
Electroreduction of CO2 toward High Current Density





Abstract
:1. Introduction
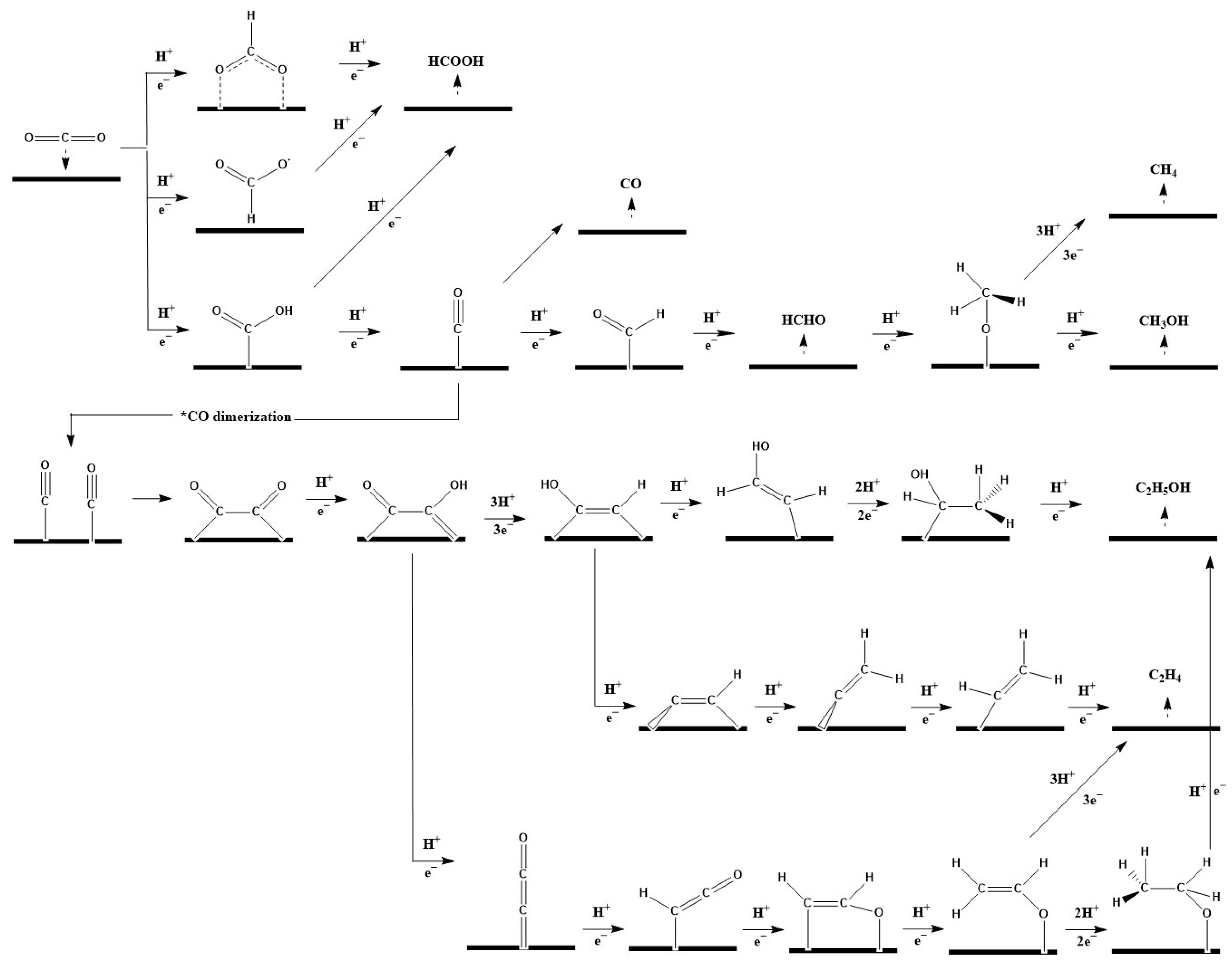
2. Mechanisms of CO2ER
3. Electrocatalysts for CO2 Electroreduction
3.1. Metal-Based Catalysts
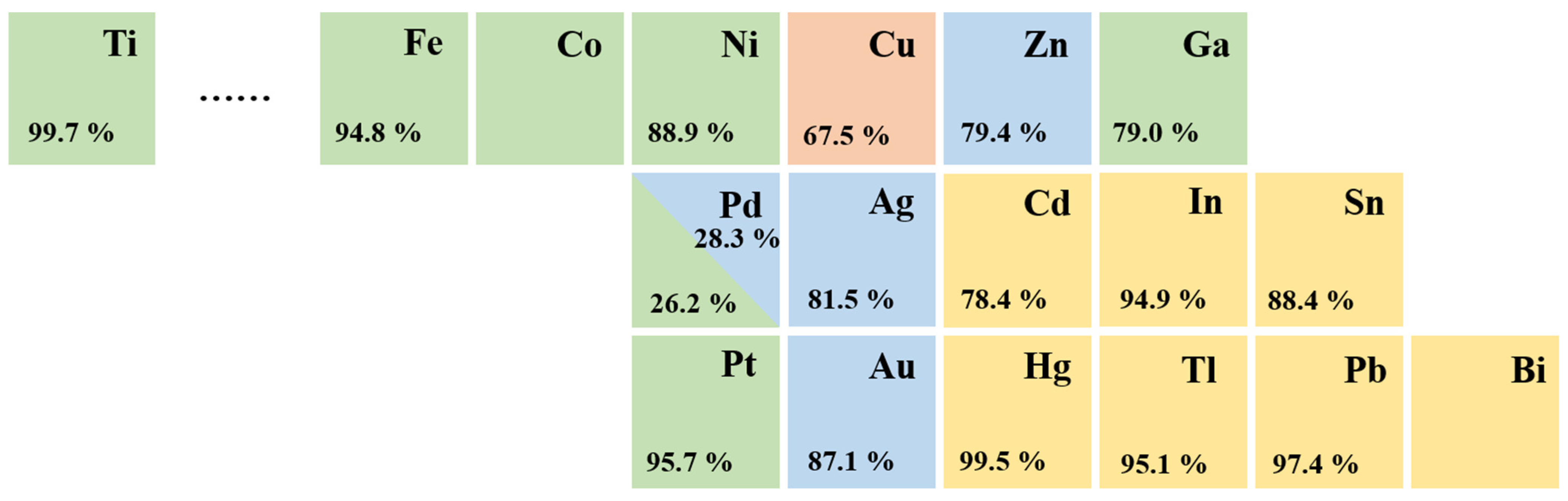
3.1.1. Noble Metals
- (I)
- Au
- (II)
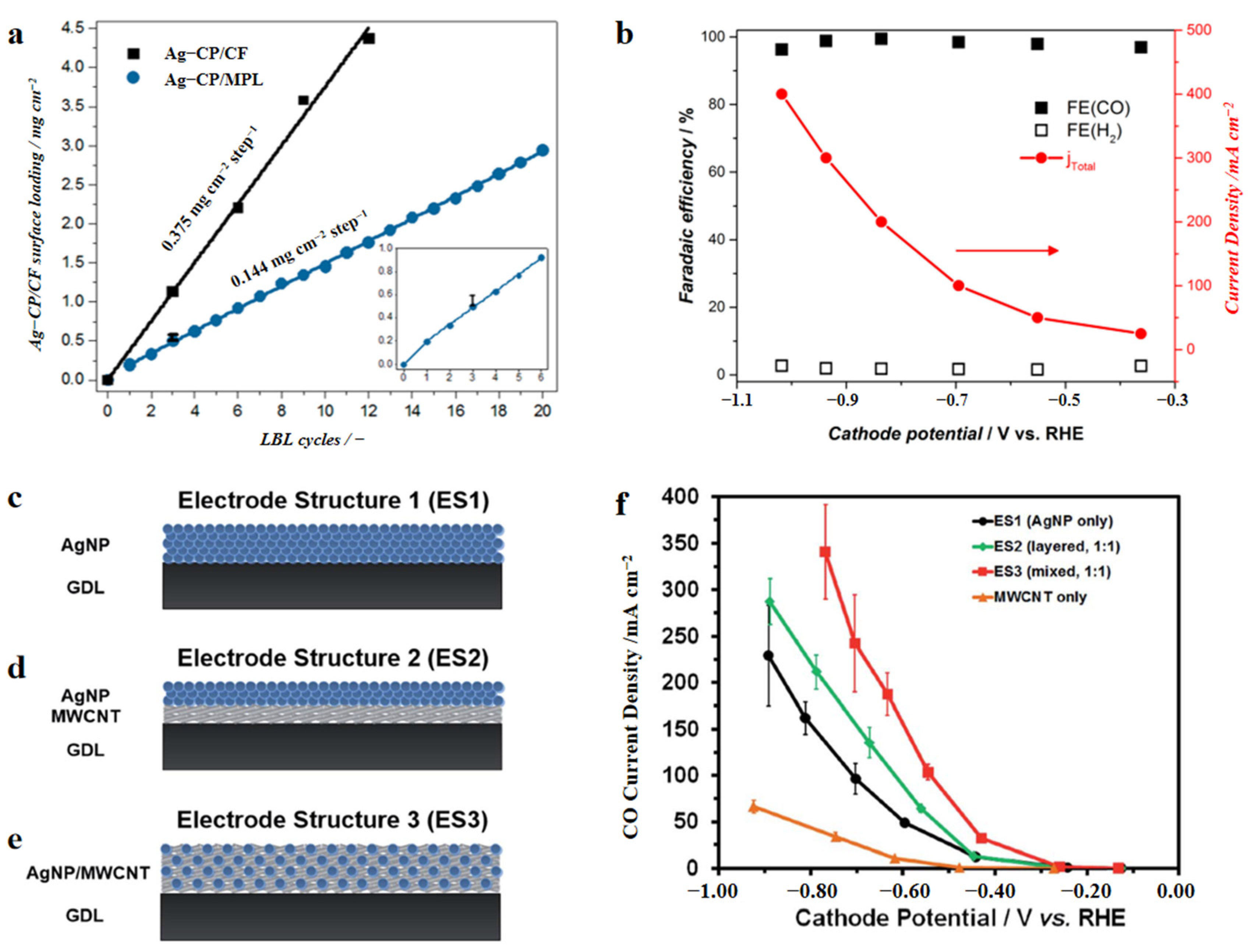
- Ag
- (III)
- Pd
3.1.2. Non-Noble Metals
- (I)
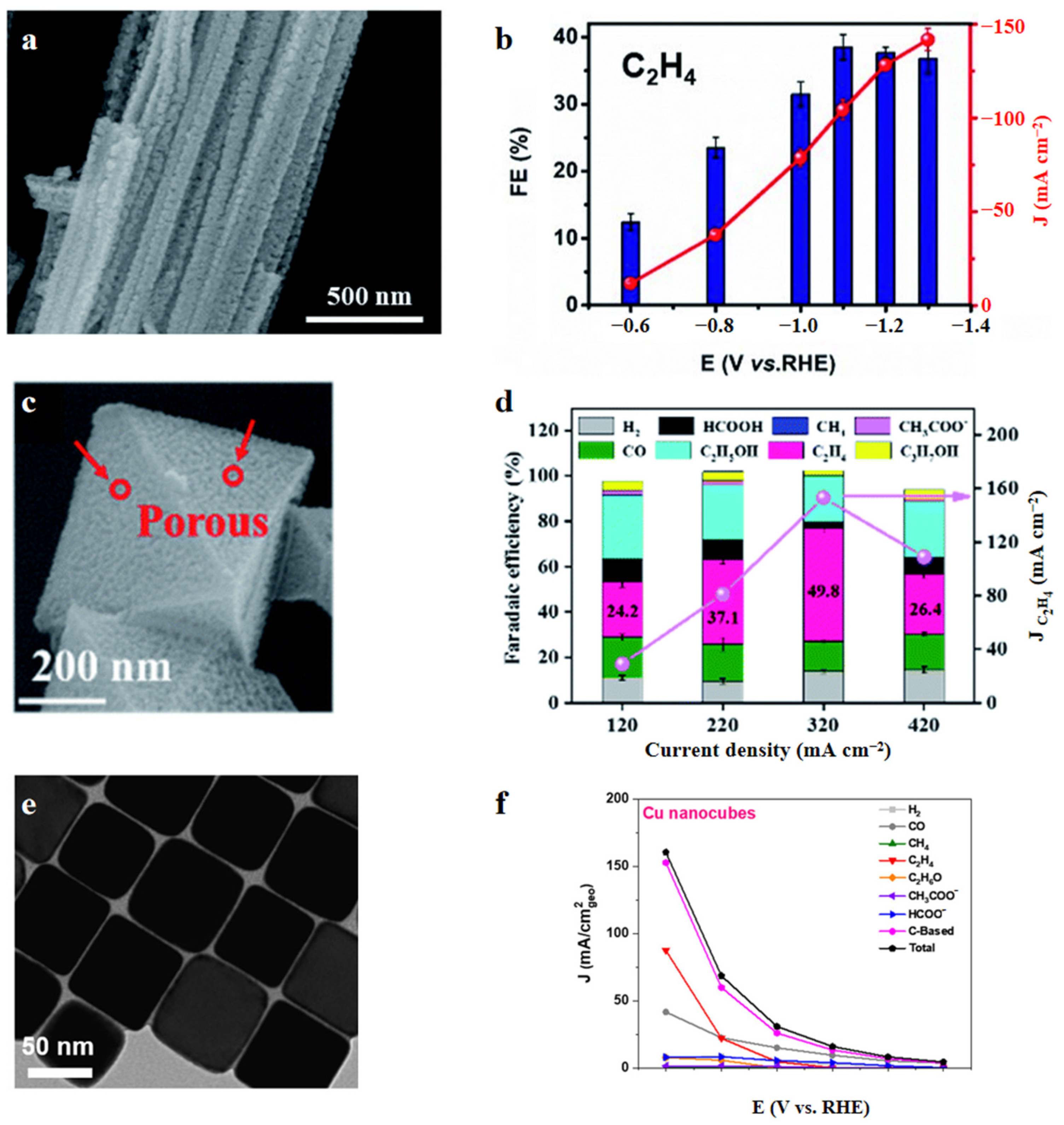
- Cu
- (II)
- Zn
- (III)
- Cd
- (IV)
- Sn
- (V)
- Bi
3.2. Metal-Free Carbon Catalysts
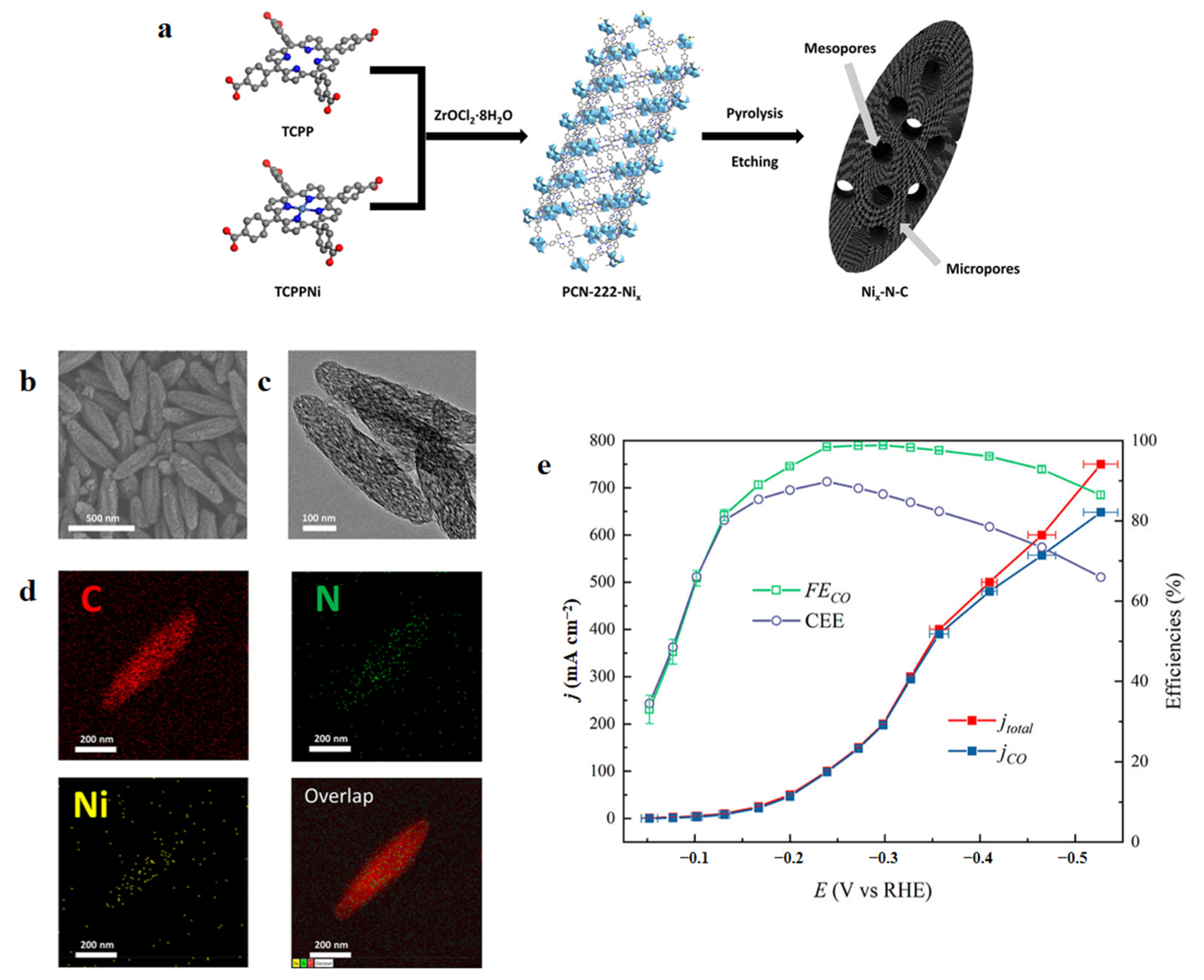
3.3. Single-Atom Catalysts
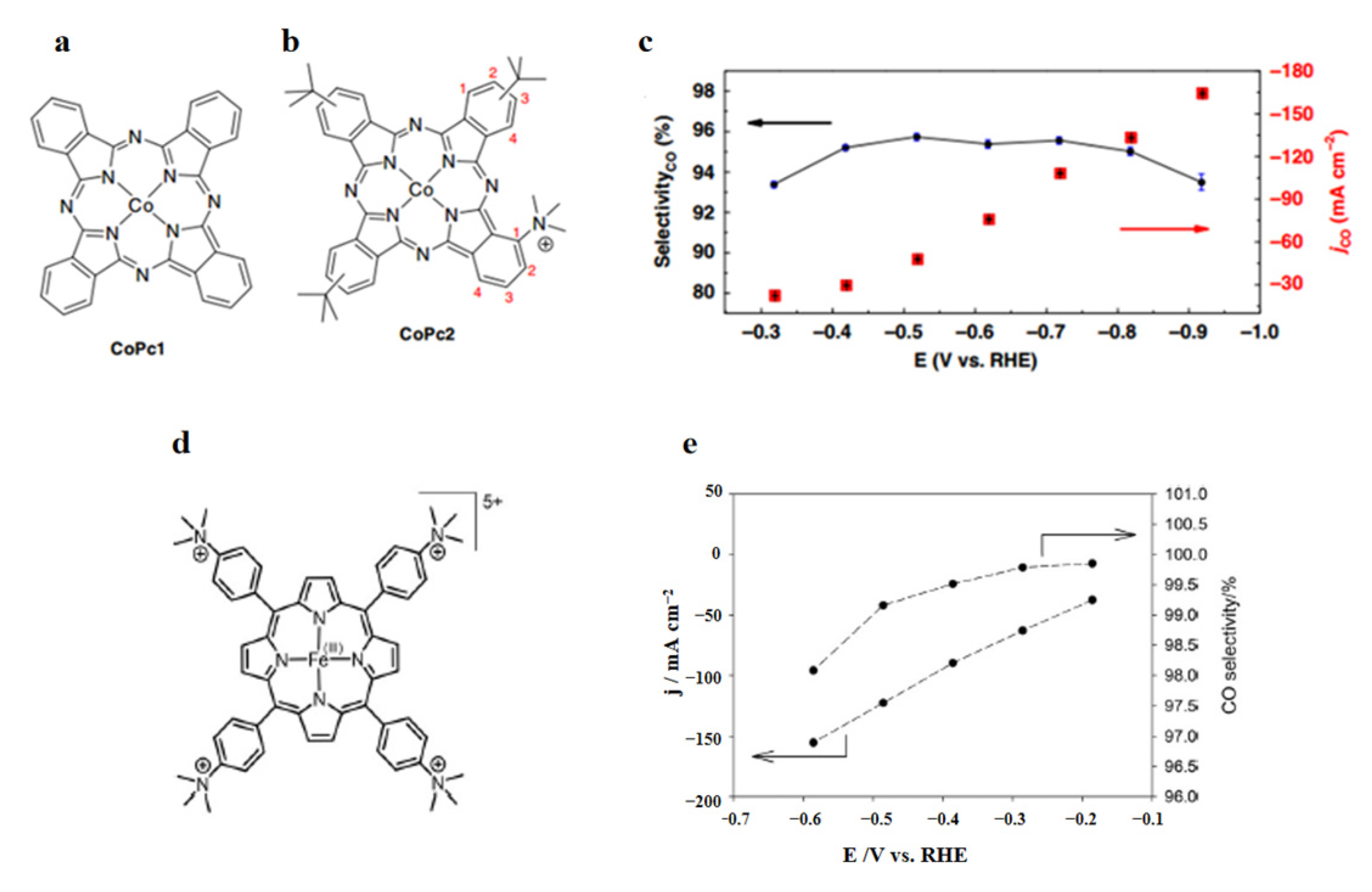
3.4. Molecular Catalysts
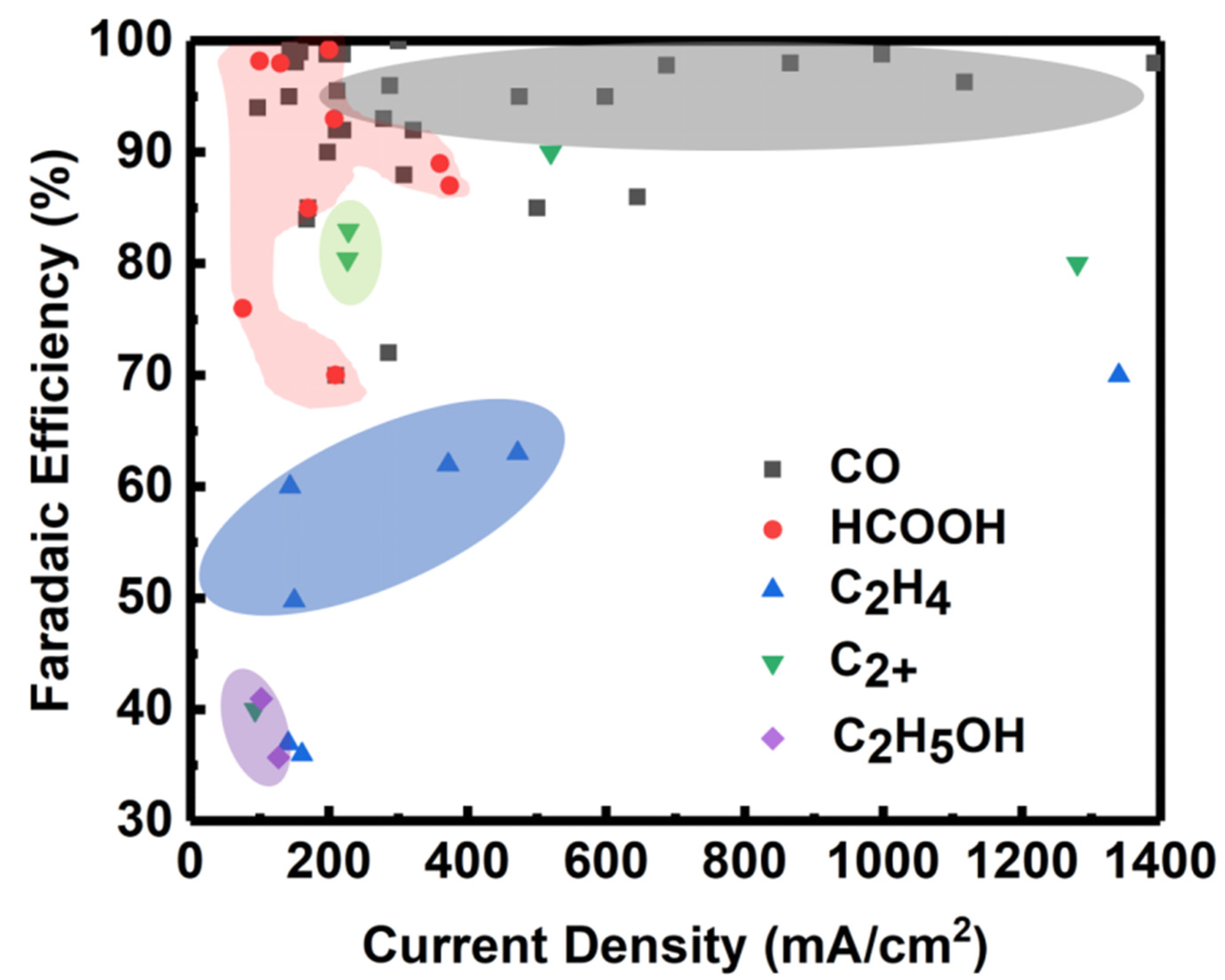
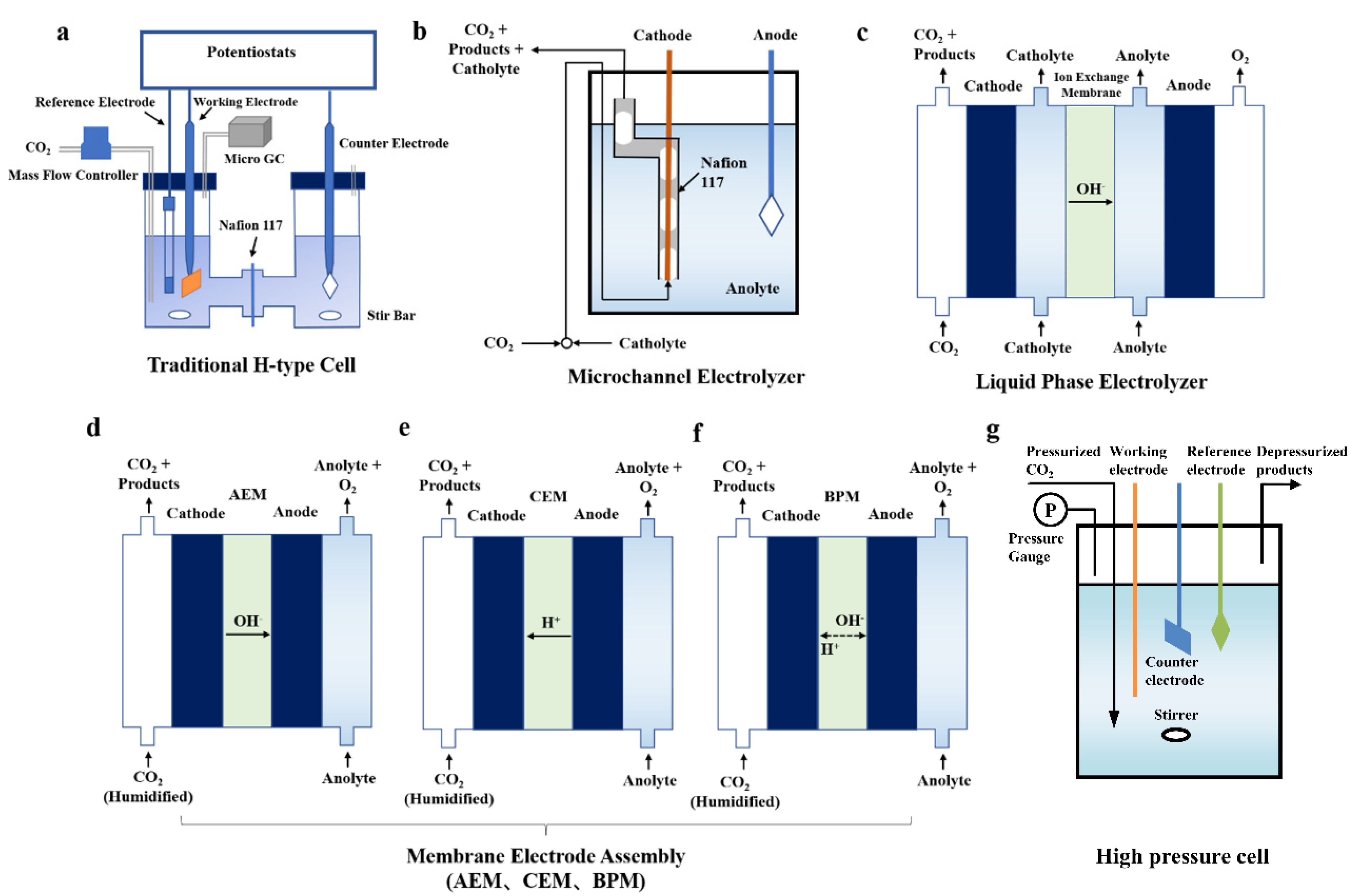
