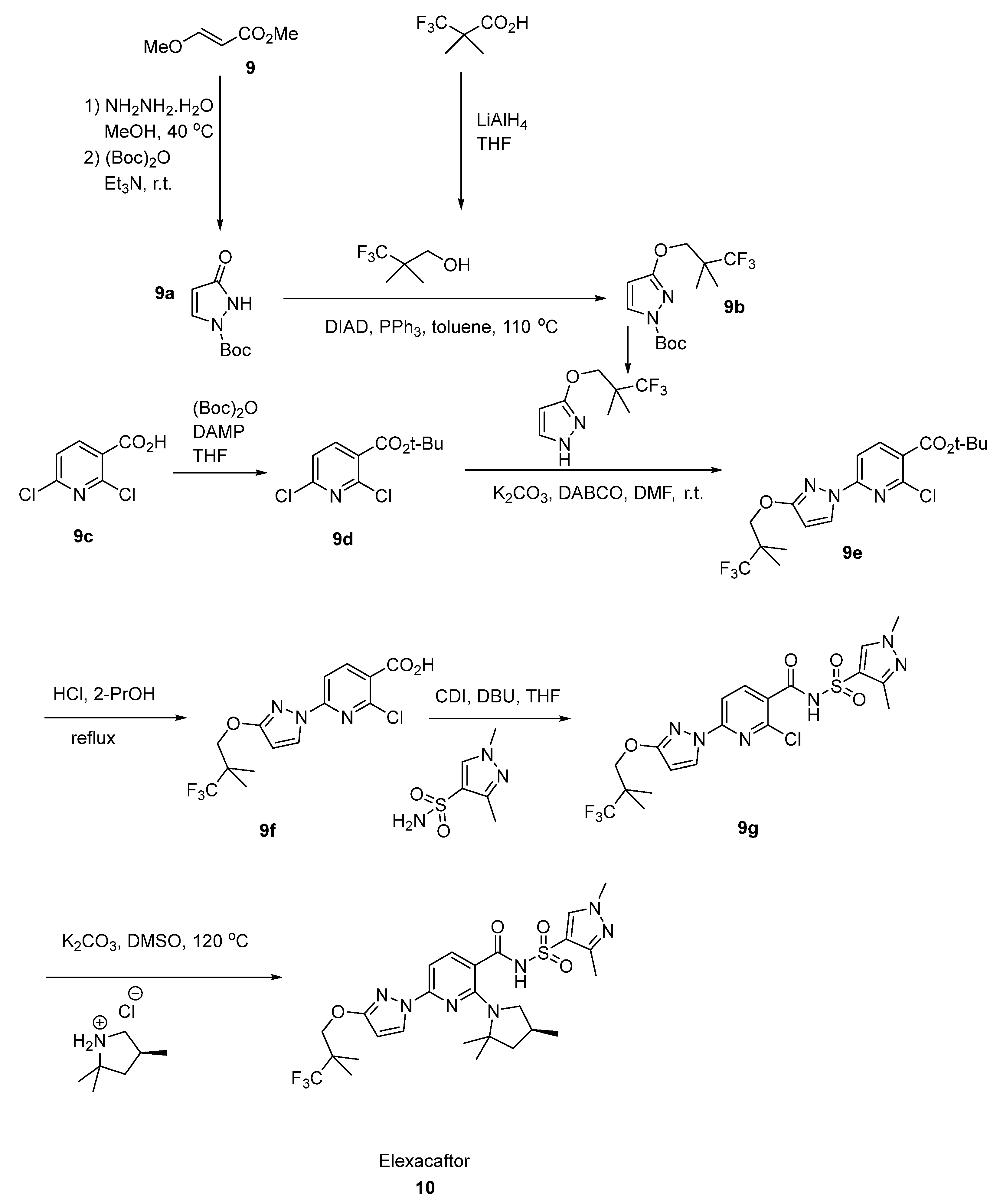
2.5. Elexacaftor
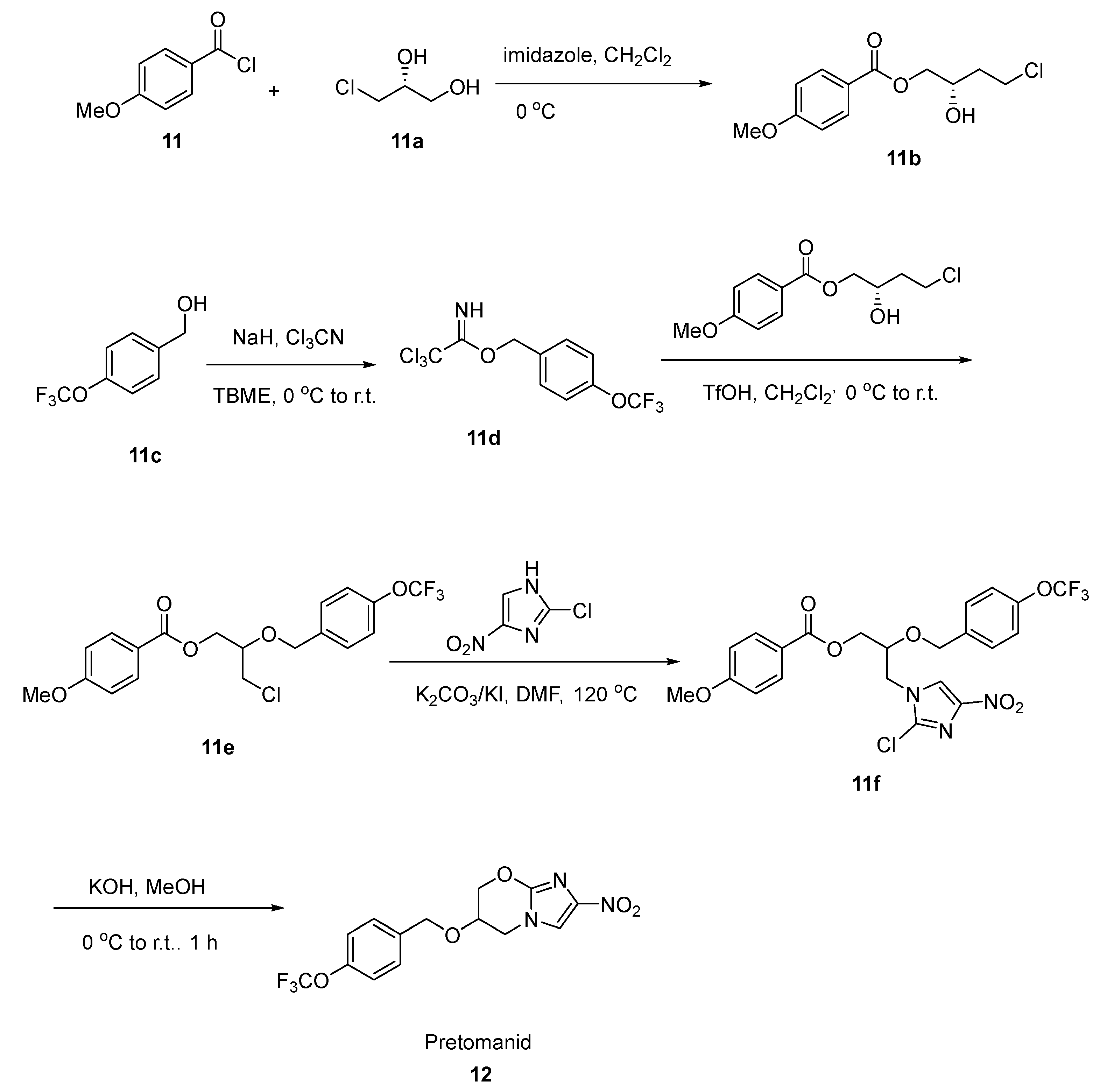
2.6. Pretomanid
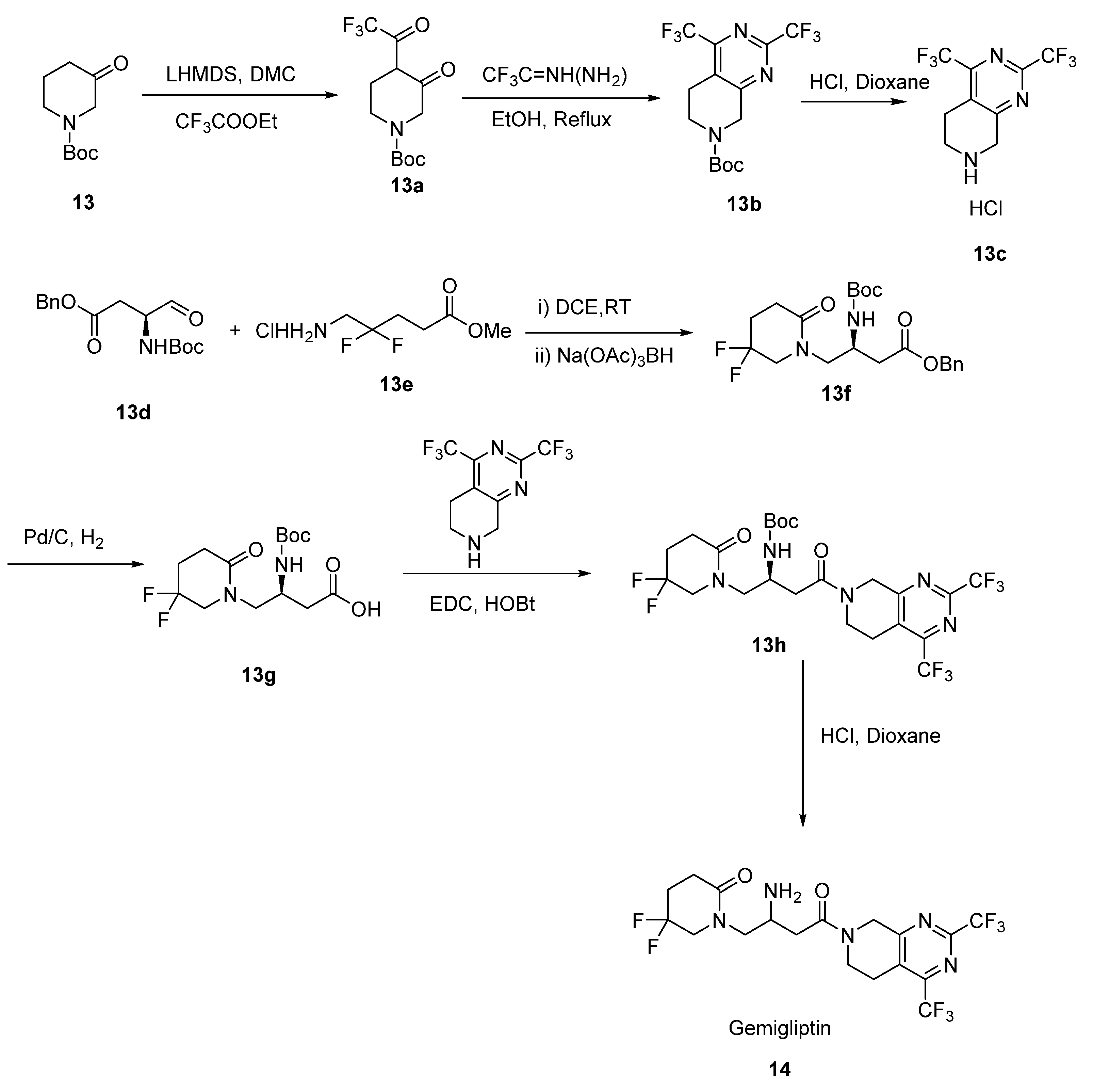
2.7. Gemigliptin
2.8. Doravirine
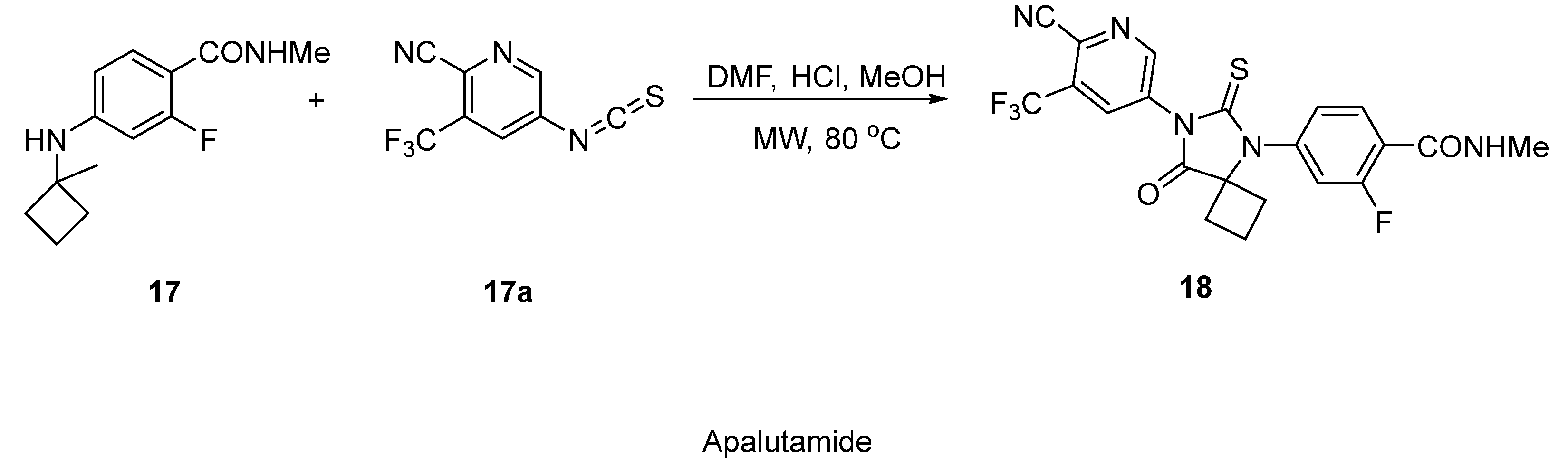
2.9. Apalutamide
2.10. Enasidenib
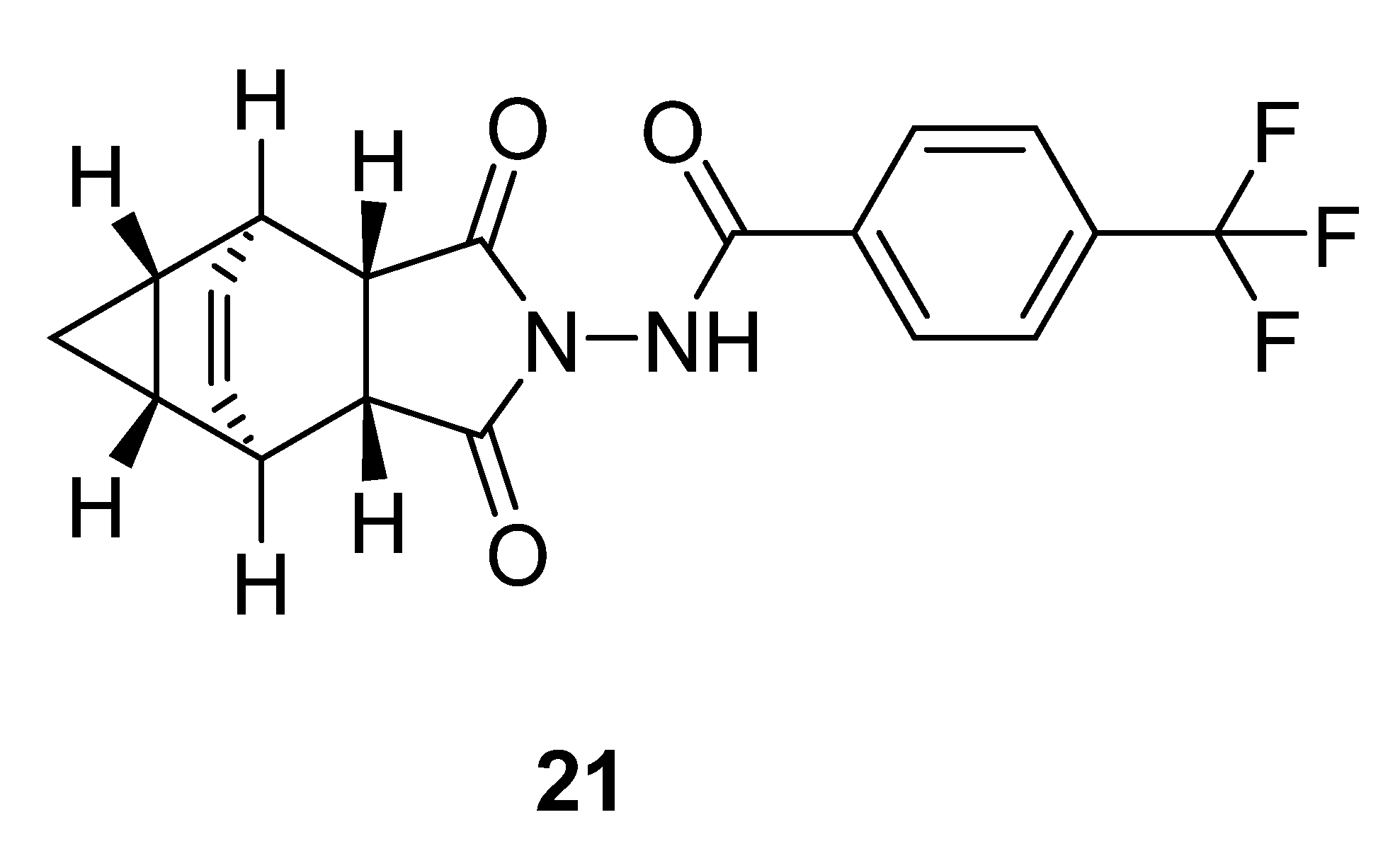
2.11. Tecovirimat
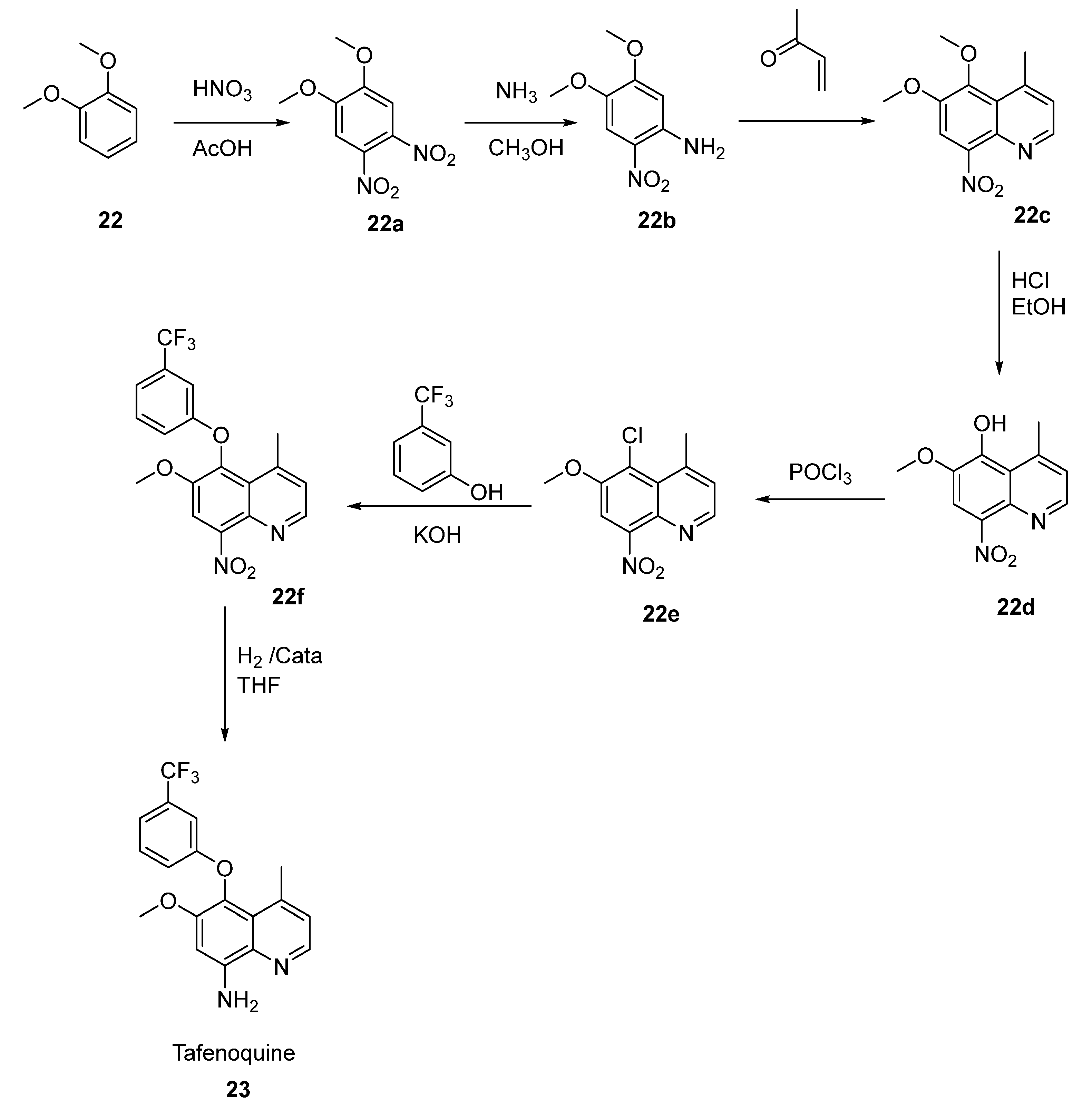
2.12. Tafenoquine
2.13. Fluoxetine
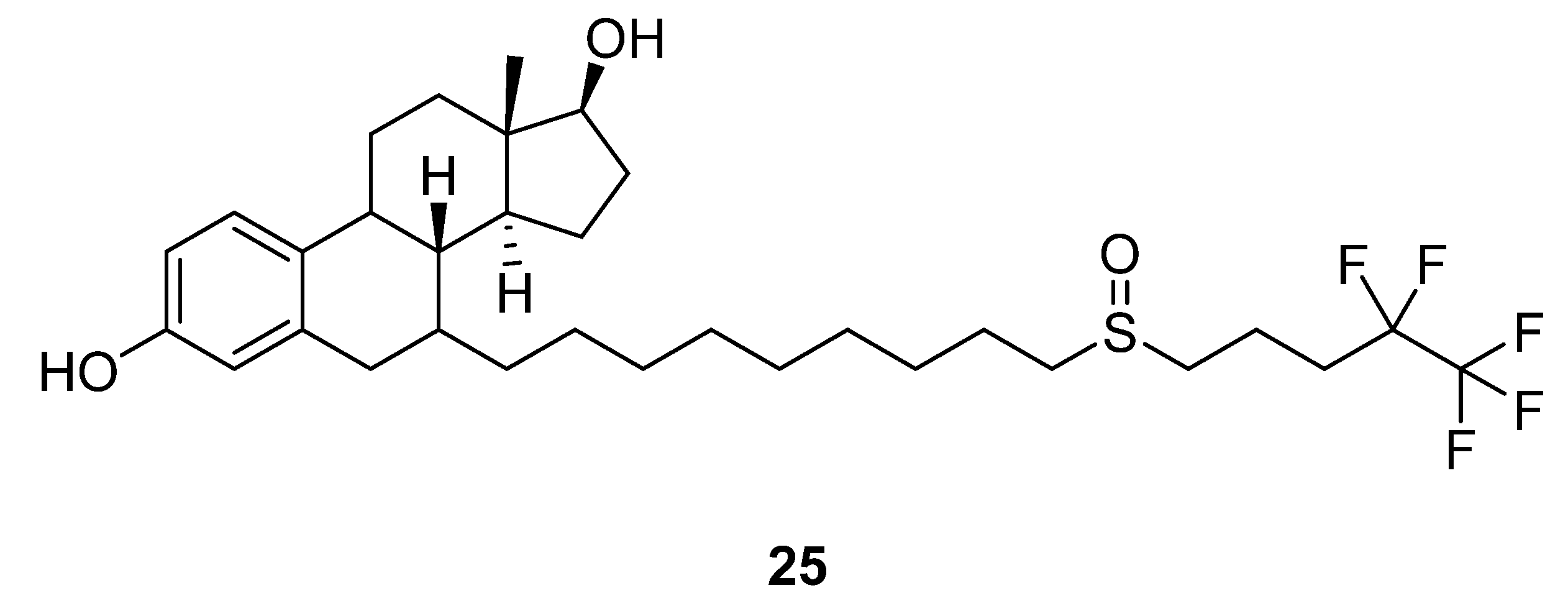
2.14. Fulvestrant
2.15. Sorafenib
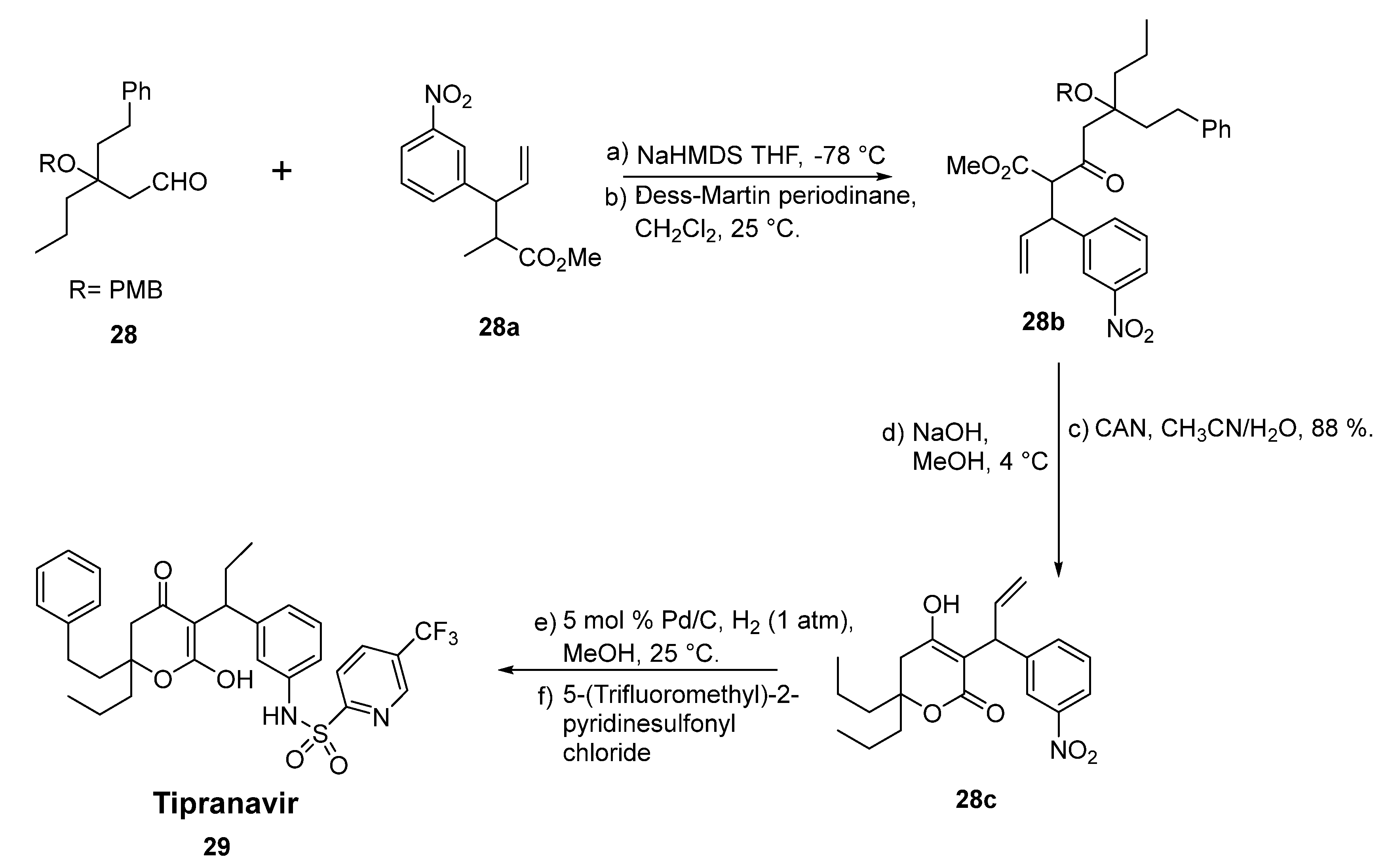
2.16. Tipranavir
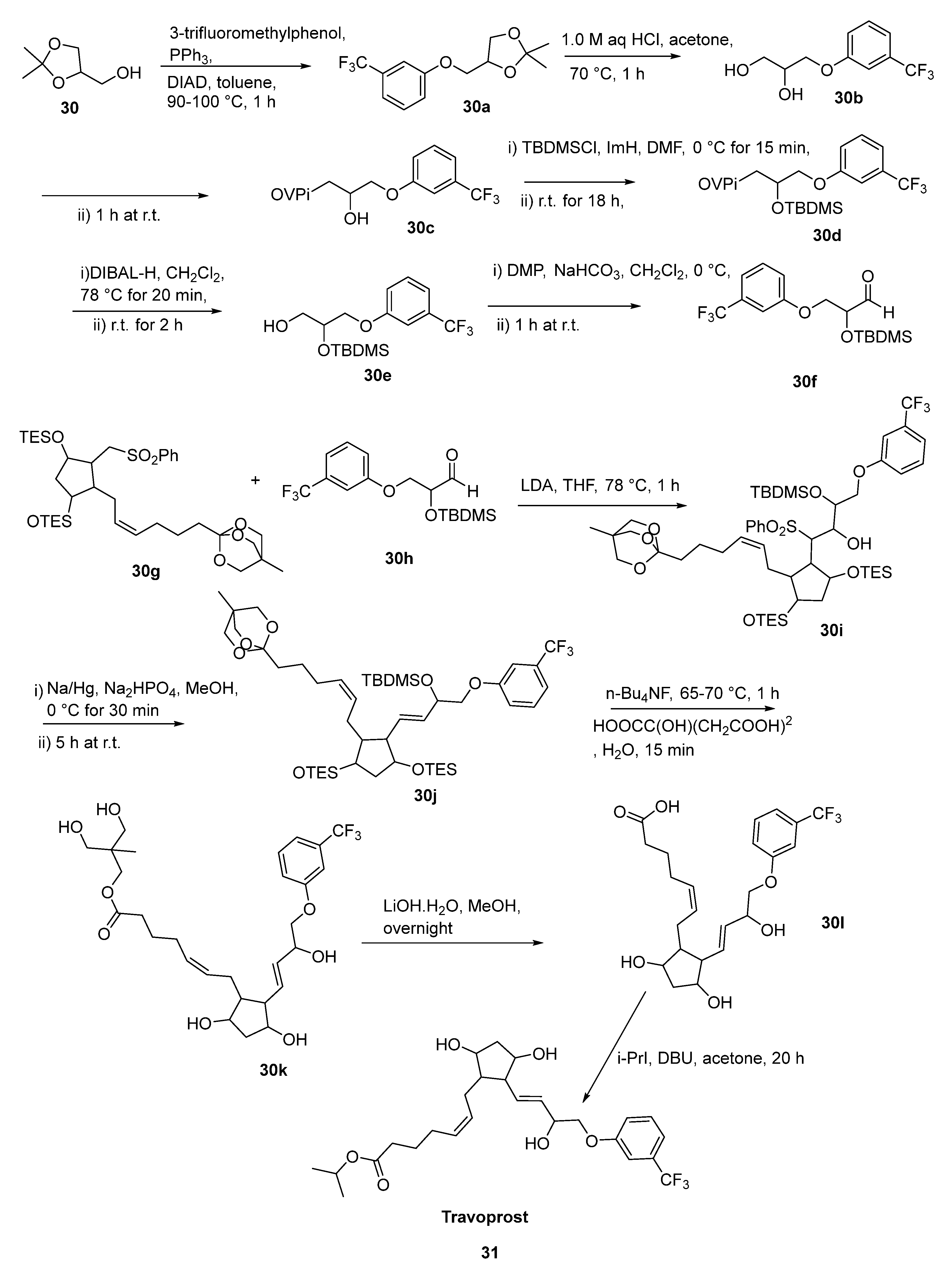
2.17. Travoprost
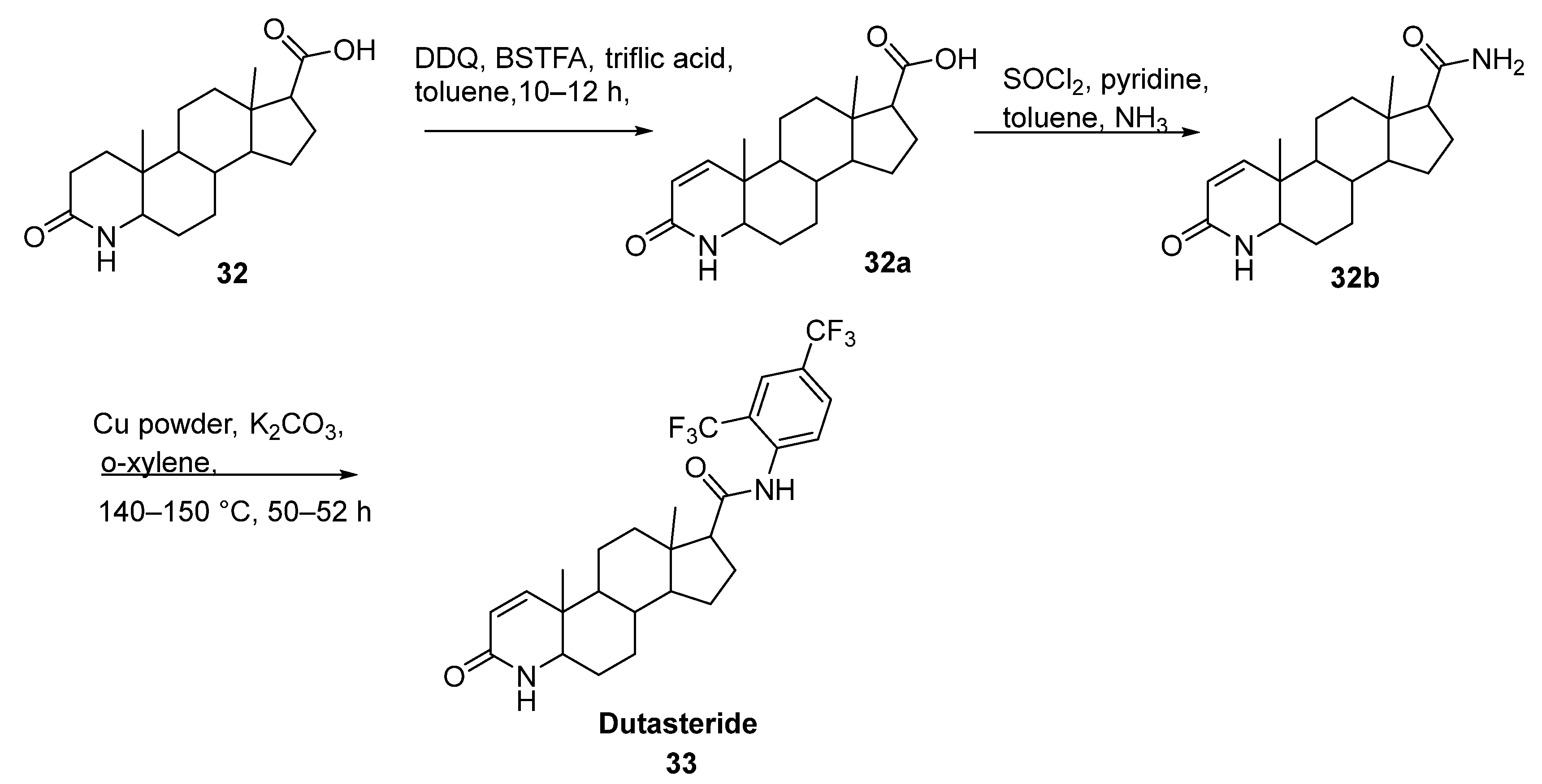
2.18. Dutasteride
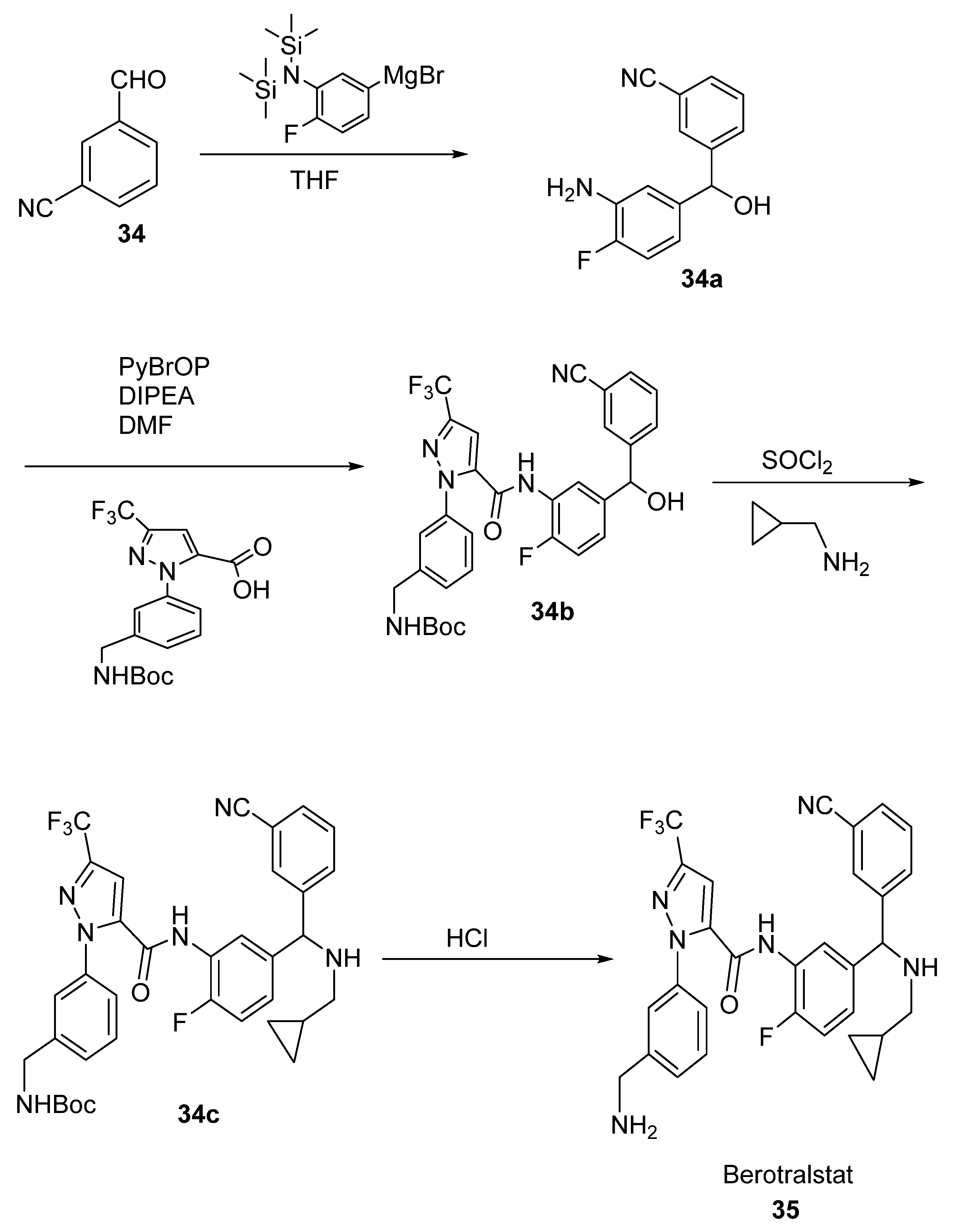
2.19. Berotralstat
3. Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Haufe, G.; Leroux, F. Fluorine in Life Sciences Progress in Fluorine Science Series; Elsevier Science & Technology: Amsterdam, The Netherlands, 2018. [Google Scholar]
- O’Hagan, D. Fluorine in Health Care: Organofluorine Containing Blockbuster Drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Swallow, S. Fluorine in Medicinal Chemistry. Prog. Med. Chem. 2015, 54, 65–133. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Maienfisch, P.; Altmann, K.H.; Schlosser, M. Special issue on “Fluorine in the Life Science. ChemBioChem 2004, 5, 559–722. [Google Scholar]
- Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in Medicinal Chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van Der Waals Volumes and Radii. Inorganic chemistry: Principles of structure and reactivity. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Kirk, K. Selective Fluorination in Drug Design and Development: An Overview of Biochemical Rationales. Curr. Top. Med. Chem. 2006, 6, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Westwell, A.D. The Role of Fluorine in Medicinal Chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current Contributions of Organofluorine Compounds to the Agrochemical Industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharm. 2001, 41, 443–470. [Google Scholar] [CrossRef]
