

Structural Behaviour and Charge-Compensation Mechanism in Li2Fe1−xCoxSeO Solid Solutions during Reversible Delithiation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Preliminary Characterization
2.2. Electrode Preparation and Electrochemical Studies
2.3. Operando Characterization at Synchrotron Radiation Facilities
3. Results and Discussion
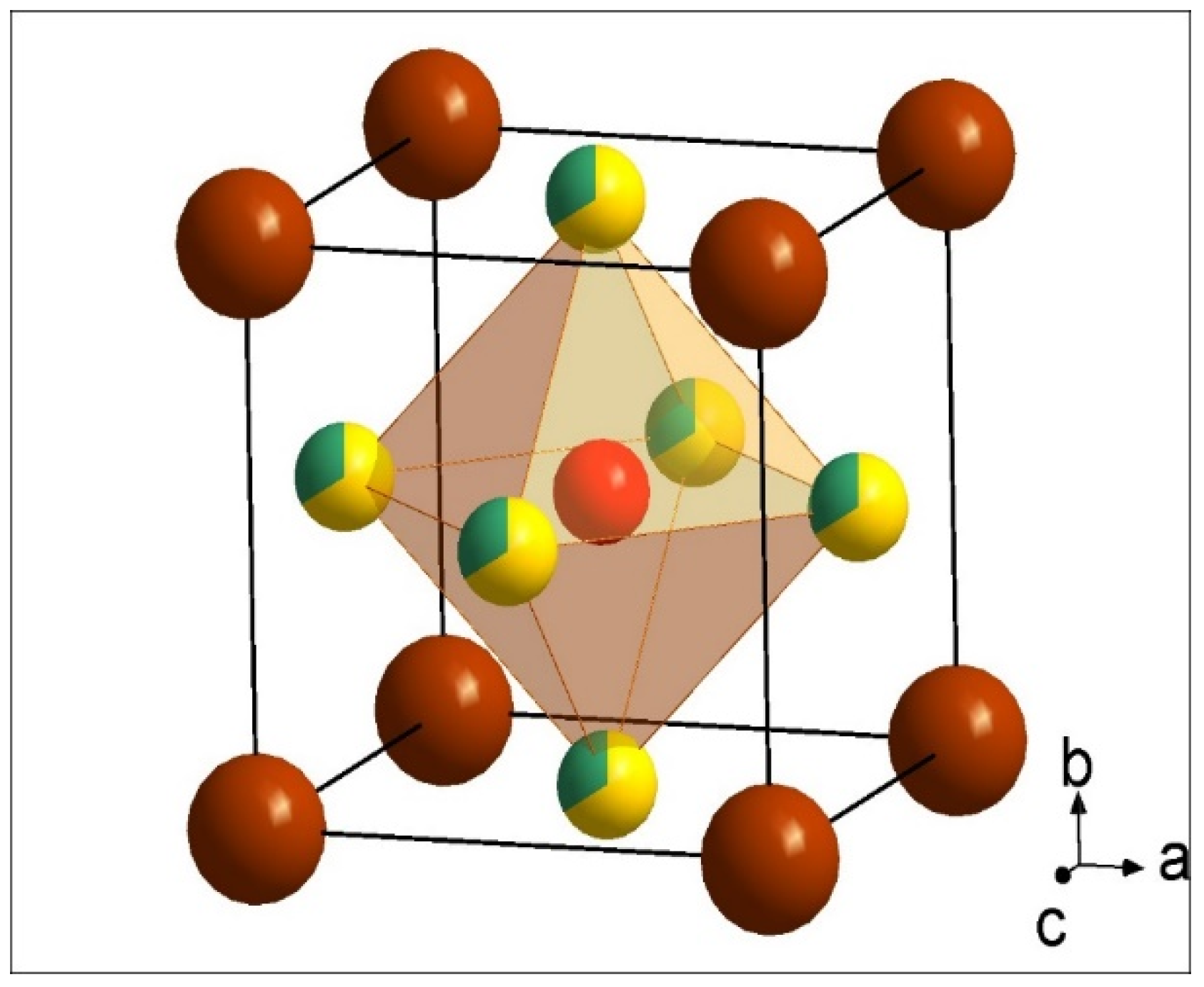
3.1. Initial Assessment of Materials Properties
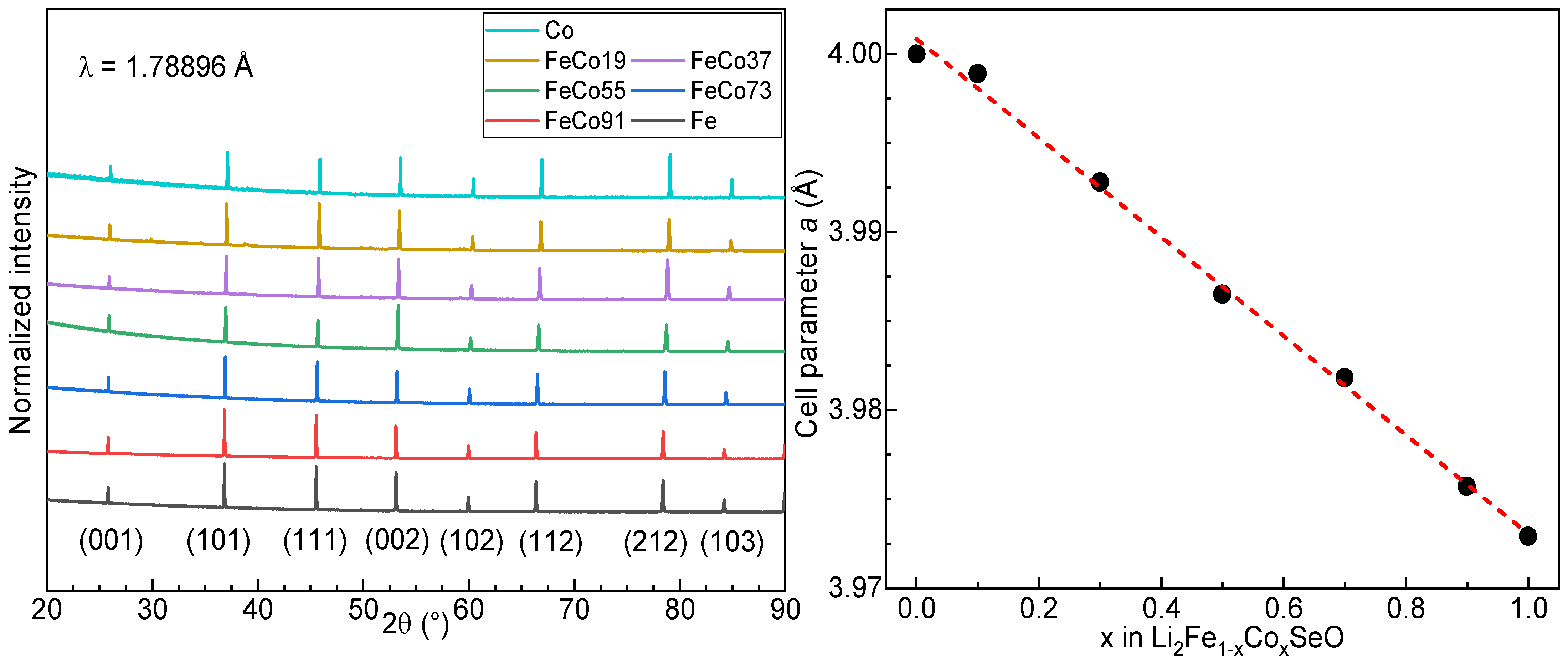
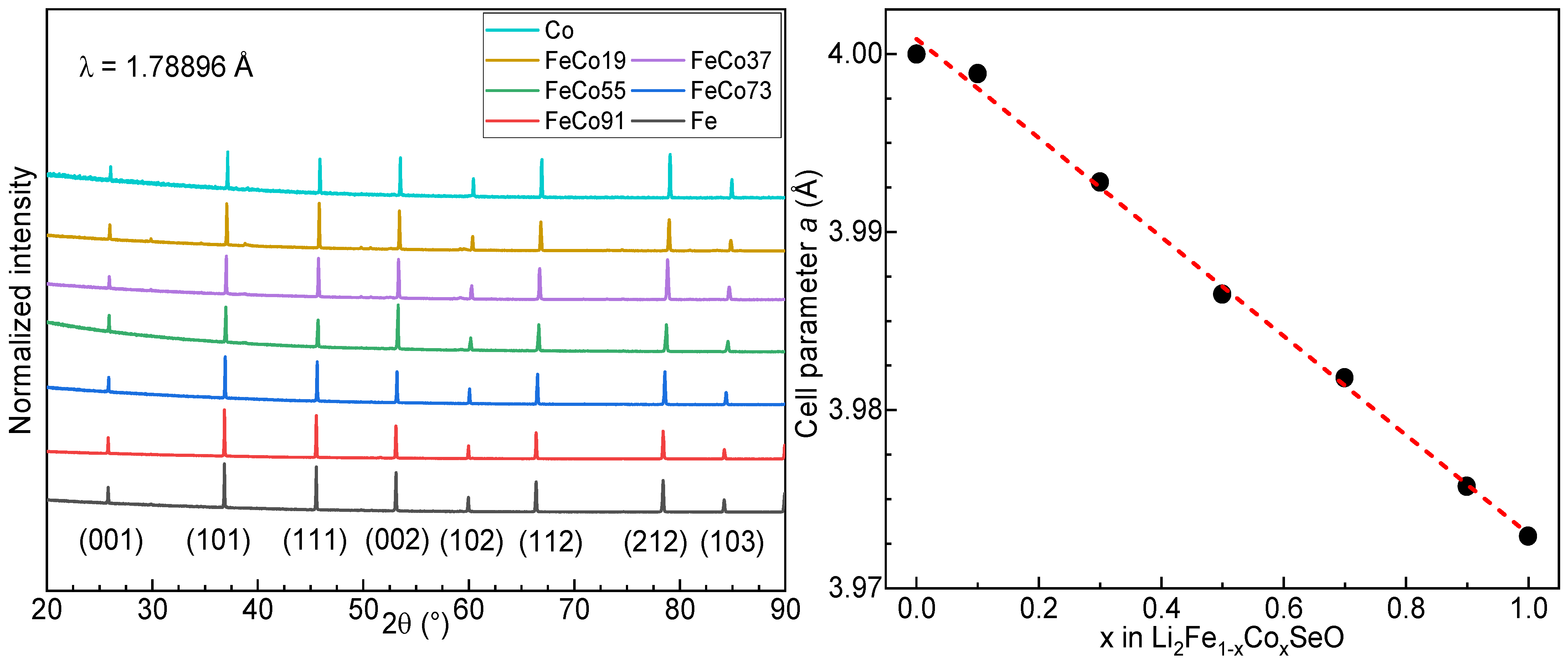
3.2. Powder X-ray Diffraction
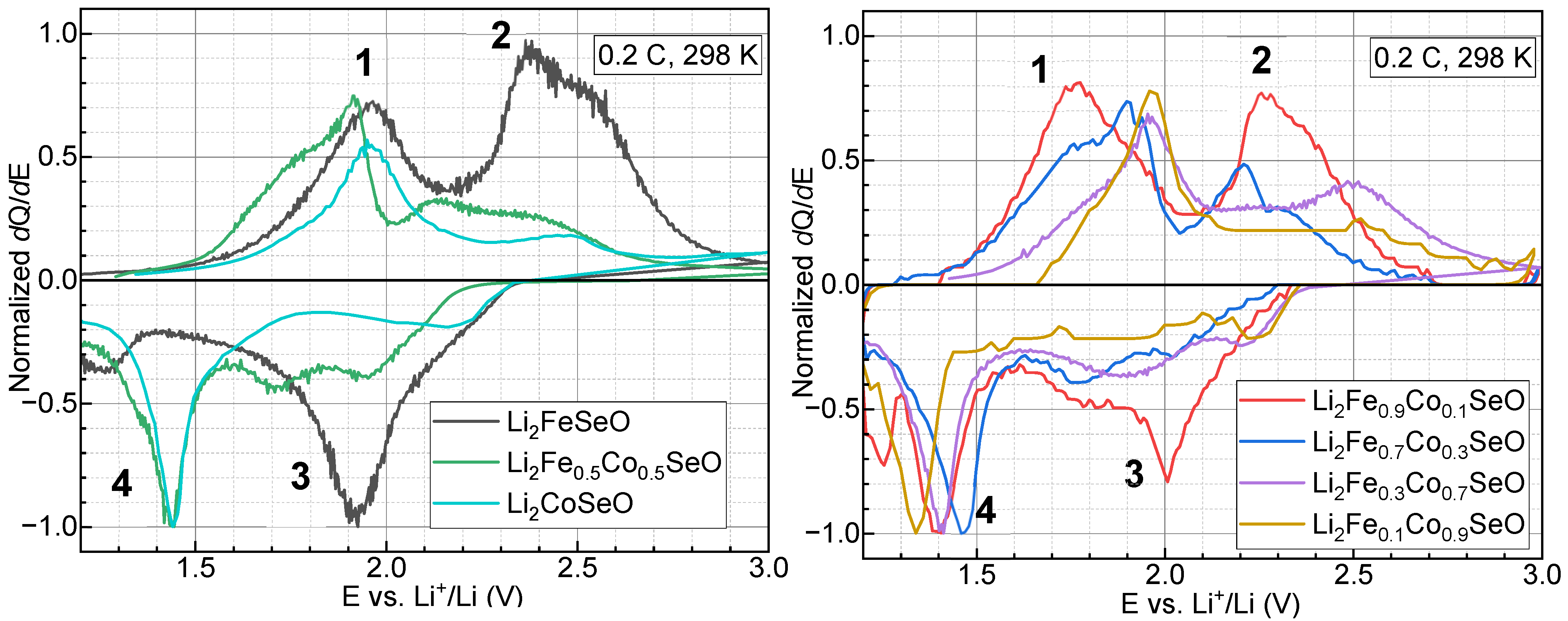