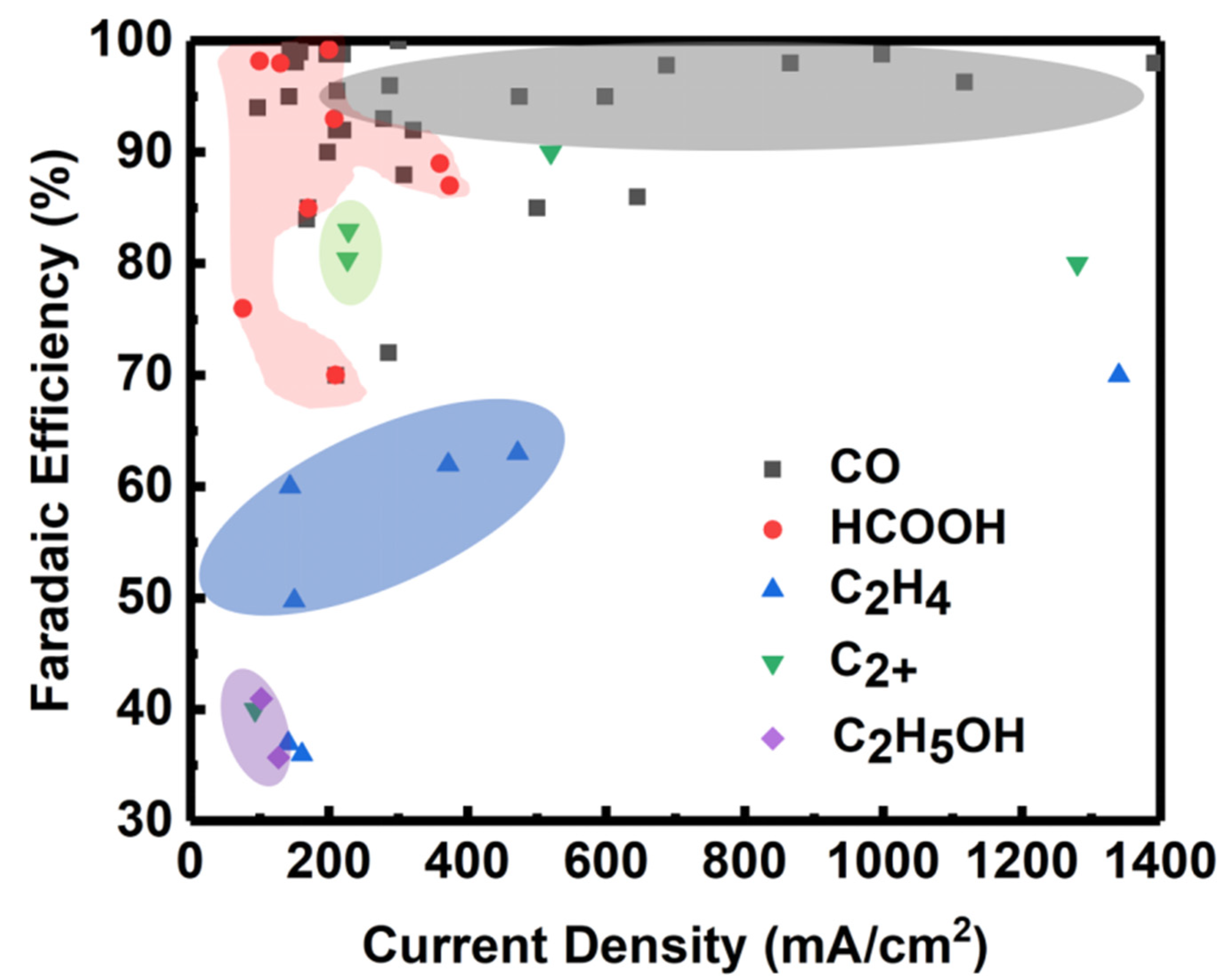
4. Electrolyzer Design
4.1. Electrolyzer Types
4.1.1. H-Type Cell
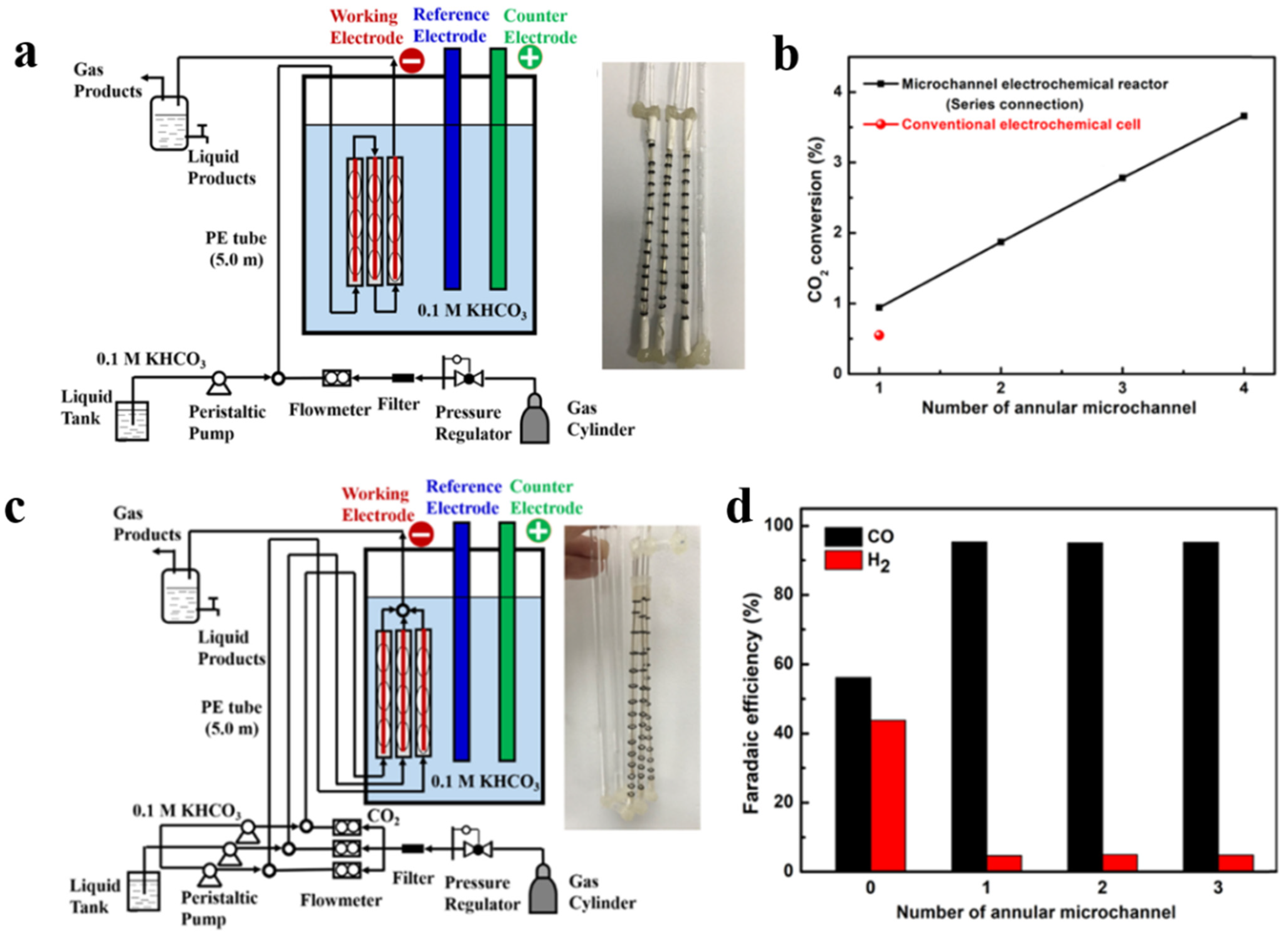
4.1.2. Microchannel Electrolyzer
4.1.3. Liquid-Phase Electrolyzer
4.1.4. Membrane Electrode Assembly (MEA)
4.1.5. High-Pressure Cell
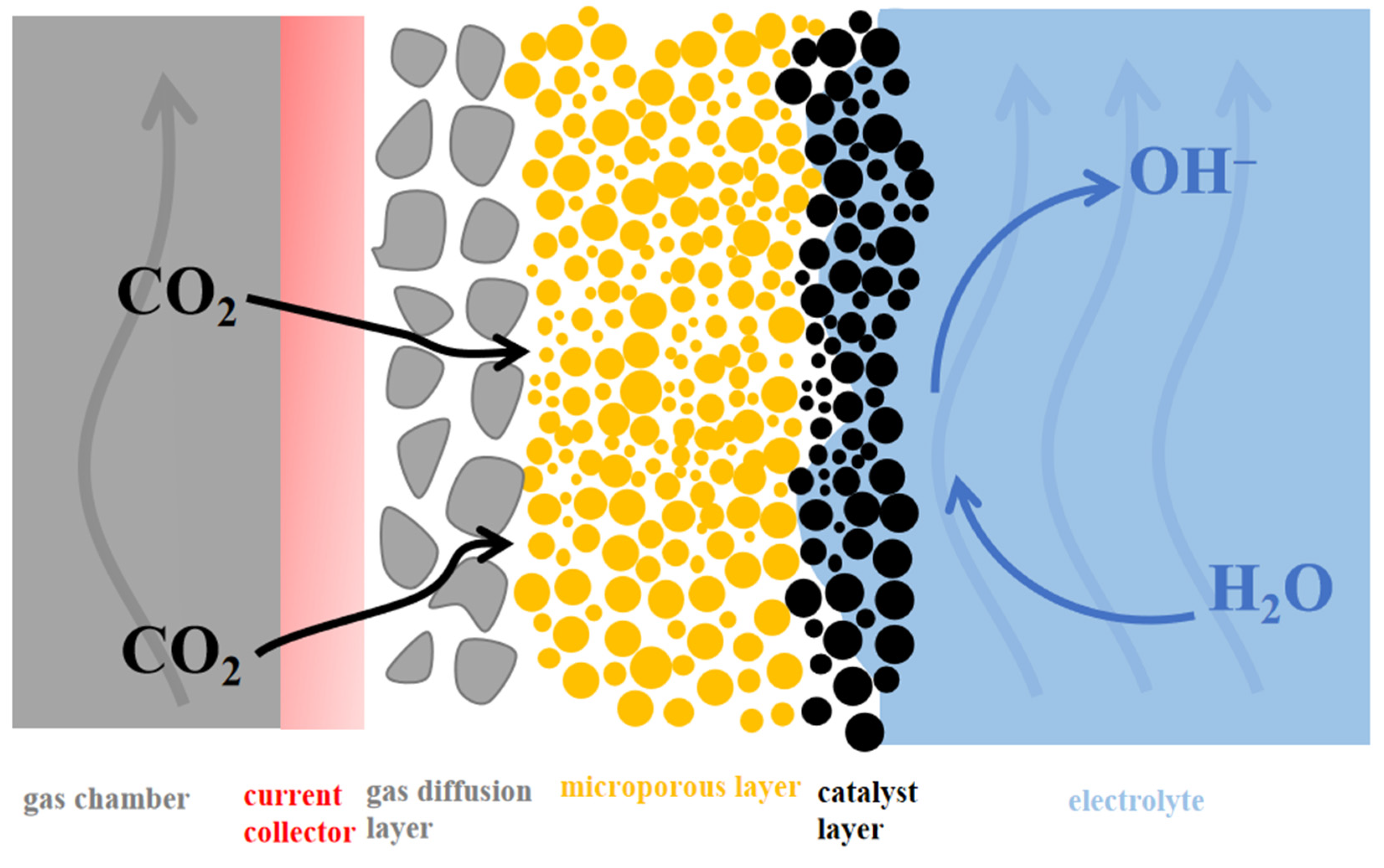
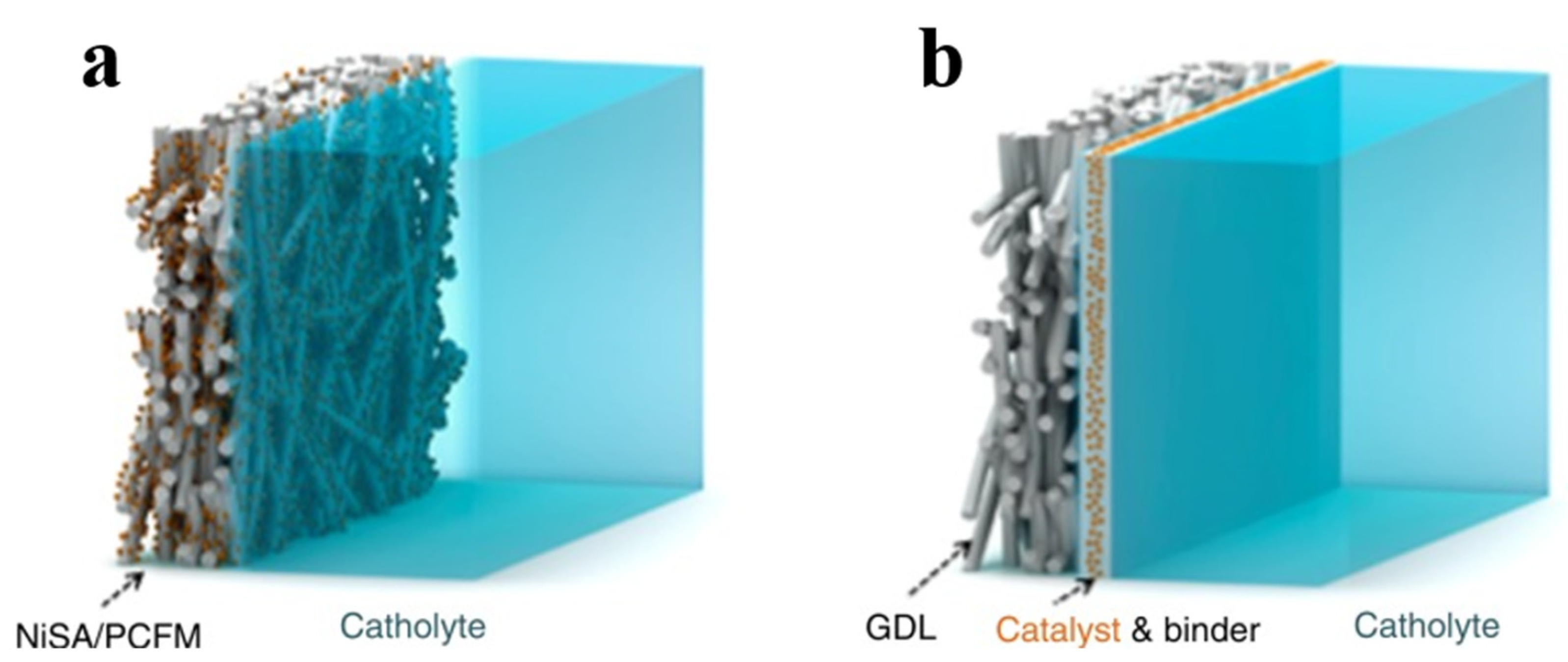
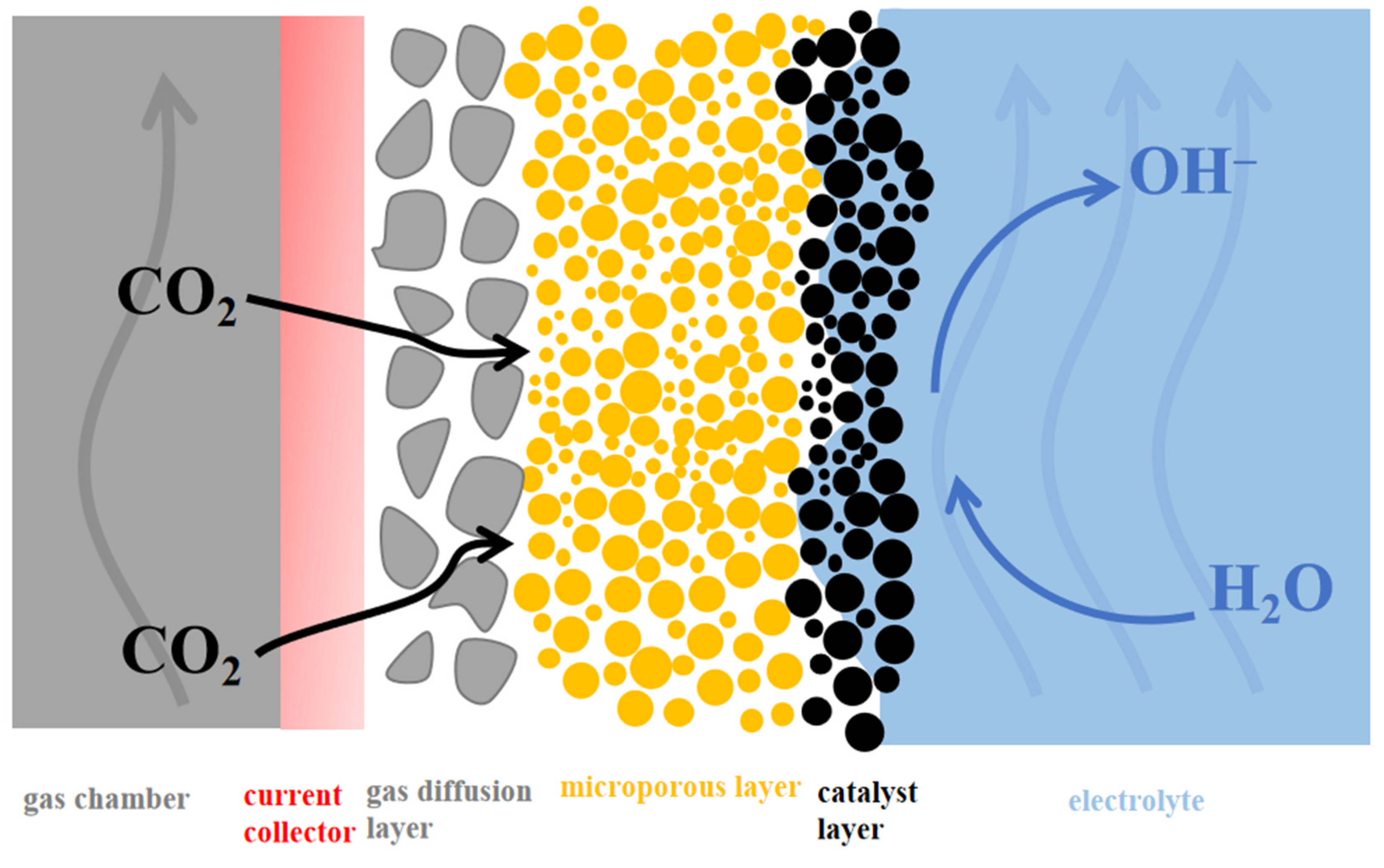
4.2. Gas Diffusion Electrodes (GDE)
4.2.1. Typical GDE
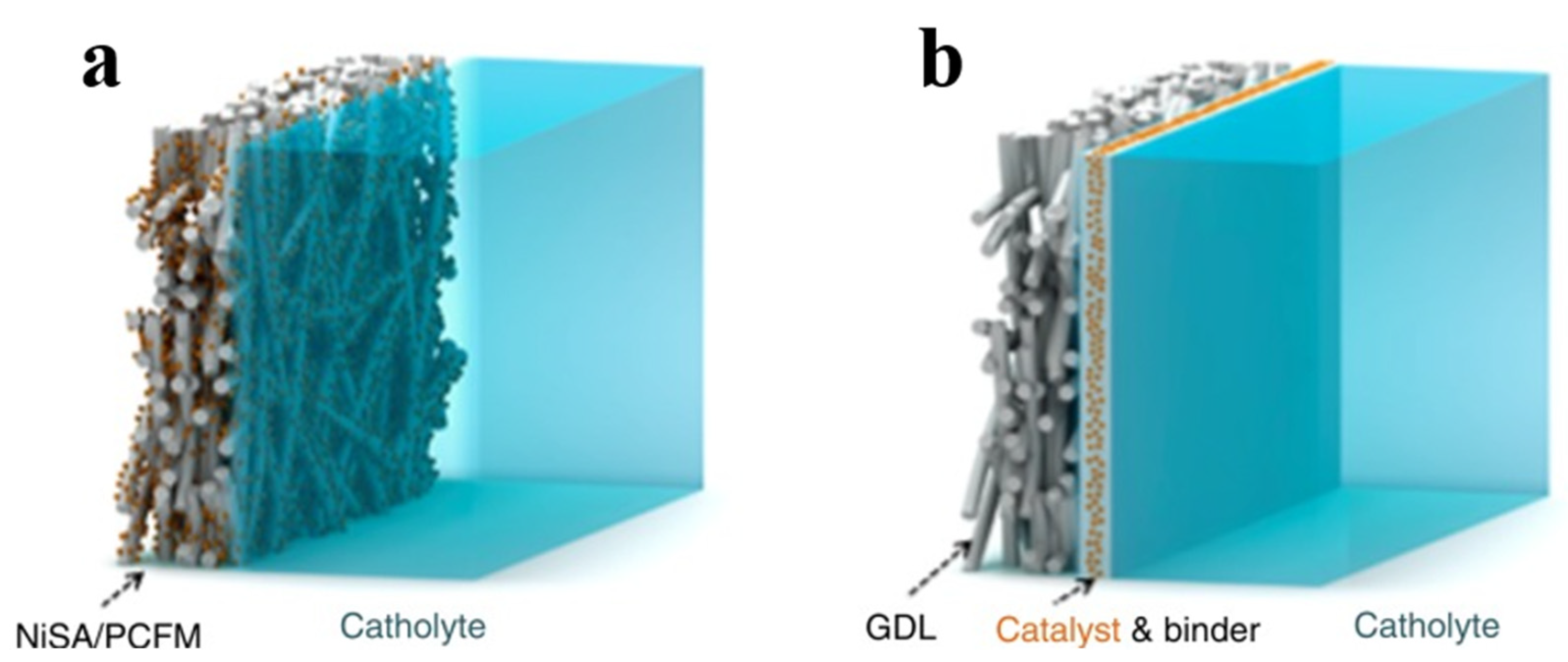
4.2.2. Integrated GDE
4.3. Hydrophobic Electrode Design
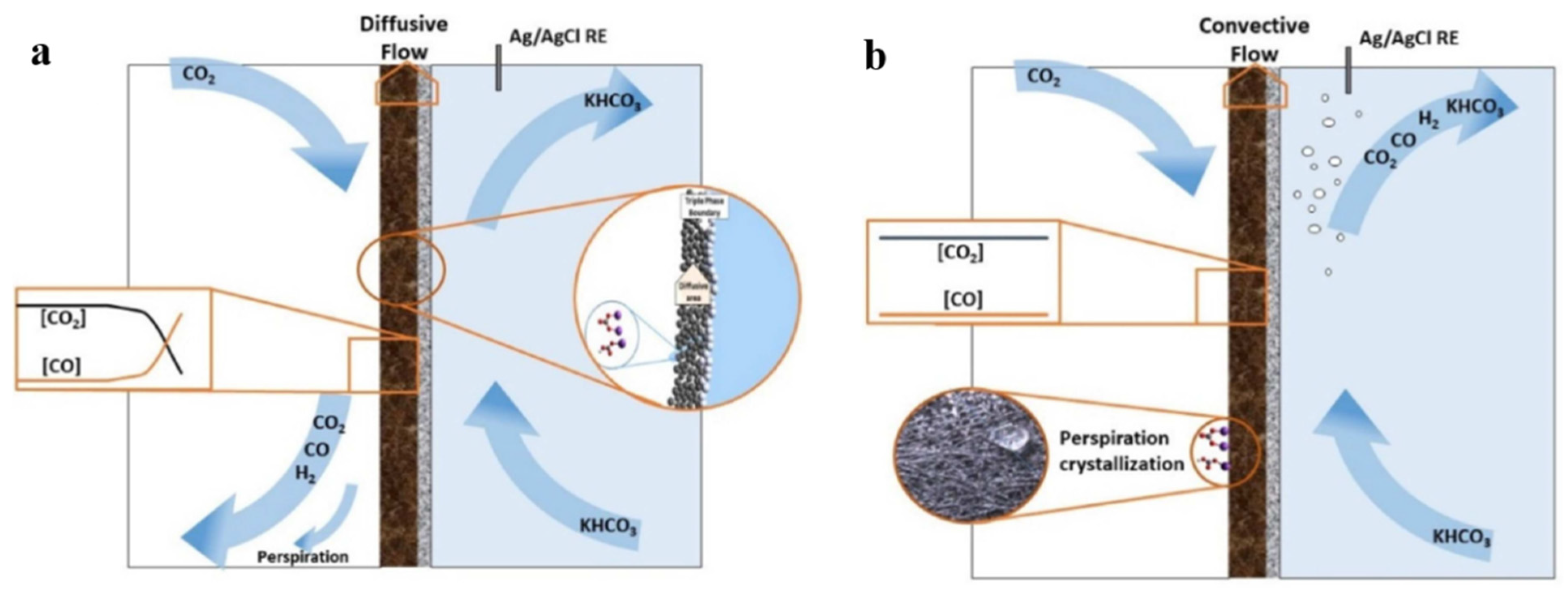
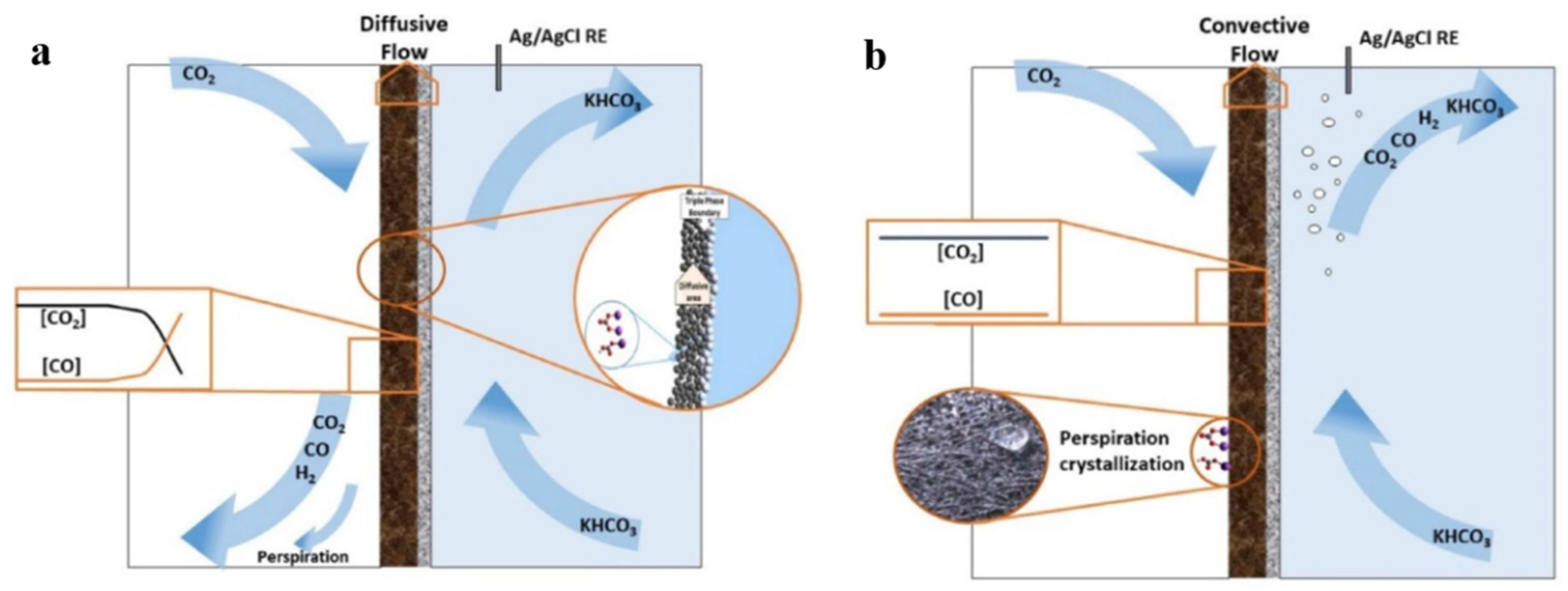
4.4. Flow Pattern
5. Electrolyte
5.1. Concentration
5.2. pH
5.3. Cation Effects
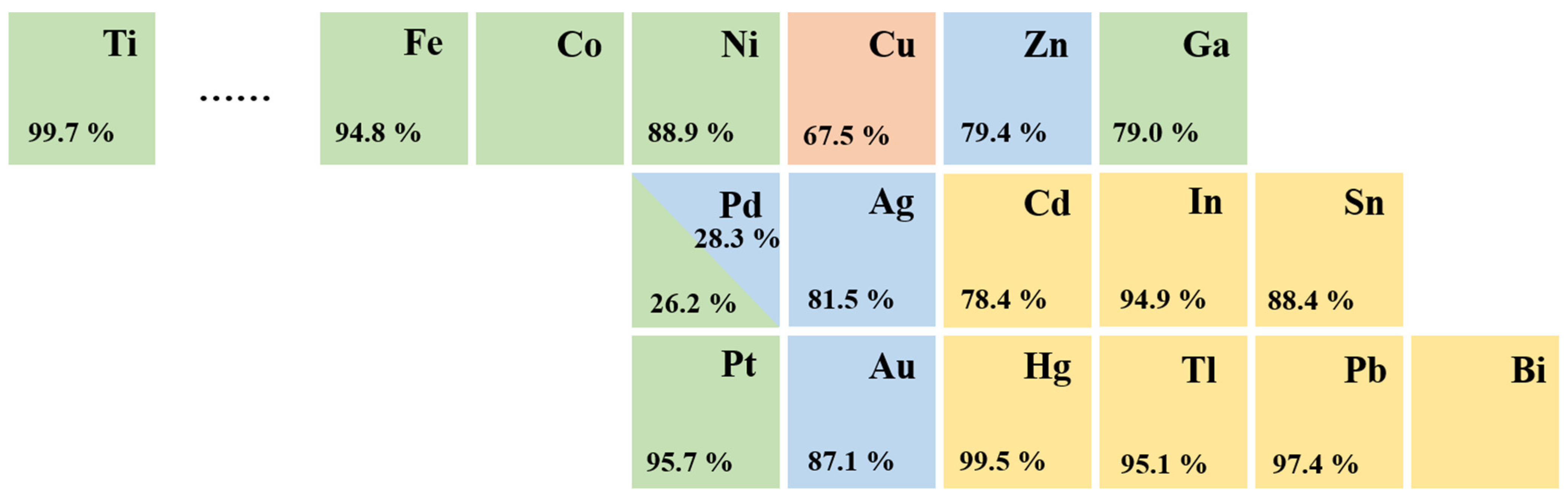