- Steiner, H. Synthesis of aromatic trifluoromethyl compounds: The potential for large scale application. Chim. Oggi 2015, 33, 26–37. [Google Scholar]
- Ruppert, I.; Schlich, K.; Volbach, W. Die Ersten CF3-Substituierten Organyl(Chlor)Silane. Tetrahedron Lett. 1984, 25, 2195–2198. [Google Scholar] [CrossRef]
- Tomashenko, O.A.; Grushin, V.V. Aromatic Trifluoromethylation with Metal Complexes. Chem. Rev. 2011, 111, 4475–4521. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Kondo, H.; Amii, H. Aromatic Trifluoromethylation Catalytic in Copper. Chem. Commun. 2009, 14, 1909. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Matsnev, A.; Cahard, D. Shelf-stable electrophilic trifluoromethylating reagents: A brief historical perspective. Beilstein J. Org. Chem. 2010, 6, 65. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, J.; Früh, N.; Togni, A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 2014, 115, 650–682. [Google Scholar] [CrossRef]
- Shibata, N.; Matsnev, A.; Mizuta, S.; Kawai, H. Recent advances in enantioselective trifluoromethylation reactions. Tetrahedron Asymmetry 2008, 19, 2633–2644. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Yang, X.; Chen, Z.; Verpoort, F.; Shibata, N. Catalytic asymmetric synthesis of enantioenriched heterocycles bearing a C-CF3 stereogenic center. Chem. Eur. J. 2015, 21, 8664–8684. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J. Fluorine-Containing Drugs Approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a Potent and Selective Phosphatidylinositol-3 Kinase Alpha Inhibitor Selected for Clinical Evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Gerspacher, M.; Fairhurst, R.A.; Mah, R.; Roehn-Carnemolla, E.; Furet, P.; Fritsch, C.; Guthy, D.A. Discovery of a Novel Tricyclic 4H-Thiazolo [5′,4′:4,5]Pyrano [2,3-c]Pyridine-2-Amino Scaffold and Its Application in a PI3Kα Inhibitor with High PI3K Isoform Selectivity and Potent Cellular Activity. Bioorg. Med. Chem. Lett. 2015, 25, 3582–3584. [Google Scholar] [CrossRef] [PubMed]