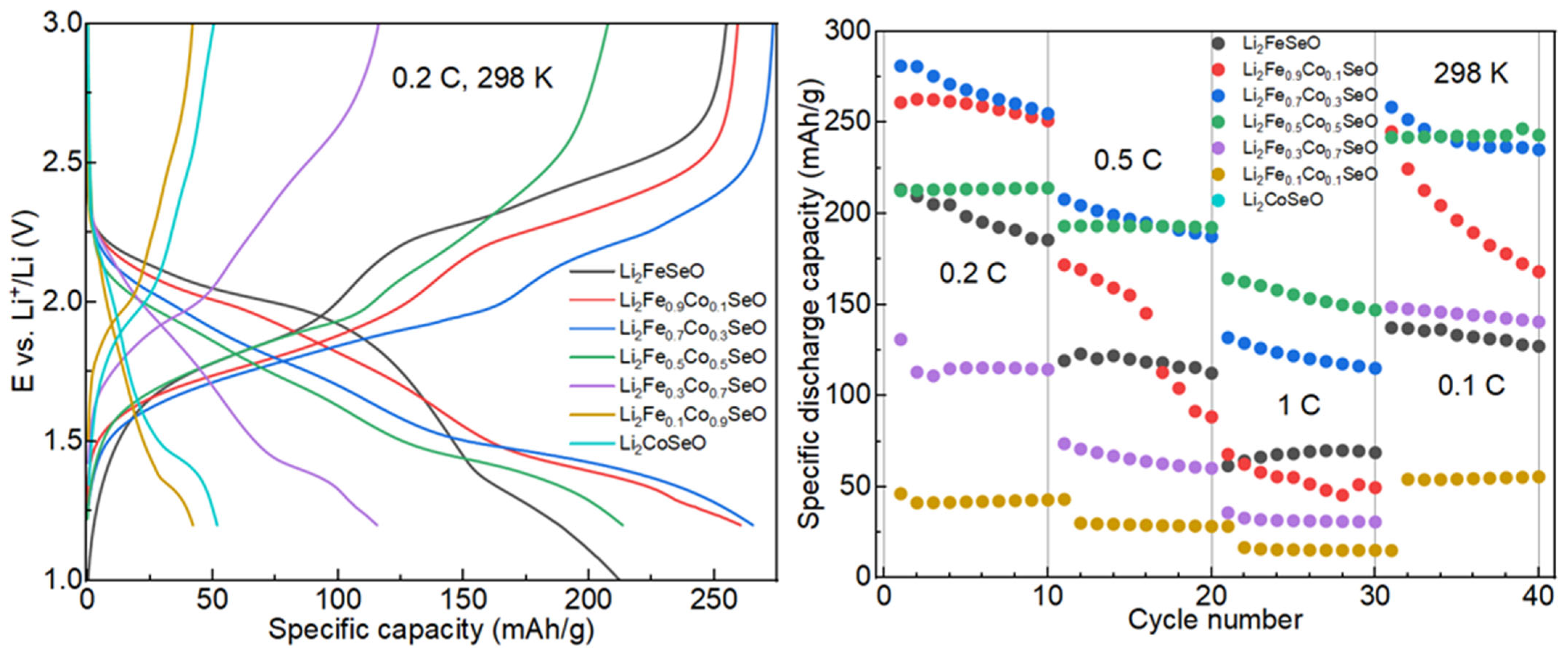
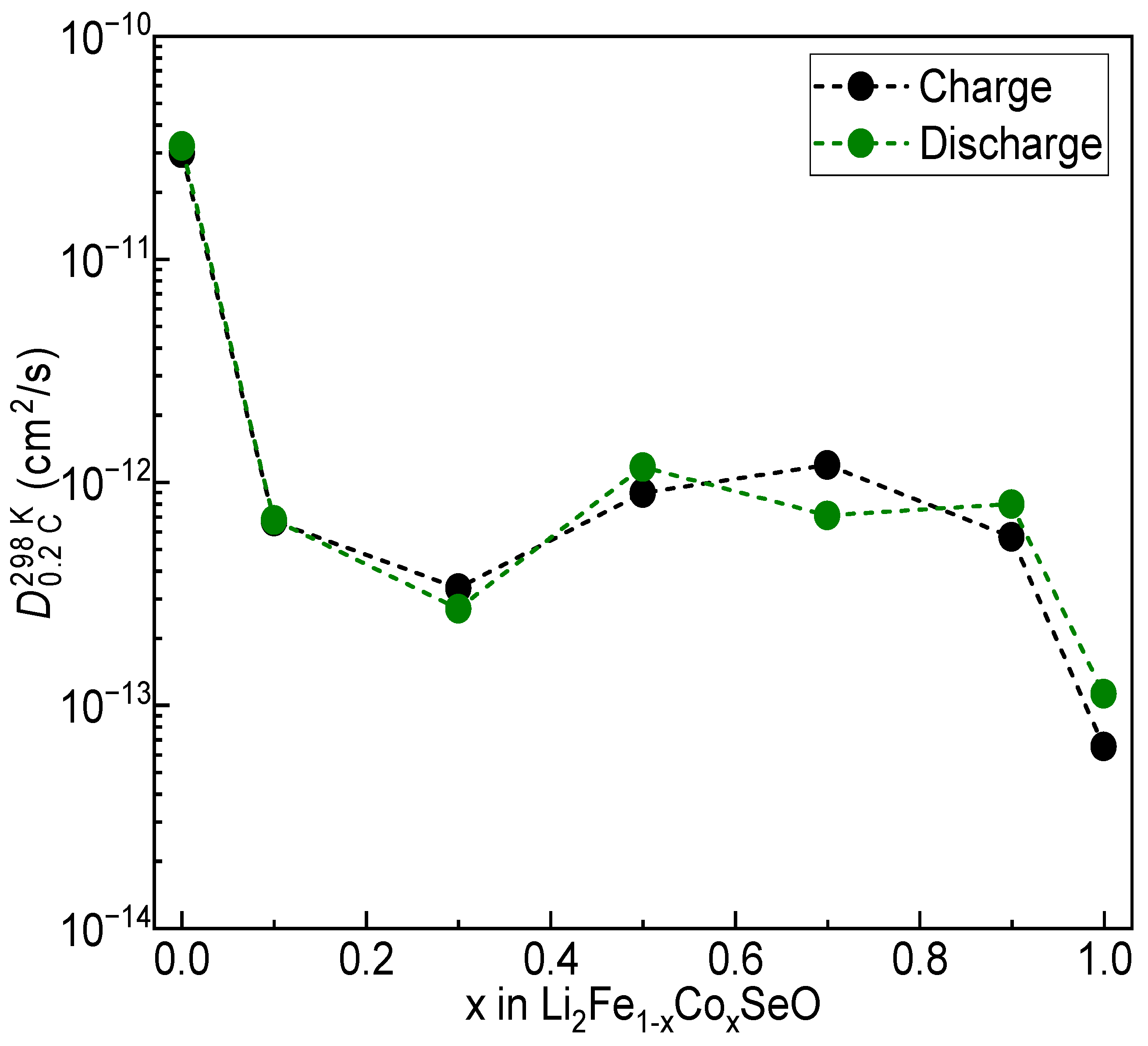
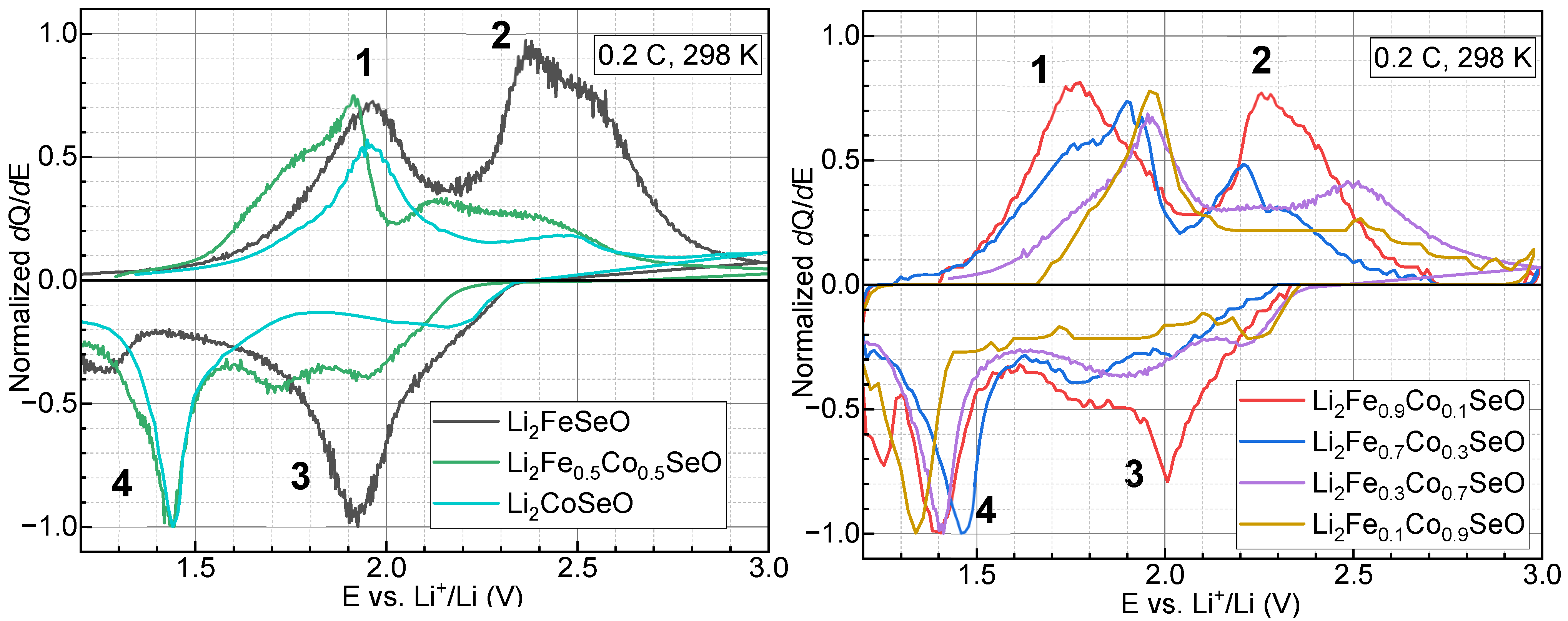
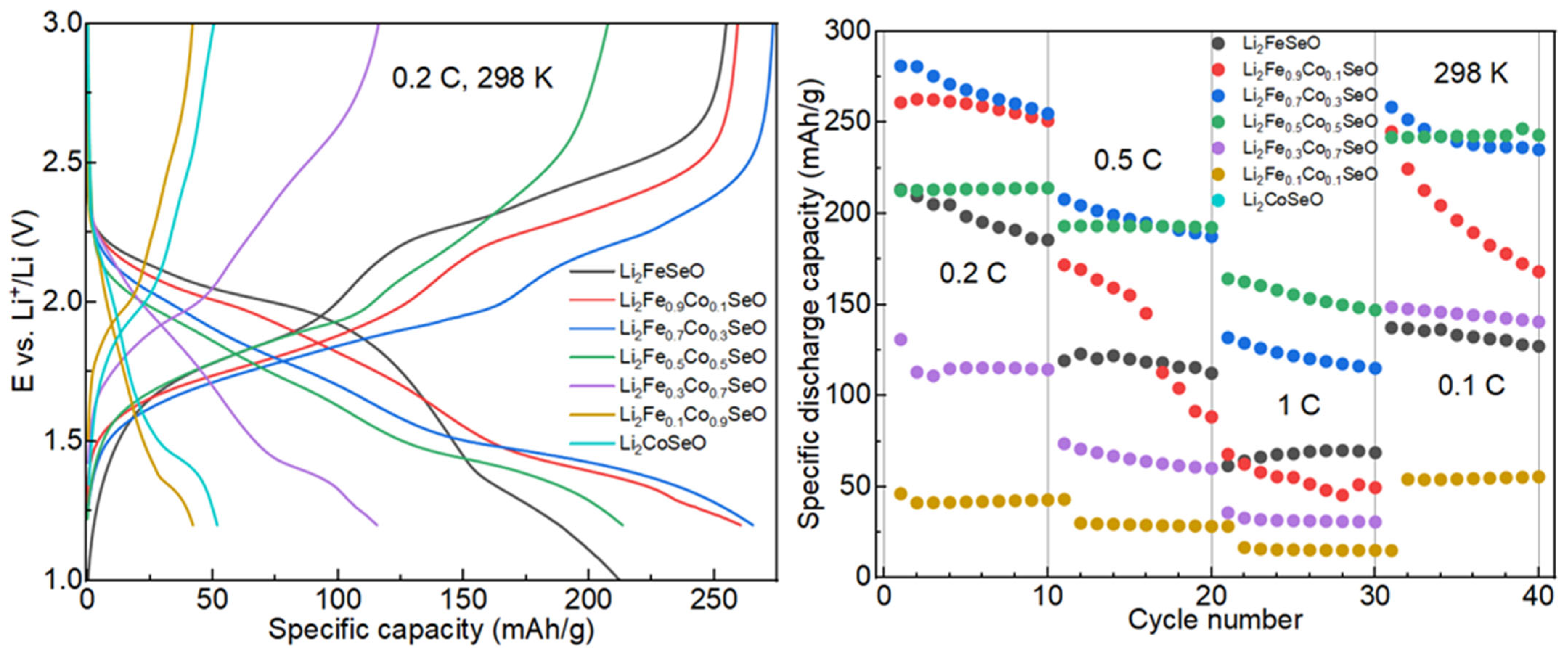
3.3. Electrochemical Properties
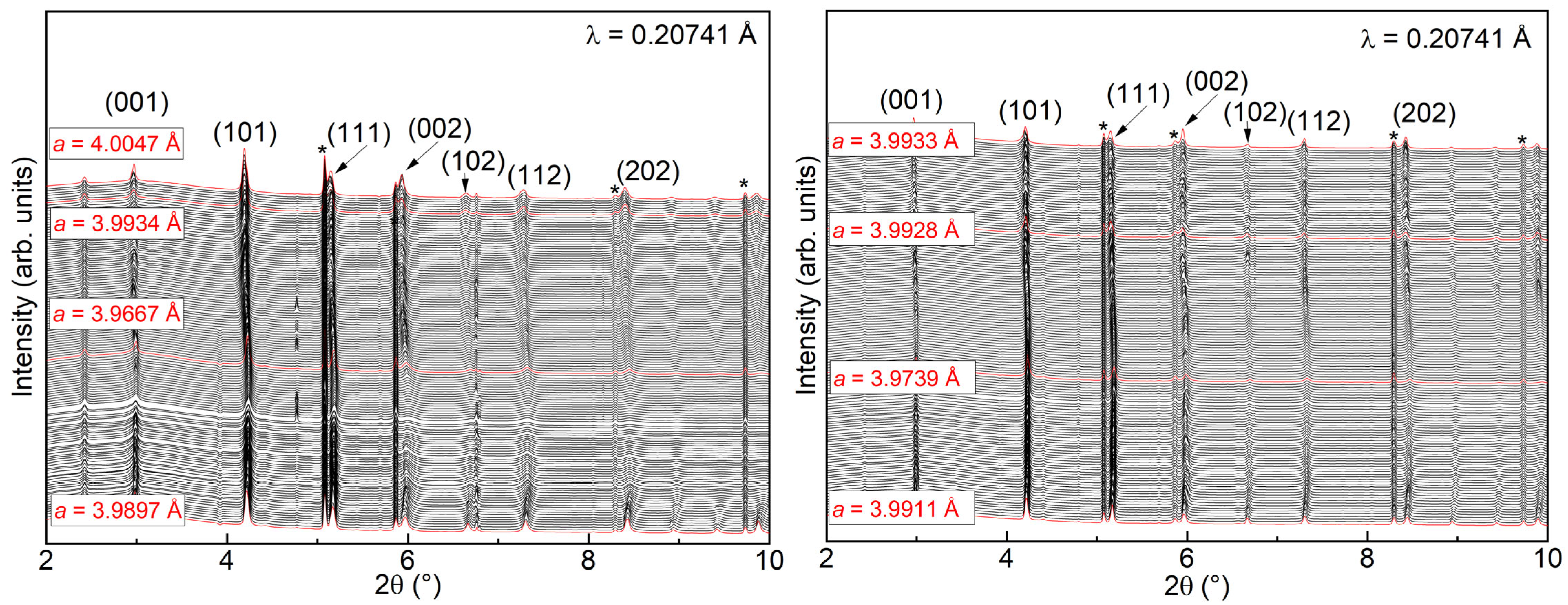
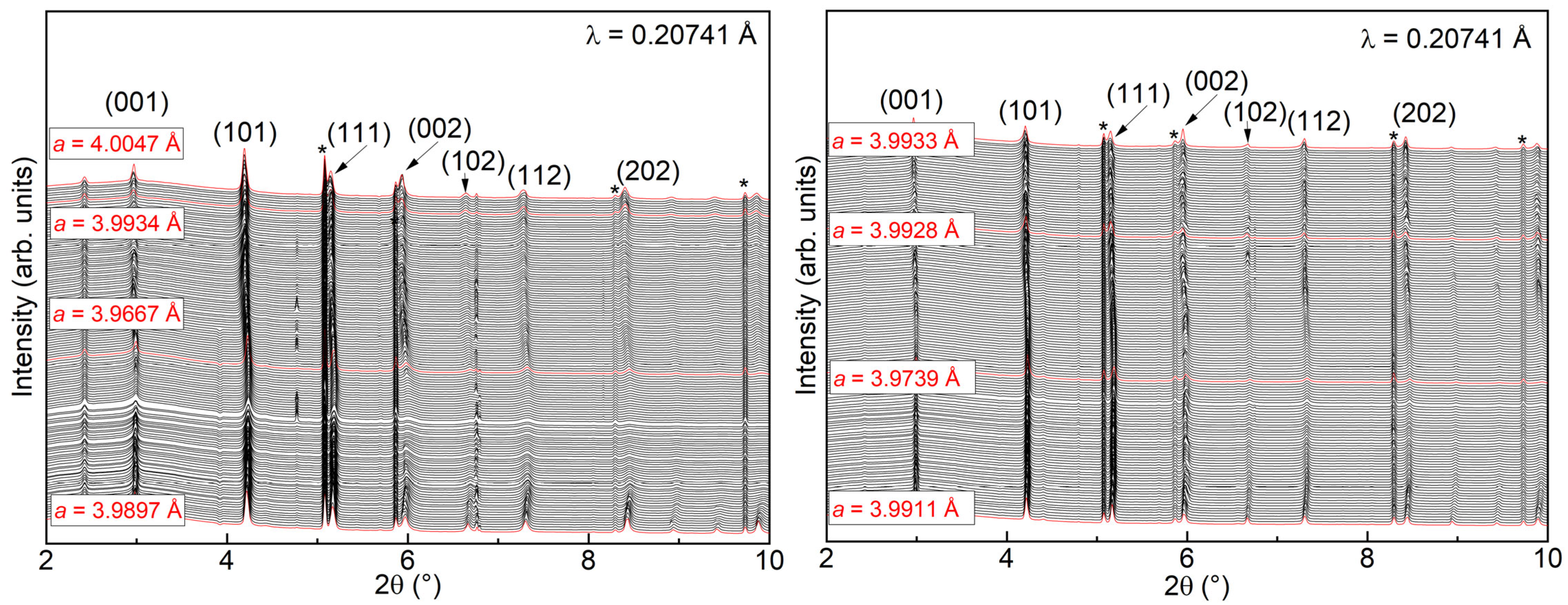
3.4. Operando X-ray Diffraction
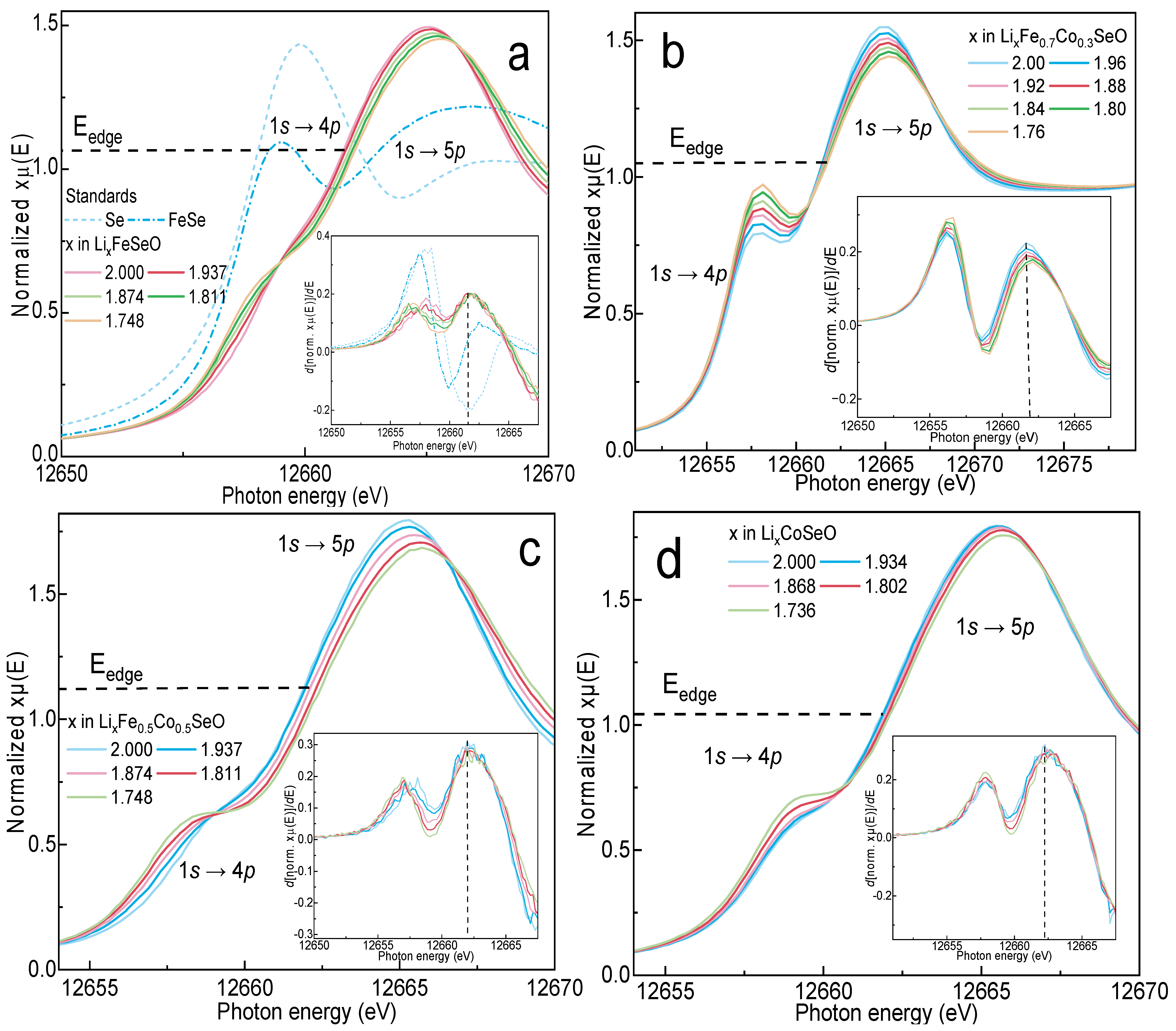
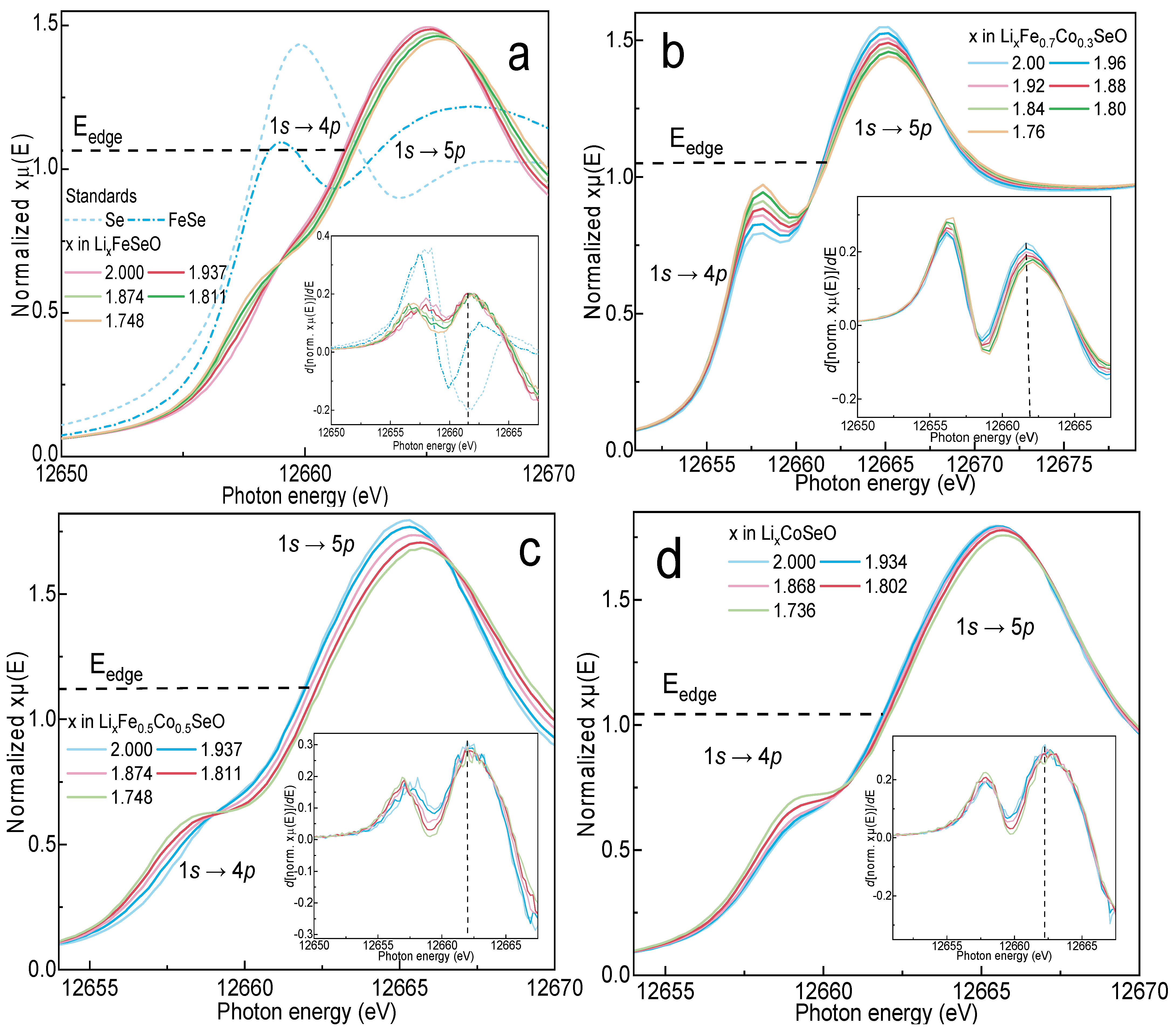
3.5. Operando X-ray Absorption Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krivovichev, S.V. Minerals with antiperovskite structure: A review. Z. Krist. 2008, 223, 109. [Google Scholar] [CrossRef]