5.4. Anion Effects
6. Conclusions and Outlook
- (1)
- The design of cost-efficient catalysts. Novel and cheap catalysts should be developed to replace or reduce the use of noble metals. Tailoring the morphology, crystal structure, and electronic distributions are three important strategies to optimize the usage of the active sites. By introducing heteroatoms (e.g., N, P, or other chalcogens), other metals, or specific functional groups, the lattice defects of metal catalysts such as vacancies and grain boundaries can be regulated.
- (2)
- Innovations in electrolyzers. Progress is also needed in the design of cheaper electrolyzers with higher efficiency. The facility should also be flexible enough to adapt to different CO2 resources such as CO2 captured from flue gas and biogas. At the same time, the use of a GDE (e.g., carbon matrix, PTFE) for stable and large-scale CO2 conversion should be optimized. In the future, better GDEs with excellent conductivity, hydrophobicity, and appropriate ventilation will be an intriguing development direction.
- (3)
- Research into non-OER anode reactions. Although the anodic OER reaction is green, it does not yield economic benefits. Coupling CO2ER with an anode oxidation reaction with more commercial value could be another industrially accessible approach. In this manner, CO2 electrolyzers could be easily integrated into other industrial processes in which the main product is formed on the anode. The existing challenge is proper product separation.