- Yale, H.L. The Trifluoromethyl Group in Medical Chemistry. J. Med. Pharm. Chem. 1959, 1, 121–133. [Google Scholar] [CrossRef]
- Jagodzinska, M.; Huguenot, F.; Candiani, G.; Zanda, M. Assessing the Bioisosterism of the Trifluoromethyl Group with a Protease Probe. ChemMedChem 2009, 4, 49–51. [Google Scholar] [CrossRef]
- Jia, H.; Häring, A.P.; Berger, F.; Zhang, L.; Ritter, T. Trifluoromethyl Thianthrenium Triflate: A Readily Available Trifluoromethylating Reagent with Formal CF3+, CF3•, and CF3– Reactivity. J. Am. Chem. Soc. 2021, 143, 7623–7628. [Google Scholar] [CrossRef]
- Yin, Z.; Hu, W.; Zhang, W.; Konno, H.; Moriwaki, H.; Izawa, K.; Han, J.; Soloshonok, V.A. Tailor-Made Amino Acid-Derived Pharmaceuticals Approved by the FDA in 2019. Amino Acids 2020, 52, 1227–1261. [Google Scholar] [CrossRef]
- Scott, L.J. Ubrogepant: First Approval. Drugs 2020, 80, 323–328. [Google Scholar] [CrossRef] [Green Version]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase α–Selective Inhibition with Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results from the First-In-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Kanojia, D.; Mayakonda, A.; Said, J.W.; Doan, N.B.; Chien, W.; Ganesan, T.S.; Huey Chuang, L.S.; Venkatachalam, N.; Baloglu, E.; et al. Molecular Mechanism and Therapeutic Implications of Selinexor (KPT-330) in Liposarcoma. Oncotarget 2016, 8, 7521–7532. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Selinexor: First Global Approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, A.R.; Kanniah, S.L.; Ravi, A.; das Tonmoy, C.; Chemate, R.P.; Singh, A.K.; Wagh, Y.D. Novel Crystalline Forms of Selinexor and Process for Their Preparation. WO2018129227, 12 July 2018. [Google Scholar]
- Podar, K.; Shah, J.; Chari, A.; Richardson, P.G.; Jagannath, S. Selinexor for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2020, 21, 399–408. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Y.; Li, J.; Liu, Y. Exploratory Process Development of Pexidartinib through the Tandem Tsuji–Trost Reaction and Heck Coupling. Synthesis 2019, 51, 2564–2571. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs 2019, 79, 1805–1812. [Google Scholar] [CrossRef]
- Middleton, P.G.; Taylor-Cousar, J.L. Development of Elexacaftor—Tezacaftor—Ivacaftor: Highly Effective CFTR Modulation for the Majority of People with Cystic Fibrosis. Expert Rev. Respir. Med. 2021, 15, 723–735. [Google Scholar] [CrossRef]
- Hughes, D.L. Patent Review of Synthetic Routes and Crystalline Forms of the CFTR-Modulator Drugs Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor. Org. Process Res. Dev. 2019, 23, 2302–2322. [Google Scholar] [CrossRef]
- Marsini, M.A.; Reider, P.J.; Sorensen, E.J. A Concise and Convergent Synthesis of PA-824. J. Org. Chem. 2010, 75, 7479–7482. [Google Scholar] [CrossRef]
- Keam, S.J. Pretomanid: First Approval. Drugs 2019, 79, 1797–1803. [Google Scholar] [CrossRef]
- Gils, T.; Lynen, L.; de Jong, B.C.; Van Deun, A.; Decroo, T. Pretomanid for tuberculosis: A systematic review. Clin. Microbiol. Infect. 2021, 28, 31–42. [Google Scholar] [CrossRef]
- Kim, S.-H.; Yoo, J.-H.; Lee, W.J.; Park, C.-Y. Gemigliptin: An Update of Its Clinical Use in the Management of Type 2 Diabetes Mellitus. Diabetes Metab. J. 2016, 40, 339. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, R.N.; Haq, W.; Katti, S.B. Discovery of 17 Gliptins in 17-Years of Research for the Treatment of Type 2 Diabetes: A Synthetic Overview. Chem. Biol. Interface 2014, 4, 137–162. [Google Scholar]
- Wang, Z.; Yu, Z.; Kang, D.; Zhang, J.; Tian, Y.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Design, Synthesis and Biological Evaluation of Novel Acetamide-Substituted Doravirine and Its Prodrugs as Potent HIV-1 NNRTIs. Bioorg. Med. Chem. 2019, 27, 447–456. [Google Scholar] [CrossRef]