- (4)
- The exploration of complicated mechanisms. The electroreduction of CO2, especially to C2+ products, involves various electron transfer processes and the formation of intermediates. Theoretical calculations can provide new insights into the structure–property relationship and the rational design of catalysts. Remarkable effort has been dedicated to obtaining a better mechanistic understanding through DFT calculations and operando/in situ techniques. However, computational models are simplified and limited at present and require further development.
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sánchez, O.G.; Birdja, Y.Y.; Bulut, M.; Vaes, J.; Breugelmans, T.; Pant, D. Recent Advances in Industrial CO2 Electroreduction. Curr. Opin. Green Sustain. Chem. 2019, 16, 47–56. [Google Scholar] [CrossRef]
- Verma, S.; Kim, B.; Jhong, H.; Ma, S.C.; Kenis, P.J.A. A Gross-Margin Model for Defining Technoeconomic Benchmarks in the Electroreduction of CO2. Chemsuschem 2016, 9, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Guo, J.; Li, X.; Patel, P.; Seifitokaldani, A. Electrochemical Reactors for CO2 Conversion. Catalysts 2020, 10, 473. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Y.J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and Selective Conversion of CO2 to CO on Ultrathin Au Nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, M.; Sharma, P.P.; Yadav, R.M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X.D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, Y.; Zhou, Z.; Cai, F.; Zhao, X.; Huang, W.; Li, Y.; Zhu, J.; Liu, P.; Yang, F.; et al. Enhancing CO2 Electroreduction with the Metal-Oxide Interface. J. Am. Chem. Soc. 2017, 139, 5652–5655. [Google Scholar] [CrossRef]
- Kortlever, R.; Peters, I.; Koper, S.; Koper, M.T.M. Electrochemical CO2 Reduction to Formic Acid at Low Overpotential and with High Faradaic Efficiency on Carbon-Supported Bimetallic Pd-Pt Nanoparticles. ACS Catal. 2015, 5, 3916–3923. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, T.; Liu, Y.Y.; Qiao, J.L. Enhancing CO2 electrolysis to formate on facilely synthesized Bi catalysts at low overpotential. Appl. Catal. B 2017, 218, 46–50. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Wan, Y.; Xie, Y.; Zhu, J.; Pan, H.; Zheng, X.; Xia, C. Perovskite Oxyfluoride Electrode Enabling Direct Electrolyzing Carbon Dioxide with Excellent Electrochemical Performances. Adv. Energy Mater. 2019, 9, 1803156. [Google Scholar] [CrossRef]
- Ren, D.; Deng, Y.L.; Handoko, A.D.; Chen, C.S.; Malkhandi, S.; Yeo, B.S. Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Ma, M.; Djanashvili, K.; Smith, W.A. Controllable Hydrocarbon Formation from the Electrochemical Reduction of CO2 over Cu Nanowire Arrays. Angew. Chem. Int. Ed. 2016, 55, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Jing, J.B.; Wu, Y.S.; Wu, Z.S.; Guo, X.T.; Materna, K.L.; Liu, W.; Batista, V.S.; Brudvig, G.W.; Wang, H.L. Electrochemical CO2 Reduction to Hydrocarbons on a Heterogeneous Molecular Cu Catalyst in Aqueous Solution. J. Am. Chem. Soc. 2016, 138, 8076–8079. [Google Scholar] [CrossRef] [PubMed]
- Birdja, Y.Y.; Vaes, J. Towards a Critical Evaluation of Electrocatalyst Stability for CO2 Electroreduction. ChemElectroChem 2020, 7, 4713–4717. [Google Scholar] [CrossRef]
- Hou, Y.T.; Wang, L.J.; Bian, L.Z.; Wang, Y.D.; Chou, K.C.; Kumar, R.V. High-performance La0.3Sr0.7Fe0.9Ti0.1O3-delta as fuel electrode for directly electrolyzing CO2 in solid oxide electrolysis cells. Electrochim. Acta 2020, 342. [Google Scholar] [CrossRef]
- Hu, X.L.; Xie, K. Active and stable Ni/Cr2O3-delta cathodes for high temperature CO2 electrolysis. J. Power Sources 2019, 430, 20–24. [Google Scholar] [CrossRef]
- Wu, M.X.; Zhou, X.L.; Xu, J.; Li, S.; Pan, L.; Zhang, N.Q. Electrochemical performance of La0.3Sr0.7Ti0.3Fe0.7O3-delta/CeO2 composite cathode for CO2 reduction in solid oxide electrolysis cells. J. Power Sources 2020, 451, 227334. [Google Scholar] [CrossRef]
- Handoko, A.D.; Wei, F.; Jenndy; Yeo, B.S.; Seh, Z.W. Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques. Nat. Catal. 2018, 1, 922–934. [Google Scholar]
- Kortlever, R.; Shen, J.; Schouten, K.J.; Calle-Vallejo, F.; Koper, M.T. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef]
- Peterson, A.A.; Norskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Montoya, J.H.; Shi, C.; Chan, K.; Norskov, J.K. Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction. J. Phys. Chem. Lett. 2015, 6, 2032–2037. [Google Scholar] [CrossRef]
- Schouten, K.J.P.; Qin, Z.S.; Gallent, E.P.; Koper, M.T.M. Two Pathways for the Formation of Ethylene in CO Reduction on Single-Crystal Copper Electrodes. J. Am. Chem. Soc. 2012, 134, 9864–9867. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Norskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Schouten, K.J.P.; Kwon, Y.; van der Ham, C.J.M.; Qin, Z.; Koper, M.T.M. A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem. Sci. 2011, 2, 1902–1909. [Google Scholar] [CrossRef]
- Kim, C.; Jeon, H.S.; Eom, T.; Jee, M.S.; Kim, H.; Friend, C.M.; Min, B.K.; Hwang, Y.J. Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Eom, T.; Jee, M.S.; Jung, H.; Kim, H.; Min, B.K.; Hwang, Y.J. Insight into Electrochemical CO2 Reduction on Surface-Molecule-Mediated Ag Nanoparticles. ACS Catal. 2016, 7, 779–785. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Liu, Y.; MacFarlane, D.R.; Wallace, G.G. Engineering Surface Amine Modifiers of Ultrasmall Gold Nanoparticles Supported on Reduced Graphene Oxide for Improved Electrochemical CO2 Reduction. Adv. Energy Mater. 2018, 8, 1801400. [Google Scholar] [CrossRef] [Green Version]
- Nam, D.H.; De Luna, P.; Rosas-Hernandez, A.; Thevenon, A.; Li, F.; Agapie, T.; Peters, J.C.; Shekhah, O.; Eddaoudi, M.; Sargent, E.H. Molecular enhancement of heterogeneous CO2 reduction. Nat. Mater. 2020, 19, 266–276. [Google Scholar] [CrossRef]
- Han, Z.; Kortlever, R.; Chen, H.Y.; Peters, J.C.; Agapie, T. CO2 Reduction Selective for C≥2 Products on Polycrystalline Copper with N-Substituted Pyridinium Additives. ACS Cent. Sci. 2017, 3, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.; Vayenas, C.G. Modern Aspects of Electrochemistry; Vayenas, C.G., White, R.E., Gamboa-Aldeco, M.E., Eds.; Springer: New York, NY, USA, 2008; Volume 42, pp. 89–190. [Google Scholar]
- Wu, M.; Xu, B.; Zhang, Y.; Qi, S.; Ni, W.; Hu, J.; Ma, J. Perspectives in emerging bismuth electrochemistry. Chem. Eng. J. 2020, 381, 122558. [Google Scholar] [CrossRef]
- Huang-Fu, Z.C.; Song, Q.T.; He, Y.H.; Wang, J.J.; Ye, J.Y.; Zhou, Z.Y.; Sun, S.G.; Wang, Z.H. Electrochemical CO2 reduction on Cu and Au electrodes studied using in situ sum frequency generation spectroscopy. Phys. Chem. Chem. Phys. 2019, 21, 25047–25053. [Google Scholar] [CrossRef]
- Roy, S.; Sharma, B.; Peaut, J.; Simon, P.; Fontecave, M.; Tran, P.D.; Derat, E.; Artero, V. Molecular Cobalt Complexes with Pendant Amines for Selective Electrocatalytic Reduction of Carbon Dioxide to Formic Acid. J. Am. Chem. Soc. 2017, 139, 3685–3696. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Murata, A.; Kikuchi, K.; Suzuki, S. Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide at a Gold Electrode in Aqueous Potassium Hydrogen Carbonate. J. Chem. Soc. Chem. Commun. 1987, 10, 728–729. [Google Scholar] [CrossRef]
- Zhu, W.; Michalsky, R.; Metin, Ö.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pang, Y.; Zhang, B.; De Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C.T.; Fan, F.; Cao, C.; et al. Enhanced Electrocatalytic CO2 Reduction via Field-induced Reagent Concentration. Nature 2016, 537, 382–386. [Google Scholar] [CrossRef]
- Jhong, H.M.; Tornow, C.E.; Kim, C.; Verma, S.; Oberst, J.L.; Anderson, P.S.; Gewirth, A.A.; Fujigaya, T.; Nakashima, N.; Kenis, P.J.A. Gold Nanoparticles on Polymer-Wrapped Carbon Nanotubes: An Efficient and Selective Catalyst for the Electroreduction of CO2. Chemphyschem 2017, 18, 3274–3279. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Hamasaki, Y.; Kim, C.; Huang, W.; Lu, S.; Jhong, H.-R.M.; Gewirth, A.A.; Fujigaya, T.; Nakashima, N.; Kenis, P.J.A. Insights into the Low Overpotential Electroreduction of CO2 to CO on a Supported Gold Catalyst in an Alkaline Flow Electrolyzer. ACS Energy Lett. 2017, 3, 193–198. [Google Scholar] [CrossRef]
- Ma, L.; Hu, W.; Pan, Q.; Zou, L.; Zou, Z.; Wen, K.; Yang, H. Polyvinyl Alcohol-modified Gold Nanoparticles with Record-high Activity for Electrochemical Reduction of CO2 to CO. J. CO2 Util. 2019, 34, 108–114. [Google Scholar] [CrossRef]
- Salehi-Khojin, A.; Jhong, H.-R.M.; Rosen, B.A.; Zhu, W.; Ma, S.; Kenis, P.J.A.; Masel, R.I. Nanoparticle Silver Catalysts That Show Enhanced Activity for Carbon Dioxide Electrolysis. J. Phys. Chem. C 2013, 117, 1627–1632. [Google Scholar] [CrossRef]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional size-dependent activity enhancement in the electroreduction of CO2 over Au nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Liu, S.; Tao, H.; Zeng, L.; Liu, Q.; Xu, Z.; Liu, Q.; Luo, J.-L. Shape-Dependent Electrocatalytic Reduction of CO2 to CO on Triangular Silver Nanoplates. J. Am. Chem. Soc. 2017, 139, 2160–2163. [Google Scholar] [CrossRef]
- Ma, S.; Lan, Y.; Perez, G.M.; Moniri, S.; Kenis, P.J. Silver Supported on Titania as an Active Catalyst for Electrochemical Carbon Dioxide Reduction. ChemSusChem 2014, 7, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Haspel, H.; Pustovarenko, A.; Dikhtiarenko, A.; Russkikh, A.; Shterk, G.; Osadchii, D.; Ould-Chikh, S.; Ma, M.; Smith, W.A.; et al. Maximizing Ag Utilization in High-Rate CO2 Electrochemical Reduction with a Coordination Polymer-Mediated Gas Diffusion Electrode. ACS Energy Lett. 2019, 4, 2024–2031. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Lee, H.; Mao, J.; Grimes, C.A.; Liu, C.; Zhang, M.; Lu, Z.; Chen, Y.; Feng, S.P. Efficient electroreduction of CO2 to CO by Ag-decorated S-doped g-C3N4/CNT nanocomposites at industrial scale current density. Mater. Today Phys. 2020, 12, 100176. [Google Scholar] [CrossRef]
- Ma, S.; Luo, R.; Gold, J.I.; Yu, A.Z.; Kim, B.; Kenis, P.J.A. Carbon nanotube containing Ag catalyst layers for efficient and selective reduction of carbon dioxide. J. Mater. Chem. A 2016, 4, 8573–8578. [Google Scholar] [CrossRef]
- Gao, D.; Zhou, H.; Cai, F.; Wang, J.; Wang, G.; Bao, X. Pd-Containing Nanostructures for Electrochemical CO2 Reduction Reaction. ACS Catal. 2018, 8, 1510–1519. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, X.G.; Jiang, K.; Wu, D.Y.; Cai, W.B. Boosting Formate Production in Electrocatalytic CO2 Reduction over Wide Potential Window on Pd Surfaces. J. Am. Chem. Soc. 2018, 140, 2880–2889. [Google Scholar] [CrossRef]
- Zhu, W.; Kattel, S.; Jiao, F.; Chen, J.G. Shape-Controlled CO2 Electrochemical Reduction on Nanosized Pd Hydride Cubes and Octahedra. Adv. Energy Mater. 2019, 9, 1802840. [Google Scholar] [CrossRef]
- Xia, R.; Zhang, S.; Ma, X.; Jiao, F. Surface-functionalized Palladium Catalysts for Electrochemical CO2 Reduction. J. Mater. Chem. A 2020, 8, 15884–15890. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, D.; Liu, H.; Sun, X.; Chen, C.; Bi, J.; Liu, J.; Wu, H.; Han, B. Hollow Metal-Organic-Framework-Mediated In Situ Architecture of Copper Dendrites for Enhanced CO2 Electroreduction. Angew Chem. Int. Ed. Engl. 2020, 59, 8896–8901. [Google Scholar] [CrossRef]
- Yang, F.; Deng, P.; Wang, Q.; Zhu, J.; Yan, Y.; Zhou, L.; Qi, K.; Liu, H.; Park, H.S.; Xia, B.Y. Metal-organic framework-derived cupric oxide polycrystalline nanowires for selective carbon dioxide electroreduction to C2 valuables. J. Mater. Chem. A 2020, 8, 12418–12423. [Google Scholar] [CrossRef]