- Dellis, A.E.; Papatsoris, A.G. Apalutamide: The Established and Emerging Roles in the Treatment of Advanced Prostate Cancer. Expert Opin. Investig. Drugs 2018, 27, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.L. Review of Synthetic Routes and Crystalline Forms of the Antiandrogen Oncology Drugs Enzalutamide, Apalutamide, and Darolutamide. Org. Process Res. Dev. 2020, 24, 347–362. [Google Scholar] [CrossRef]
- Galkin, M.; Jonas, B.A. Enasidenib in the Treatment of Relapsed/Refractory Acute Myeloid Leukemia: An Evidence-Based Review of Its Place in Therapy. Core Evid. 2019, 14, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Mucker, E.M.; Goff, A.J.; Shamblin, J.D.; Grosenbach, D.W.; Damon, I.K.; Mehal, J.M.; Holman, R.C.; Carroll, D.; Gallardo, N.; Olson, V.A.; et al. Efficacy of Tecovirimat (ST-246) in Nonhuman Primates Infected with Variola Virus (Smallpox). Antimicrob. Agents Chemother. 2013, 57, 6246–6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, N.J. Tafenoquine—A Radical Improvement? N. Engl. J. Med. 2019, 380, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Prashar, L.; Paul, R. Tafenoquine: A New 8- Aminoquinoline. Med. J. Zamb. 2009, 36, 187–190. [Google Scholar] [CrossRef]
- Waters, N.C.; Edstein, M.D. 8-Aminoquinolines: Primaquine and Tafenoquine. In Treatment and Prevention of Malaria; Springer: Basel, Switzerland, 2011; pp. 69–94. [Google Scholar] [CrossRef]
- Wenthur, C.J.; Bennett, M.R.; Lindsley, C.W. Classics in Chemical Neuroscience: Fluoxetine (Prozac). ACS Chem. Neurosci. 2013, 5, 14–23. [Google Scholar] [CrossRef]
- Kakei, H.; Nemoto, T.; Ohshima, T.; Shibasaki, M. Efficient Synthesis of Chiralα- Andβ-Hydroxy Amides: Application to the Synthesis of (R)-Fluoxetine. Angew. Int. Ed. 2004, 43, 317–320. [Google Scholar] [CrossRef]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An Oestrogen Receptor Antagonist with a Novel Mechanism of Action. Br. J. Cancer 2004, 90, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Caprioglio, D.; Fletcher, S.P. An Alternative Synthesis of the Breast Cancer Drug Fulvestrant (Faslodex®): Catalyst Control over C–c Bond Formation. Chem. Commun. 2015, 51, 14866–14868. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chen, J.; He, Z.; Sun, W.; Xu, W. Design, Synthesis and Biological Activities of Thiourea Containing Sorafenib Analogs as Antitumor Agents. Bioorg. Med. Chem. 2012, 20, 2923–2929. [Google Scholar] [CrossRef]
- Chen, F.; Fang, Y.; Zhao, R.; Le, J.; Zhang, B.; Huang, R.; Chen, Z.; Shao, J. Evolution in Medicinal Chemistry of Sorafenib Derivatives for Hepatocellular Carcinoma. Eur. J. Med. Chem. 2019, 179, 916–935. [Google Scholar] [CrossRef]
- Rahman, S.; Sarker, M.S.; Aralaguppe, S.G.; Sarwar, G.; Khan, S.I.; Rahman, M. Drug Resistance Pattern among ART-Naive Clients Attending an HIV Testing and Counseling Center in Dhaka, Bangladesh. J. Med. Virol. 2021, 94, 787–790. [Google Scholar] [CrossRef]
- Xiong, J.; Li, Y.; Tan, X.; Chen, T.; Liu, B.; Fu, L. The Anti-HIV Drug Tipranavir Induces Gastric Cancer Stem Cell Apoptosis and Exerts Anticancer Activity via the PRSS23–IL24 Pathway. Res. Sq. 2022, in press. [Google Scholar] [CrossRef]
- Turner, S.R.; Strohbach, J.W.; Tommasi, R.A.; Aristoff, P.A.; Johnson, P.D.; Skulnick, H.I.; Dolak, L.A.; Seest, E.P.; Tomich, P.K.; Bohanon, M.J.; et al. Tipranavir (PNU-140690): A Potent, Orally Bioavailable Nonpeptidic HIV Protease Inhibitor of the 5,6-Dihydro-4-hydroxy-2-pyrone Sulfonamide Class∇. J. Med. Chem. 1998, 41, 3467–3476. [Google Scholar] [CrossRef] [PubMed]
- Waugh, J.; Jarvis, B. Travoprost. Drugs Aging 2002, 19, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Boulton, L.T.; Brick, D.; Fox, M.E.; Jackson, M.; Lennon, I.C.; McCague, R.; Parkin, N.; Rhodes, D.; Ruecroft, G. Synthesis of the Potent Antiglaucoma Agent, Travoprost. Org. Process Res. Dev. 2002, 6, 138–145. [Google Scholar] [CrossRef]