- Yao, K.; Xia, Y.; Li, J.; Wang, N.; Han, J.; Gao, C.; Han, M.; Shen, G.; Liu, Y.; Seifitokaldani, A.; et al. Metal-organic Framework Derived Copper Catalysts for CO2 to Ethylene Conversion. J. Mater. Chem. A 2020, 8, 11117–11123. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, H.; Livi, K.J.T.; Raciti, D.; Zong, H.; Gregg, J.; Onadeko, M.; Wan, Y.; Watson, A.; Wang, C. Copper Nanocubes for CO2 Reduction in Gas Diffusion Electrodes. Nano Lett. 2019, 19, 8461–8468. [Google Scholar] [CrossRef] [PubMed]
- Baruch, M.F.; Pander, J.E.; White, J.L.; Bocarsly, A.B. Mechanistic Insights into the Reduction of CO2 on Tin Electrodes using in Situ ATR-IR Spectroscopy. ACS Catal. 2015, 5, 3148–3156. [Google Scholar] [CrossRef]
- Ju, W.; Jiang, F.; Ma, H.; Pan, Z.; Zhao, Y.-B.; Pagani, F.; Rentsch, D.; Wang, J.; Battaglia, C. Electrocatalytic reduction of gaseous CO2 to CO on SnCu-nanofiber-based gas diffusion electrodes. Adv. Energy Mater. 2019, 9, 1901514. [Google Scholar] [CrossRef]
- Xiang, H.; Rasul, S.; Hou, B.; Portoles, J.; Cumpson, P.; Yu, E.H. Copper-Indium Binary Catalyst on a Gas Diffusion Electrode for High-Performance CO2 Electrochemical Reduction with Record CO Production Efficiency. ACS Appl. Mater. Interfaces 2020, 12, 601–608. [Google Scholar] [CrossRef]
- Luc, W.; Collins, C.; Wang, S.; Xin, H.; He, K.; Kang, Y.; Jiao, F. Ag-Sn Bimetallic Catalyst with a Core-Shell Structure for CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Zhou, Z.; Shao, J.; Lin, L.; Gao, D.; Ta, N.; Si, R.; Wang, G.; Bao, X. In Situ Reconstruction of a Hierarchical Sn-Cu/SnOx Core/Shell Catalyst for High-Performance CO2 Electroreduction. Angew Chem. Int. Ed. Engl. 2020, 59, 4814–4821. [Google Scholar] [CrossRef]
- Li, Y.C.; Wang, Z.; Yuan, T.; Nam, D.H.; Luo, M.; Wicks, J.; Chen, B.; Li, J.; Li, F.; de Arquer, F.P.G.; et al. Binding Site Diversity Promotes CO2 Electroreduction to Ethanol. J. Am. Chem. Soc. 2019, 141, 8584–8591. [Google Scholar] [CrossRef]
- Ma, W.; Xie, S.; Liu, T.; Fan, Q.; Ye, J.; Sun, F.; Jiang, Z.; Zhang, Q.; Cheng, J.; Wang, Y. Electrocatalytic Reduction of CO2 to Ethylene and Ethanol through Hydrogen-assisted C–C Coupling over Fluorine-modified Copper. Nat. Catal. 2020, 3, 478–487. [Google Scholar] [CrossRef]
- Lee, J.-C.; Kim, J.-Y.; Joo, W.-H.; Hong, D.; Oh, S.-H.; Kim, B.; Lee, G.-D.; Kim, M.; Oh, J.; Joo, Y.-C. Thermodynamically Driven Self-formation of Copper-embedded Nitrogen-doped Carbon Nanofiber Catalysts for a Cascade Electroreduction of Carbon Dioxide to Ethylene. J. Mater. Chem. A 2020, 8, 11632–11641. [Google Scholar] [CrossRef]
- Chen, C.; Yan, X.; Liu, S.; Wu, Y.; Wan, Q.; Sun, X.; Zhu, Q.; Liu, H.; Ma, J.; Zheng, L.; et al. Highly Efficient Electroreduction of CO2 to C2+ Alcohols on Heterogeneous Dual Active Sites. Angew Chem. Int. Ed. Engl. 2020, 59, 16459–16464. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Shen, H.; Guan, A.; Liu, J.; Li, T.; Ji, Y.; Al-Enizi, A.M.; Zhang, L.; Qian, L.; Zheng, G. Fast Cooling Induced Grain-boundary-rich Copper Oxide for Electrocatalytic Carbon Dioxide Reduction to Ethanol. J. Colloid Interface Sci. 2020, 570, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Takahashi, I.; Koga, O.; Hoshi, N. Electrochemical Reduction of Carbon Dioxide at Various Series of Copper Single Crystal Electrodes. J. Mol. Catal. A Chem. 2003, 199, 39–47. [Google Scholar] [CrossRef]
- Schouten, K.J.P.; Pérez Gallent, E.; Koper, M.T.M. Structure Sensitivity of the Electrochemical Reduction of Carbon Monoxide on Copper Single Crystals. ACS Catal. 2013, 3, 1292–1295. [Google Scholar] [CrossRef]
- Perez-Gallent, E.; Figueiredo, M.C.; Calle-Vallejo, F.; Koper, M.T. Spectroscopic Observation of a Hydrogenated CO Dimer Intermediate During CO Reduction on Cu(100) Electrodes. Angew Chem. Int. Ed. Engl. 2017, 56, 3621–3624. [Google Scholar] [CrossRef]
- Roberts, F.S.; Kuhl, K.P.; Nilsson, A. High Selectivity for Ethylene from Carbon Dioxide Reduction over Copper Nanocube Electrocatalysts. Angew Chem. Int. Ed. Engl. 2015, 54, 5179–5182. [Google Scholar] [CrossRef]
- Jiang, K.; Sandberg, R.B.; Akey, A.J.; Liu, X.; Bell, D.C.; Nørskov, J.K.; Chan, K.; Wang, H. Metal Ion Cycling of Cu Foil for Selective C–C Coupling in Electrochemical CO2 Reduction. Nat. Catal. 2018, 1, 111–119. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Dinh, C.-T.; Li, J.; Ozden, A.; Golam Kibria, M.; Seifitokaldani, A.; Tan, C.-S.; Gabardo, C.M.; Luo, M.; et al. Catalyst synthesis under CO2 electroreduction favours faceting and promotes renewable fuels electrosynthesis. Nat. Catal. 2019, 3, 98–106. [Google Scholar] [CrossRef]
- Zhang, T.; Bui, J.C.; Li, Z.; Bell, A.T.; Weber, A.Z.; Wu, J. Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes. Nat. Catal. 2022, 5, 202–211. [Google Scholar] [CrossRef]
- Luo, W.; Zhang, J.; Li, M.; Züttel, A. Boosting CO Production in Electrocatalytic CO2 Reduction on Highly Porous Zn Catalysts. ACS Catal. 2019, 9, 3783–3791. [Google Scholar] [CrossRef] [Green Version]
- Lamaison, S.; Wakerley, D.; Montero, D.; Rousse, G.; Taverna, D.; Giaume, D.; Mercier, D.; Blanchard, J.; Tran, H.N.; Fontecave, M.; et al. Zn-Cu Alloy Nanofoams as Efficient Catalysts for the Reduction of CO2 to Syngas Mixtures with a Potential-Independent H2/CO Ratio. ChemSusChem 2019, 12, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Lamaison, S.; Wakerley, D.; Blanchard, J.; Montero, D.; Rousse, G.; Mercier, D.; Marcus, P.; Taverna, D.; Giaume, D.; Mougel, V.; et al. High-Current-Density CO2-to-CO Electroreduction on Ag-Alloyed Zn Dendrites at Elevated Pressure. Joule 2020, 4, 395–406. [Google Scholar] [CrossRef]
- Saberi Safaei, T.; Mepham, A.; Zheng, X.; Pang, Y.; Dinh, C.T.; Liu, M.; Sinton, D.; Kelley, S.O.; Sargent, E.H. High-Density Nanosharp Microstructures Enable Efficient CO2 Electroreduction. Nano Lett. 2016, 16, 7224–7228. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.Y.; Hu, S.J.; Zhang, X.L.; Zheng, Y.R.; Wang, H.J.; Niu, Z.Z.; Yang, P.P.; Bao, R.C.; Ma, T.; Dang, Z.; et al. High-Curvature Transition-Metal Chalcogenide Nanostructures with a Pronounced Proximity Effect Enable Fast and Selective CO2 Electroreduction. Angew Chem. Int. Ed. Engl. 2020, 59, 8706–8712. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, J.; Cheng, F.; Chen, J. Mn-doped atomic SnO2 layers for highly efficient CO2 electrochemical reduction. J. Mater. Chem. A 2019, 7, 19651–19656. [Google Scholar] [CrossRef]
- Löwe, A.; Rieg, C.; Hierlemann, T.; Salas, N.; Kopljar, D.; Wagner, N.; Klemm, E. Influence of Temperature on the Performance of Gas Diffusion Electrodes in the CO2 Reduction Reaction. ChemElectroChem 2019, 6, 4497–4506. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Miller, H.A.; Bellini, M.; Christensen, H.; Scott, K.; Rasul, S.; Yu, E.H. Production of Formate by CO2 Electrochemical Reduction and Its Application in Energy Storage. Sustain. Energy Fuels 2020, 4, 277–284. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, M.; Liu, J.; Zhou, J.; Cen, K. A Cu foam cathode used as a Pt–RGO catalyst matrix to improve CO2 reduction in a photoelectrocatalytic cell with a TiO2 photoanode. J. Mater. Chem. A 2015, 3, 12947–12957. [Google Scholar] [CrossRef]
- Wang, J.; Zou, J.; Hu, X.; Ning, S.; Wang, X.; Kang, X.; Chen, S. Heterostructured intermetallic CuSn catalysts high performance towards the electrochemical reduction of CO2 to formate. J. Mater. Chem. A 2019, 7, 27514–27521. [Google Scholar] [CrossRef]
- Díaz-Sainz, G.; Alvarez-Guerra, M.; Solla-Gullón, J.; García-Cruz, L.; Montiel, V.; Irabien, A. CO2 Electroreduction to Formate: Continuous Single-pass Operation in a Filter-press Reactor at High Current Densities Using Bi Gas Diffusion Electrodes. J. CO2 Util. 2019, 34, 12–19. [Google Scholar] [CrossRef]
- Deng, P.; Yang, F.; Wang, Z.; Chen, S.; Zhou, Y.; Zaman, S.; Xia, B.Y. Metal-Organic Framework-Derived Carbon Nanorods Encapsulating Bismuth Oxides for Rapid and Selective CO2 Electroreduction to Formate. Angew Chem. Int. Ed. Engl. 2020, 59, 10807–10813. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Zhu, P.; Jiang, Q.; Pan, Y.; Liang, W.; Stavitski, E.; Alshareef, H.N.; Wang, H. Continuous Production of Pure Liquid Fuel Solutions via Electrocatalytic CO2 Reduction Using Solid-electrolyte Devices. Nat. Energy 2019, 4, 776–785. [Google Scholar] [CrossRef]
- Yang, J.; Wang, X.; Qu, Y.; Wang, X.; Huo, H.; Fan, Q.; Wang, J.; Yang, L.M.; Wu, Y. Bi-Based Metal-Organic Framework Derived Leafy Bismuth Nanosheets for Carbon Dioxide Electroreduction. Adv. Energy Mater. 2020, 2001709. [Google Scholar] [CrossRef]
- Cao, C.; Ma, D.D.; Gu, J.F.; Xie, X.; Zeng, G.; Li, X.; Han, S.G.; Zhu, Q.L.; Wu, X.T.; Xu, Q. Metal-Organic Layers Leading to Atomically Thin Bismuthene for Efficient Carbon Dioxide Electroreduction to Liquid Fuel. Angew Chem. Int. Ed. Engl. 2020, 59, 15014–15020. [Google Scholar] [CrossRef]
- Han, P.; Yu, X.; Yuan, D.; Kuang, M.; Wang, Y.; Al-Enizi, A.M.; Zheng, G. Defective Graphene for Electrocatalytic CO2 Reduction. J. Colloid Interface Sci. 2019, 534, 332–337. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J.; Zhang, F.; Zheng, L.; Mo, G.; Han, B.; Yang, G. Selenium-Doped Hierarchically Porous Carbon Nanosheets as an Effcient Metal-Free Electrocatalyst for CO2 Reduction. Adv. Funct. Mater. 2020, 30, 1906194. [Google Scholar] [CrossRef]
- Yang, H.; Wu, Y.; Lin, Q.; Fan, L.; Chai, X.; Zhang, Q.; Liu, J.; He, C.; Lin, Z. Composition Tailoring via N and S Co-doping and Structure Tuning by Constructing Hierarchical Pores: Metal-Free Catalysts for High-Performance Electrochemical Reduction of CO2. Angew Chem. Int. Ed. Engl. 2018, 57, 15476–15480. [Google Scholar] [CrossRef]
- Chen, C.; Sun, X.; Yan, X.; Wu, Y.; Liu, H.; Zhu, Q.; Bediako, B.B.A.; Han, B. Boosting CO2 Electroreduction on N, P-Co-doped Carbon Aerogels. Angew Chem. Int. Ed. Engl. 2020, 59, 11123–11129. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhao, S.; Johannessen, B.; Veder, J.P.; Saunders, M.; Rowles, M.R.; Cheng, M.; Liu, C.; Chisholm, M.F.; De Marco, R.; et al. Atomically Dispersed Transition Metals on Carbon Nanotubes with Ultrahigh Loading for Selective Electrochemical Carbon Dioxide Reduction. Adv. Mater. 2018, 30, 1706287. [Google Scholar] [CrossRef] [PubMed]
- Jhong, H.-R.M.; Brushett, F.R.; Kenis, P.J.A. The Effects of Catalyst Layer Deposition Methodology on Electrode Performance. Adv. Energy Mater. 2013, 3, 589–599. [Google Scholar] [CrossRef]
- Yang, H.; Lin, Q.; Zhang, C.; Yu, X.; Cheng, Z.; Li, G.; Hu, Q.; Ren, X.; Zhang, Q.; Liu, J.; et al. Carbon Dioxide Electroreduction on Single-atom Nickel Decorated Carbon Membranes with Industry Compatible Current Densities. Nat. Commun. 2020, 11, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lin, Q.; Wu, Y.; Li, G.; Hu, Q.; Chai, X.; Ren, X.; Zhang, Q.; Liu, J.; He, C. Highly efficient utilization of single atoms via constructing 3D and free-standing electrodes for CO2 reduction with ultrahigh current density. Nano Energy 2020, 70, 104454. [Google Scholar] [CrossRef]
- Yang, H.B.; Hung, S.-F.; Liu, S.; Yuan, K.; Miao, S.; Zhang, L.; Huang, X.; Wang, H.-Y.; Cai, W.; Chen, R.; et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 2018, 3, 140–147. [Google Scholar] [CrossRef]
- Chen, Y.; Zou, L.; Liu, H.; Chen, C.; Wang, Q.; Gu, M.; Yang, B.; Zou, Z.; Fang, J.; Yang, H. Fe and N Co-Doped Porous Carbon Nanospheres with High Density of Active Sites for Efficient CO2 Electroreduction. J. Phys. Chem. C 2019, 123, 16651–16659. [Google Scholar] [CrossRef]
- Wu, S.; Lv, X.; Ping, D.; Zhang, G.; Wang, S.; Wang, H.; Yang, X.; Guo, D.; Fang, S. Highly exposed atomic Fe–N active sites within carbon nanorods towards electrocatalytic reduction of CO2 to CO. Electrochim. Acta 2020, 340, 135930. [Google Scholar] [CrossRef]
- Yan, C.; Ye, Y.; Lin, L.; Wu, H.; Jiang, Q.; Wang, G.; Bao, X. Improving CO2 Electroreduction over ZIF-derived Carbon Doped with Fe-N Sites by an Additional Ammonia Treatment. Catal. Today 2019, 330, 252–258. [Google Scholar] [CrossRef]