- Keam, S.J.; Scott, L.J. Dutasteride. Drugs 2008, 68, 463–485. [Google Scholar] [CrossRef]
- Evans, H.C.; Goa, K.L. Dutasteride. Drugs Aging 2003, 20, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Bork, K. A Decade of Change: Recent Developments in Pharmacotherapy of Hereditary Angioedema (HAE). Clin. Rev. Allergy Immunol. 2016, 51, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.R.; Hwang, G.; Johri, A.; Craig, T. Oral Plasma Kallikrein Inhibitor BCX7353 for Treatment of Hereditary Angioedema. Immunotherapy 2019, 11, 1439–1444. [Google Scholar] [CrossRef]
- Zuraw, B.; Lumry, W.R.; Johnston, D.T.; Aygören-Pürsün, E.; Banerji, A.; Bernstein, J.A.; Christiansen, S.C.; Jacobs, J.S.; Sitz, K.V.; Gower, R.G.; et al. Oral Once-Daily Berotralstat for the Prevention of Hereditary Angioedema Attacks: A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial. J. Allergy Clin. Immunol. 2020, 148, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Harilal, S.; Al-Sehemi, A.G.; Mathew, G.E.; Carradori, S.; Mathew, B. The Chronicle of COVID-19 and Possible Strategies to Curb the Pandemic. Curr. Med. Chem. 2021, 28, 2852–2886. [Google Scholar] [CrossRef]
- Mathew, B.; Carradori, S.; Guglielmi, P.; Uddin, M.S.; Kim, H. New Aspects of Monoamine Oxidase B Inhibitors: The Key Role of Halogens to Open the Golden Door. Curr. Med. Chem. 2021, 28, 266–283. [Google Scholar] [CrossRef]
- Esterhuysen, C.; Heßelmann, A.; Clark, T. Trifluoromethyl: An Amphiphilic Noncovalent Bonding Partner. Chemphyschem 2017, 18, 772–784. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
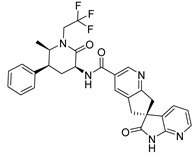
| Structure | Name | Therapeutic Use | Ref. |
|---|---|---|---|
 | Ubrogepant | Migraine | [29] |
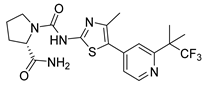 | Alpelisib | Breast Cancer | [32] |
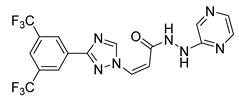 | Selinexor | Anticancer | [36] |
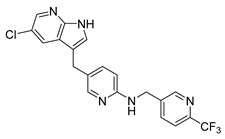 | Pexidartinib | Anticancer | [38] |
 | Elexacaftor | Cystic fibrosis | [41] |