- Wang, X.; Pan, Y.; Ning, H.; Wang, H.; Guo, D.; Wang, W.; Yang, Z.; Zhao, Q.; Zhang, B.; Zheng, L.; et al. Hierarchically micro- and meso-porous Fe-N4O-doped carbon as robust electrocatalyst for CO2 reduction. Appl. Catal. B 2020, 266, 118630. [Google Scholar] [CrossRef]
- Gu, J.; Hsu, C.-S.; Bai, L.; Chen, H.M.; Hu, X. Atomically dispersed Fe3+ sitescatalyze efficient CO2 electroreduction to CO. Science 2019, 364, 1091–1094. [Google Scholar] [CrossRef]
- Hou, Y.; Liang, Y.-L.; Shi, P.-C.; Huang, Y.-B.; Cao, R. Atomically dispersed Ni species on N-doped carbon nanotubes for electroreduction of CO2 with nearly 100% CO selectivity. Appl. Catal. B 2020, 271, 118929. [Google Scholar] [CrossRef]
- Möller, T.; Ju, W.; Bagger, A.; Wang, X.; Luo, F.; Ngo Thanh, T.; Varela, A.S.; Rossmeisl, J.; Strasser, P. Efficient CO2 to CO Electrolysis on Solid Ni–N–C Catalysts at Industrial Current Densities. Energy Environ. Sci. 2019, 12, 640–647. [Google Scholar] [CrossRef]
- Zheng, T.; Jiang, K.; Ta, N.; Hu, Y.; Zeng, J.; Liu, J.; Wang, H. Large-Scale and Highly Selective CO2 Electrocatalytic Reduction on Nickel Single-Atom Catalyst. Joule 2019, 3, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.-Y.; Balamurugan, M.; Choutipalli, V.S.K.; Jeong, E.-s.; Subramanian, V.; Sim, U.; Nam, K.T. Achieving Highly Efficient CO2 to CO Electroreduction Exceeding 300 mA cm−2 with Single-atom Nickel Electrocatalysts. J. Mater. Chem. A 2019, 7, 10651–10661. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, Z.; Zhang, X.; Niu, Z.; Zhou, Q.; Wang, X.; Li, H.; Lin, Z.; Zheng, H.; Liang, Y. Metal Phthalocyanine-Derived Single-Atom Catalysts for Selective CO2 Electroreduction under High Current Densities. ACS Appl. Mater. Interfaces 2020, 12, 33795–33802. [Google Scholar] [CrossRef]
- Guo, J.-H.; Zhang, X.-Y.; Dao, X.-Y.; Sun, W.-Y. Nanoporous Metal–Organic Framework-Based Ellipsoidal Nanoparticles for the Catalytic Electroreduction of CO2. ACS Appl. Nano Mater. 2020, 3, 2625–2635. [Google Scholar] [CrossRef]
- Zhang, T.; Lin, L.; Li, Z.; He, X.; Xiao, S.; Shanov, V.N.; Wu, J. Nickel Nitrogen Carbon Molecular Catalysts for High Rate CO2 Electro-reduction to CO On the Role of Carbon Substrate and Reaction Chemistry. ACS Appl. Nano Mater. 2020, 3, 1617–1626. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Gu, M.; Wang, M.; Zhang, Z.; Pan, W.; Jiang, Z.; Zheng, H.; Lucero, M.; Wang, H.; et al. Molecular engineering of dispersed nickel phthalocyanines on carbon nanotubes for selective CO2 reduction. Nat. Energy 2020, 5, 684–692. [Google Scholar] [CrossRef]
- Wen, C.F.; Mao, F.; Liu, Y.; Zhang, X.Y.; Fu, H.Q.; Zheng, L.R.; Liu, P.F.; Yang, H.G. Nitrogen-stabilized low-valent Ni motifs for efficient CO2 electrocatalysis. ACS Catal. 2020, 10, 1086–1093. [Google Scholar] [CrossRef]
- Hou, P.; Song, W.; Wang, X.; Hu, Z.; Kang, P. Well-Defined Single-Atom Cobalt Catalyst for Electrocatalytic Flue Gas CO2 Reduction. Small 2020, 16, 2001896. [Google Scholar] [CrossRef]
- Fujinuma, N.; Ikoma, A.; Lofland, S.E. Highly Efficient Electrochemical CO2 Reduction Reaction to CO with One-Pot Synthesized Co-Pyridine-Derived Catalyst Incorporated in a Nafion-Based Membrane Electrode Assembly. Adv. Energy Mater. 2020, 10, 2001645. [Google Scholar] [CrossRef]
- Roy, A.; Hursán, D.; Artyushkova, K.; Atanassov, P.; Janáky, C.; Serov, A. Nanostructured Metal-N-C Electrocatalysts for CO2 Reduction and Hydrogen Evolution Reactions. Appl. Catal. B 2018, 232, 512–520. [Google Scholar] [CrossRef]
- Yang, H.; Wu, Y.; Li, G.; Lin, Q.; Hu, Q.; Zhang, Q.; Liu, J.; He, C. Scalable Production of Efficient Single-Atom Copper Decorated Carbon Membranes for CO2 Electroreduction to Methanol. J. Am. Chem. Soc. 2019, 141, 12717–12723. [Google Scholar] [CrossRef]
- Ren, S.; Joulié, D.; Salvatore, D.; Torbensen, K.; Wang, M.; Robert, M.; Berlinguette, C.P. Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science 2019, 365, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Torbensen, K.; Salvatore, D.; Ren, S.; Joulie, D.; Dumoulin, F.; Mendoza, D.; Lassalle-Kaiser, B.; Isci, U.; Berlinguette, C.P.; et al. CO2 Electrochemical Catalytic Reduction with a Highly Active Cobalt Phthalocyanine. Nat. Commun. 2019, 10, 3602. [Google Scholar] [CrossRef] [PubMed]
- Torbensen, K.; Han, C.; Boudy, B.; von Wolff, N.; Bertail, C.; Braun, W.; Robert, M. Iron Porphyrin Allows Fast and Selective Electrocatalytic Conversion of CO2 to CO in a Flow Cell. Chemistry 2020, 26, 3034–3038. [Google Scholar] [CrossRef]
- Arquer, F.P.G.d.; Dinh, C.-T.; Ozden, A.; Wicks, J.; McCallum, C.; Kirmani, A.R.; Nam, D.-H.; Gabardo, C.; Seifitokaldani, A.; Wang, X.; et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 2020, 367, 661–666. [Google Scholar] [CrossRef]
- Bhargava, S.S.; Proietto, F.; Azmoodeh, D.; Cofell, E.R.; Henckel, D.A.; Verma, S.; Brooks, C.J.; Gewirth, A.A.; Kenis, P.J.A. System Design Rules for Intensifying the Electrochemical Reduction of CO2 to CO on Ag Nanoparticles. ChemElectroChem 2020, 7, 2001–2011. [Google Scholar] [CrossRef]
- Chen, L.; Xu, J.; Wang, X.; Xie, K. Sr2Fe1.5+xMo0.5O6-δ Cathode with Exsolved Fe Nanoparticles for Enhanced CO2 Electrolysis. Int. J. Hydrogen Energ. 2020, 45, 11901–11907. [Google Scholar] [CrossRef]
- De Luna, P.; Quintero-Bermudez, R.; Dinh, C.-T.; Ross, M.B.; Bushuyev, O.S.; Todorović, P.; Regier, T.; Kelley, S.O.; Yang, P.; Sargent, E.H. Catalyst Electro-redeposition Controls Morphology and Oxidation State for Selective Carbon Dioxide Reduction. Nat. Catal. 2018, 1, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; Arquer, F.P.G.d.; Kiani, A.; Edwards, J.P.; Luna, P.D.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, M.; De Mot, B.; Hereijgers, J.; Breugelmans, T. Electrochemical Reduction of CO2: Effect of Convective CO2 Supply in Gas Diffusion Electrodes. ChemElectroChem 2019, 6, 5596–5602. [Google Scholar] [CrossRef]
- Dufek, E.J.; Lister, T.E.; Stone, S.G.; McIlwain, M.E. Operation of a Pressurized System for Continuous Reduction of CO2. J. Electrochem. Soc. 2012, 159, F514–F517. [Google Scholar] [CrossRef]
- Gabardo, C.M.; Seifitokaldani, A.; Edwards, J.P.; Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; O’Brien, C.P.; Sargent, E.H.; Sinton, D. Combined High Alkalinity and Pressurization Enable Efficient CO2 Electroreduction to CO. Energy Environ. Sci. 2018, 11, 2531–2539. [Google Scholar] [CrossRef]
- Haas, T.; Krause, R.; Weber, R.; Demler, M.; Schmid, G. Technical photosynthesis involving CO2 electrolysis and fermentation. Nat. Catal. 2018, 1, 32–39. [Google Scholar] [CrossRef]
- Huang, Z.; Qi, H.; Zhao, Z.; Shang, L.; Tu, B.; Cheng, M. Efficient CO2 electroreduction on a solid oxide electrolysis cell with La0.6Sr0.4Co0.2Fe0.8O3−δ-Gd0.2Ce0.8O2-δ infiltrated electrode. J. Power Sources 2019, 434, 226730. [Google Scholar] [CrossRef]
- Lee, J.; Lim, J.; Roh, C.-W.; Whang, H.S.; Lee, H. Electrochemical CO2 Reduction Using Alkaline Membrane Electrode Assembly on Various Metal Electrodes. J. CO2 Util. 2019, 31, 244–250. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.; Han, H.; Chung, Y.S.; Yoon, W.; Choi, J.; Kim, W.B. In Situ Exsolved Co Nanoparticles on Ruddlesden-Popper Material as Highly Active Catalyst for CO2 Electrolysis to CO. Appl. Catal B-Environ. 2019, 248, 147–156. [Google Scholar] [CrossRef]
- Tian, Y.F.; Zhang, L.L.; Jia, L.C.; Wang, X.; Yang, J.; Chi, B.; Pu, J.; Li, J. Novel Quasi-symmetrical Solid Oxide Electrolysis Cells with In-situ Exsolved Cathode for CO2 Electrolysis. J. CO2 Util. 2019, 31, 43–50. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.; Noh, Y.; Kim, T.; Han, H.; Yoon, W.; Choi, J.; Yi, S.H.; Leec, W.J.; Kim, W.B. A Sulfur-tolerant Cathode Catalyst Fabricated with in Situ Exsolved CoNi Alloy Nanoparticles Anchored on a Ruddlesden-Popper Support for CO2 Electrolysis. J. Mater. Chem. A 2020, 8, 138–148. [Google Scholar] [CrossRef]
- Yin, Z.; Peng, H.; Wei, X.; Zhou, H.; Gong, J.; Huai, M.; Xiao, L.; Wang, G.; Lu, J.; Zhuang, L. An Alkaline Polymer Electrolyte CO2 Electrolyzer Operated with Pure Water. Energy Environ. Sci. 2019, 12, 2455–2462. [Google Scholar] [CrossRef]
- Xiang, H.; Rasul, S.; Scott, K.; Portoles, J.; Cumpson, P.; Yu, E.H. Enhanced Selectivity of Carbonaceous Products from Electrochemical Reduction of CO2 in Aqueous Media. J. CO2 Util. 2019, 30, 214–221. [Google Scholar] [CrossRef]
- Mot, B.D.; Hereijgers, J.; Duarte, M.; Breugelmans, T. Influence of flow and pressure distribution inside a gas diffusion electrode on the performance of a flow-by CO2 electrolyzer. Chem. Eng. J. 2019, 378, 122224. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, B.; Zhong, J.; Cheng, Z. Selective electrochemical CO2 reduction over highly porous gold films. J. Mater. Chem. A 2017, 5, 21955–21964. [Google Scholar] [CrossRef]
- Zhang, F.; Jin, Z.; Chen, C.; Tang, Y.; Mahyoub, S.A.; Yan, S.; Cheng, Z. Electrochemical Conversion of CO2 to CO into a Microchannel Reactor System in the Case of Aqueous Electrolyte. Ind. Eng. Chem. Res. 2020, 59, 5664–5674. [Google Scholar] [CrossRef]
- Kibria, M.G.; Edwards, J.P.; Gabardo, C.M.; Dinh, C.T.; Seifitokaldani, A.; Sinton, D.; Sargent, E.H. Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design. Adv. Mater. 2019, 31, 1807166. [Google Scholar] [CrossRef]
- Tan, J.; Lu, Y.C.; Xu, J.H.; Luo, G.S. Mass transfer characteristic in the formation stage of gas–liquid segmented flow in microchannel. Chem. Eng. J. 2012, 185–186, 314–320. [Google Scholar] [CrossRef]
- Yue, J.; Chen, G.; Yuan, Q.; Luo, L.; Gonthier, Y. Hydrodynamics and mass transfer characteristics in gas–liquid flow through a rectangular microchannel. Chem. Eng. Sci. 2007, 62, 2096–2108. [Google Scholar] [CrossRef]
- Yue, J.; Luo, L.; Gonthier, Y.; Chen, G.; Yuan, Q. An experimental study of air–water Taylor flow and mass transfer inside square microchannels. Chem. Eng. Sci. 2009, 64, 3697–3708. [Google Scholar] [CrossRef]
- Chen, J.-F.; Chen, G.-Z.; Wang, J.-X.; Shao, L.; Li, P.-F. High-throughput microporous tube-in-tube microreactor as novel gas-liquid contactor: Mass transfer study. AIChE J. 2011, 57, 239–249. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, C.; Tang, Y.; Cheng, Z. CO2 reduction in a microchannel electrochemical reactor with gas-liquid segmented flow. Chem. Eng. J. 2020, 392, 124798. [Google Scholar] [CrossRef]
- Naqiuddin, N.H.; Saw, L.H.; Yew, M.C.; Yusof, F.; Ng, T.C.; Yew, M.K. Overview of micro-channel design for high heat flux application. Renew. Sust. Energy Rev. 2018, 82, 901–914. [Google Scholar] [CrossRef]
- Kamitani, A.; Morishita, S.; Kotaki, H.; Arscott, S. Improved Fuel Use Efficiency in Microchannel Direct Methanol Fuel Cells Using a Hydrophilic Macroporous Layer. J. Power Sources 2009, 187, 148–155. [Google Scholar] [CrossRef]
- Wu, G.; Constantinou, A.; Cao, E.; Kuhn, S.; Morad, M.; Sankar, M.; Bethell, D.; Hutchings, G.J.; Gavriilidis, A. Continuous Heterogeneously Catalyzed Oxidation of Benzyl Alcohol Using a Tube-in-Tube Membrane Microreactor. Ind. Eng. Chem. Res. 2015, 54, 4183–4189. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Siri-Nguan, N.; Sornchamni, T.; Jovanovic, G.N.; Yokochi, A.F. CO2 Reduction in Wet Ionic Liquid Solution in Microscale-based Electrochemical Reactor. Chem. Eng. J. 2018, 333, 300–309. [Google Scholar] [CrossRef]
- Zong, X.; Zhang, J.; Zhang, J.; Luo, W.; Züttel, A.; Xiong, Y. Synergistic Cu/CeO2 carbon nanofiber catalysts for efficient CO2 electroreduction. Electrochem. Commun. 2020, 114, 106716. [Google Scholar] [CrossRef]
- Zhang, J.; Luo, W.; Züttel, A. Self-supported Copper-based Gas Diffusion Electrodes for CO2 Electrochemical Reduction. J. Mater. Chem. A 2019, 7, 26285–26292. [Google Scholar] [CrossRef]
- Krause, R.; Reinisch, D.; Reller, C.; Eckert, H.; Hartmann, D.; Taroata, D.; Wiesner-Fleischer, K.; Bulan, A.; Lueken, A.; Schmid, G. Industrial Application Aspects of the Electrochemical Reduction of CO2 to CO in Aqueous Electrolyte. Chem. Ing. Tech. 2020, 92, 53–61. [Google Scholar] [CrossRef]
- Reyes, A.; Jansonius, R.P.; Mowbray, B.A.W.; Cao, Y.; Wheeler, D.G.; Chau, J.; Dvorak, D.J.; Berlinguette, C.P. Managing Hydration at the Cathode Enables Efficient CO2 Electrolysis at Commercially Relevant Current Densities. ACS Energy Lett. 2020, 5, 1612–1618. [Google Scholar] [CrossRef]
- Li, Y.C.; Lee, G.; Yuan, T.; Wang, Y.; Nam, D.-H.; Wang, Z.; García de Arquer, F.P.; Lum, Y.; Dinh, C.-T.; Voznyy, O.; et al. CO2 Electroreduction from Carbonate Electrolyte. ACS Energy Lett. 2019, 4, 1427–1431. [Google Scholar] [CrossRef]
- Larrazabal, G.O.; Strom-Hansen, P.; Heli, J.P.; Zeiter, K.; Therkildsen, K.T.; Chorkendorff, I.; Seger, B. Analysis of Mass Flows and Membrane Cross-over in CO2 Reduction at High Current Densities in an MEA-Type Electrolyzer. ACS Appl. Mater. Interfaces 2019, 11, 41281–41288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, W. III. Experiments on the Quantity of Gases Absorbed by Water, at Different Temperatures, and under Different Pressures. Philos. Trans. R. Soc. London 1803, 93, 14. [Google Scholar]
- Kohjiro Hara, A.K. Tadayoshi Sakata Electrochemical Reduction of Carbon Dioxide under High Pressure on Various Electrodes in an Aqueous Electrolyte. J. Electroanal. Chem. 1995, 391, 141–147. [Google Scholar] [CrossRef]
- Makoto Todoroki, K.H. Akihiko Kudo, Tadayoshi Sakata Electrochemical Reduction of High Pressure CO2 at Pb, Hg and In Electrodes in an Aqueous KHCO3 Solution. J. Electroanal. Chem. 1995, 394, 199–203. [Google Scholar] [CrossRef]
- Kohjiro Hara, T.S. Large Current Density CO2 Reduction under High Pressure Using Gas Diffusion Electrodes. Bull. Chem. Soc. Jpn. 1997, 70, 571–576. [Google Scholar] [CrossRef]
- Lin, J.; Yan, S.; Zhang, C.; Hu, Q.; Cheng, Z. Hydrophobic Electrode Design for CO2 Electroreduction in a Microchannel Reactor. ACS Appl. Mater. Interfaces 2022, 14, 8623–8632. [Google Scholar] [CrossRef]
- Yan, S.; Mahyoub, S.A.; Lin, J.; Zhang, C.; Hu, Q.; Zhong, J.; Chen, C.; Zhang, F.; Cheng, Z. Controllable Growth of Branched Silver Crystals over a Rod of the Same Material as an Efficient Electrode in CO2 Reduction at High Current Densities. J. Catal. 2022, 405, 224–235. [Google Scholar] [CrossRef]
- Mahyoub, S.A.; Qaraah, F.A.; Yan, S.; Hezam, A.; Zhong, J.; Cheng, Z. Rational design of low loading Pd-alloyed Ag nanocorals for high current density CO2-to-CO electroreduction at elevated pressure. Mater. Today Energy 2022, 24, 100923. [Google Scholar] [CrossRef]
- Fan, M.; Prabhudev, S.; Garbarino, S.; Qiao, J.; Botton, G.A.; Harrington, D.A.; Tavares, A.C.; Guay, D. Uncovering the nature of electroactive sites in nano architectured dendritic Bi for highly efficient CO2 electroreduction to formate. Appl. Catal. B 2020, 274, 119031. [Google Scholar] [CrossRef]
- Burdyny, T.; Smith, W.A. CO2 Reduction on Gas-diffusion Electrodes and Why Catalytic Performance must be assessed at Commercially-relevant Conditions. Energy Environ. Sci. 2019, 12, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Zhou, D.; Yan, Z.; Gonçalves, R.H.; Salvatore, D.A.; Berlinguette, C.P.; Mallouk, T.E. Electrolysis of CO2 to Syngas in Bipolar Membrane-Based Electrochemical Cells. ACS Energy Lett. 2016, 1, 1149–1153. [Google Scholar] [CrossRef]
- Hori, Y.; Konishi, H.; Futamura, T.; Murata, A.; Koga, O.; Sakurai, H.; Oguma, K. “Deactivation of Copper Electrode” in Electrochemical Reduction of CO2. Electrochim. Acta 2005, 50, 5354–5369. [Google Scholar] [CrossRef]
- Wuttig, A.; Surendranath, Y. Impurity Ion Complexation Enhances Carbon Dioxide Reduction Catalysis. ACS Catal. 2015, 5, 4479–4484. [Google Scholar] [CrossRef] [Green Version]
- Jeanty, P.; Scherer, C.; Magori, E.; Wiesner-Fleischer, K.; Hinrichsen, O.; Fleischer, M. Upscaling and continuous operation of electrochemical CO2 to CO conversion in aqueous solutions on silver gas diffusion electrodes. J. CO2 Util. 2018, 24, 454–462. [Google Scholar] [CrossRef]
- Lv, K.L.; Teng, C.; Shi, M.H.; Yuan, Y.; Zhu, Y.; Wang, J.R.; Kong, Z.; Lu, X.Y.; Zhu, Y. Hydrophobic and Electronic Properties of the E-MoS2 Nanosheets Induced by FAS for the CO2 Electroreduction to Syngas with a Wide Range of CO/H2 Ratios. Adv. Funct. Mater. 2018, 28, 1802339. [Google Scholar] [CrossRef]
- Xing, Z.; Hu, L.; Ripatti, D.S.; Hu, X.; Feng, X.F. Enhancing carbon dioxide gas-diffusion electrolysis by creating a hydrophobic catalyst microenvironment. Nat. Commun. 2021, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.Z.; Gao, F.Y.; Zhang, X.L.; Yang, P.P.; Liu, R.; Chi, L.P.; Wu, Z.Z.; Qin, S.; Yu, X.X.; Gao, M.R. Hierarchical Copper with Inherent Hydrophobicity Mitigates Electrode Flooding for High-Rate CO2 Electroreduction to Multicarbon Products. J. Am. Chem. Soc. 2021, 143, 8011–8021. [Google Scholar] [CrossRef]
- Liu, S.Q.; Shahini, E.; Gao, M.R.; Gong, L.; Sui, P.F.; Tang, T.; Zeng, H.B.; Luo, J.L. Bi2O3 Nanosheets Grown on Carbon Nanofiber with Inherent Hydrophobicity for High-Performance CO2 Electroreduction in a Wide Potential Window. ACS Nano 2021, 15, 17757–17768. [Google Scholar] [CrossRef]
- Vedharathinam, V.; Qi, Z.; Horwood, C.; Bourcier, B.; Stadermann, M.; Biener, J.; Biener, M. Using a 3D Porous Flow-Through Electrode Geometry for High-Rate Electrochemical Reduction of CO2 to CO in Ionic Liquid. ACS Catal. 2019, 9, 10605–10611. [Google Scholar] [CrossRef]
- Saeki, T.; Hashimoto, K.; Kimura, N.; Omata, K.; Fujishima, A. Electrochemical reduction of CO2 with high current density in a CO2 + methanol medium II. CO formation promoted by tetrabutylammonium cation. J. Electroanal. Chem. 1995, 390, 77–82. [Google Scholar] [CrossRef]
- Verma, S.; Lu, X.; Ma, S.C.; Masel, R.I.; Kenis, P.J.A. The Effect of Electrolyte Composition on the Electroreduction of CO2 to CO on Ag Based Gas Diffusion Electrodes. Phys. Chem. Chem. Phys. 2016, 18, 7075–7084. [Google Scholar] [CrossRef] [PubMed]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Gupta, N.; Gattrell, M.; MacDougall, B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 2005, 36, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Pinsent, B.R.W.; Pearson, L.; Roughto, F.J.W. The kinetics of combination of carbon dioxide with hydroxide ions. Trans. Faraday Soc. 1956, 52, 1512–1520. [Google Scholar] [CrossRef]
- Varela, A.S.; Kroschel, M.; Reier, T.; Strasser, P. Controlling the Selectivity of CO2 Electroreduction on Copper: The Effect of the Electrolyte Concentration and the Importance of the Local pH. Catal. Today 2016, 260, 8–13. [Google Scholar] [CrossRef]
- Thorson, M.R.; Siil, K.I.; Kenis, P.J.A. Effect of Cations on the Electrochemical Conversion of CO2 to CO. J. Electrochem. Soc. 2013, 160, F69–F74. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.E.; Li, F.W.; Ozden, A.; Rasouli, A.S.; de Arquer, F.P.G.; Liu, S.J.; Zhang, S.Z.; Luo, M.C.; Wang, X.; Lum, Y.W.; et al. CO2 Electrolysis to Multicarbon Products in Strong Acid. Science 2021, 372, 1074–1078. [Google Scholar] [CrossRef]
- Edwards, J.P.; Xu, Y.; Gabardo, C.M.; Dinh, C.-T.; Li, J.; Qi, Z.; Ozden, A.; Sargent, E.H.; Sinton, D. Efficient electrocatalytic conversion of carbon dioxide in a low-resistance pressurized alkaline electrolyzer. Appl. Energy 2020, 261, 114305. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | E0/(V vs. RHE) |
|---|---|
| 2H+ + 2e− → H2 | 0.00 |
| CO2 + 2H++ 2e− → CO + H2O | −0.10 |
| CO2 + 2H+ + 2e− → HCOOH | −0.12 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | +0.03 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | +0.17 |
| 2CO2 + 8H+ + 8e− → CH3COOH + 2H2O | +0.11 |
| 2CO2 + 10H+ + 10e− → CH3CHO + 3H2O | +0.06 |
| 2CO2 + 12H+ + 12e− → CH3CH2OH + 3H2O | +0.09 |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | +0.08 |
| 3CO2 + 18H+ + 18e− → CH3CH2CH2OH + 5H2O | +0.10 |
| Catalyst | jmain product /mA cm−2 | Main Product | Electrolyzer | Reference |
|---|---|---|---|---|
| Au/PyPBI/MWNTs | 160 | CO | Microfluidic cell | [36] |
| Au/PyPBI/MWNTs | 158 | CO | Flow cell | [37] |
| PVA-Au/C | 98.6 | CO | Two-component cell | [38] |
| Ag/TiO2 | 101 | CO | Flow cell | [42] |
| Ag-CP/MPL-nC | 385 | CO | Zero-gap flow cell | [43] |
| Ag-S-C3N4/CNT | 303 | CO | Flow cell | [44] |
| Mixed AgNP/MWCNT | 350 | CO | Flow cell | [45] |
| Pd octahedra (111) | 220 | CO | Flow cell | [48] |
| Pd/C-PDDA | ~279 | CO | Microfluidic flow cell | [49] |
| Catalyst | jmain product /mA cm−2 | Main Product | Electrolyzer | Reference |
|---|---|---|---|---|
| d-Cu-1 | 100.3 | HCOOH | H-cell | [50] |
| OD-Cu | 141 | C2H4 | Flow cell | [51] |
| Cu@CuxO | 150 | C2H4 | Flow cell | [52] |
| Cu nanocube | 144 | C2H4 | Flow cell | [53] |
| Sn/Cu-PVDF | >100 | CO | Flow cell | [55] |
| Cu–In/GDE | ~173 | CO | Flow cell | [56] |
| Sn2.7Cu | 397.88 | CO + HCOOH | Flow cell | [58] |
| Ag0.14/Cu0.86 | 102.5 | C2H5OH | Flow cell | [59] |
| F–Cu | 1280 | mainly C2H4 and C2H5OH | Flow cell | [60] |
| Cu/N-CNF | 372 | C2H4 | Flow cell | [61] |
| NGQ/Cu-nr | 147.8 | C2 + C2H5OH | Flow cell | [62] |
| CuO-FC | 231 | C2 (mainly C2H5OH) | Flow cell | [64] |
| Cu-CO2 | 520 | C2+ (mainly C2H4) | Flow cell | [70] |
| Cu/Fe–N–C s-GDE | >1000 | C2+ | MEA | [71] |
| Catalyst | jmain product /mA cm−2 | Main Product | Electrolyzer | Reference |
|---|---|---|---|---|
| P–Zn | 168 | CO | Flow cell | [72] |
| Ag–Zn | 286 | CO | High-pressure cell | [74] |
| CdS needle | 212 | CO | Flow cell | [76] |
| SnO2-GDE | 800 | Formate | Semi-batch cell | [79] |
| SnO2/C | ~211 | Formate | Flow cell | [80] |
| CuSn–C | 148 | Formate | Flow cell | [82] |
| Bi-GDEs | 210 | Formate | Filter press reactor | [83] |
| Bi2O3@C | >200 | Formate | Flow cell | [84] |
| 2D-Bi | 172.2 | Formate | Flow cell | [85] |
| Bi NSs | 374 | HCOOH | Flow cell | [86] |
| Bi-ene | ~200 | HCOOH | Flow cell | [87] |
| Catalyst | jmain product /mA cm−2 | Main Product | Electrolyzer | Reference |
|---|---|---|---|---|
| NSHCF | 96.82 | CO | H-cell | [90] |
| NPCA | 143.6 | CO | H-cell | [91] |
| Catalyst | jmain product /mA cm−2 | Main Product | Electrolyzer | Reference |
|---|---|---|---|---|
| NiSA/PCFM | 308.4 | CO | Flow cell | [94] |
| CoSA/HCNFs | 211 | CO | Flow cell | [95] |
| Fe3+–N–C | 94 | CO | Flow cell | [101] |
| Ni–NCB | 130 | CO | MEA cell | [104] |
| Ni–SA–NCs | 380 | CO | MEA cell | [105] |
| Ni-SAC(Pc) | 200 | CO | Flow cell | [106] |
| Ni20−N−C | 645 | CO | Flow cell | [107] |
| NiPc–OMe MDE | >300 | CO | Flow cell | [109] |
| Ni(NC)-1 | 158.4 | CO | Flow cell | [110] |
| Catalyst | jmain product /mA cm−2 | Main Product | Electrolyzer | Reference |
|---|---|---|---|---|
| CoPc1 | 175 | CO | Zero-gap membrane flow cell | [115] |
| CoPc2 | 165 | CO | Flow cell | [116] |
| FeP | 152 | CO | Flow cell | [117] |
| Catalyst | jmain product /mA cm−2 | Main Product | Pressure/Bar | Reference |
|---|---|---|---|---|
| Ag | 123.22 | CO | 30 | [154] |
| Pb | 200.8 | HCOOH | ~60 | [155] |
| Hg | 201.8 | HCOOH | ~20 | [155] |
| In | 215.2 | HCOOH | ~60 | [155] |
| Ag-alloyed Zn | 286 | CO | 9.5 | [74] |
| Zn@Ag–2PTFE | 106.76 | CO | 9 | [157] |
| Ag dendrites foam | 288.68 | CO | 9.5 | [158] |
| Ag–Pd | 318 | CO | 9.5 | [159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; Yan, S.; Zhang, C.; Hu, Q.; Cheng, Z. Electroreduction of CO2 toward High Current Density. Processes 2022, 10, 826. https://doi.org/10.3390/pr10050826
Lin J, Yan S, Zhang C, Hu Q, Cheng Z. Electroreduction of CO2 toward High Current Density. Processes. 2022; 10(5):826. https://doi.org/10.3390/pr10050826
Chicago/Turabian StyleLin, Jing, Shenglin Yan, Chunxiao Zhang, Qing Hu, and Zhenmin Cheng. 2022. "Electroreduction of CO2 toward High Current Density" Processes 10, no. 5: 826. https://doi.org/10.3390/pr10050826
APA StyleLin, J., Yan, S., Zhang, C., Hu, Q., & Cheng, Z. (2022). Electroreduction of CO2 toward High Current Density. Processes, 10(5), 826. https://doi.org/10.3390/pr10050826