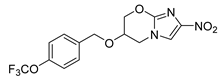 | Pretomanid | Tuberculosis | [43] |
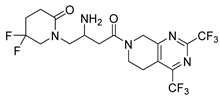 | Gemigliptin | Antihyper Glycemia | [45] |
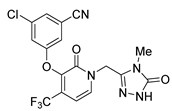 | Doravirine | Anti-HIV | [46] |
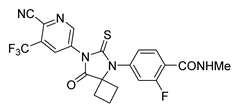 | Apalutamide | Prostate Cancer | [48] |
 | Enasidenib | Leukemia | [50] |
 | Tecovirimat | Antiviral | [51] |
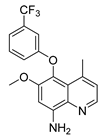 | Tafenoquine | Malaria | [54] |
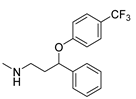 | Fluoxetine | Antidepressant | [56] |
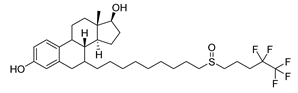 | Fulvestrant | Breast Cancer | [58] |
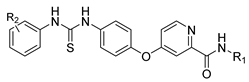 | Sorafenib | Antineoplastic | [60] |
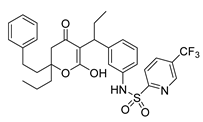 | Tipranavir | Anti-HIV | [63] |
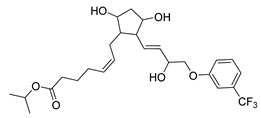 | Travoprost | Glaucoma | [65] |
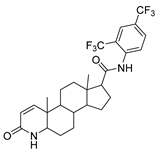 | Dutasteride | Benign Prostatetic Hyperplasia | [67] |
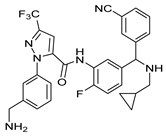 | Berotralstat | Hereditary Angioedema | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, A.S.; Singh, A.K.; Kumar, A.; Kumar, S.; Sukumaran, S.; Koyiparambath, V.P.; Pappachen, L.K.; Rangarajan, T.M.; Kim, H.; Mathew, B. FDA-Approved Trifluoromethyl Group-Containing Drugs: A Review of 20 Years. Processes 2022, 10, 2054. https://doi.org/10.3390/pr10102054
Nair AS, Singh AK, Kumar A, Kumar S, Sukumaran S, Koyiparambath VP, Pappachen LK, Rangarajan TM, Kim H, Mathew B. FDA-Approved Trifluoromethyl Group-Containing Drugs: A Review of 20 Years. Processes. 2022; 10(10):2054. https://doi.org/10.3390/pr10102054
Chicago/Turabian StyleNair, Aathira Sujathan, Ashutosh Kumar Singh, Astik Kumar, Sunil Kumar, Sunitha Sukumaran, Vishal Payyalot Koyiparambath, Leena K. Pappachen, T. M. Rangarajan, Hoon Kim, and Bijo Mathew. 2022. "FDA-Approved Trifluoromethyl Group-Containing Drugs: A Review of 20 Years" Processes 10, no. 10: 2054. https://doi.org/10.3390/pr10102054