- Pabst, A. The Crystal Structure of Sulphohalite. Z. Krist. 1934, 89, 514. [Google Scholar] [CrossRef]
- Dawson, J.A.; Farmprikis, T.; Johnston, K.E. Anti-perovskites for solid-state batteries: Recent developments, current challenges and future prospects. J. Mater. Chem. A 2021, 9, 18746. [Google Scholar] [CrossRef]
- He, T.; Huang, Q.; Ramirez, A.P.; Wang, Y.; Regan, K.A.; Rogado, N.; Hayward, M.A.; Haas, M.K.; Slusky, J.S.; Inumara, K.; et al. Superconductivity in the non-oxide perovskite MgCNi3. Nature 2001, 411, 54. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Li, L.; Cai, F.; Xu, X.; Qian, L. Low-temperature negative thermal expansion of the antiperovskite manganese nitride Mn3CuN codoped with Ge and Si. Appl. Phys. Lett. 2008, 93, 81902. [Google Scholar] [CrossRef]
- Kamishima, K.; Goto, T.; Nakagawa, H.; Miura, N.; Ohashi, M.; Mori, N.; Sasaki, T.; Kanomata, T. Giant magnetoresistance in the intermetallic compound Mn3GaC. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 63, 24426. [Google Scholar] [CrossRef]
- Lukashev, P.; Sabirianov, R.F.; Belashchenko, K. Theory of the piezomagnetic effect in Mn-based antiperovskites. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 78, 184414. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, G.; Zhong, Y.; Chen, Y.; Ma, N.; Zhou, W.; Shao, Z. A surface-modified antiperovskite as an electrocatalyst for water oxidation. Nat. Commun. 2018, 9, 2326. [Google Scholar] [CrossRef]
- Zhao, Y.; Daemen, L.L. Superionic Conductivity in Lithium-Rich Anti-Perovskites. J. Am. Chem. Soc. 2012, 134, 15042. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, Y.; Sun, X.; Xie, X.; Guan, X.; Chen, C.; Lu, P.; Ma, X. Theoretical design of Na-rich anti-perovskite as solid electrolyte: The effect of cluster anion in stability and ionic conductivity. J. Solid State Chem. 2022, 316, 123643. [Google Scholar] [CrossRef]
- Lai, K.T.; Antonyshyn, I.; Prots, Y.; Valldor, M. Anti-Perovskite Li-Battery Cathode Materials. J. Am. Chem. Soc. 2017, 139, 9645. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.T.; Antonyshyn, I.; Prots, Y.; Valldor, M. Extended Chemical Flexibility of Cubic Anti-Perovskite Lithium Battery Cathode Materials. Inorg. Chem. 2018, 57, 13296. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ciucci, F. Anti-perovskite cathodes for lithium batteries. J. Mater. Chem. A 2018, 6, 5185. [Google Scholar] [CrossRef]
- Mikhailova, D.; Giebeler, L.; Maletti, S.; Oswald, S.; Sarapulova, A.; Indris, S.; Hu, Z.; Bednarcik, J.; Valldor, M. Operando Studies of Antiperovskite Lithium Battery Cathode Material (Li2Fe)SO. ACS Appl. Energy Mater. 2018, 1, 6593. [Google Scholar] [CrossRef]
- Wang, J.; Dreyer, S.L.; Wang, K.; Ding, Z.; Diemant, T.; Karkera, G.; Ma, Y.; Sarkar, A.; Zhou, B.; Gorbunov, M.V.; et al. P2-type layered high-entropy oxides as sodium-ion cathode materials. Mater. Futures 2022, 1, 035104. [Google Scholar] [CrossRef]
- Gorbunov, M.V.; Carocci, S.; Gonzalez Martinez, I.G.; Baran, V.; Mikhailova, D. Studies of Li2Fe0.9M0.1SO Antiperovskite Materials for Lithium-Ion Batteries: The Role of Partial Fe2+ to M2+ Substitution. Front. Energy Res. 2021, 9, 657962. [Google Scholar] [CrossRef]
- Gorbunov, M.V.; Carrocci, S.; Maletti, S.; Valldor, M.; Doert, T.; Hampel, S.; Gonzalez Martinez, I.G.; Mikhailova, D.; Gräßler, N. Synthesis of (Li2Fe1–yMny)SO Antiperovskites with Comprehensive Investigations of (Li2Fe0.5Mn0.5)SO as Cathode in Li-ion Batteries. Inorg. Chem. 2020, 59, 15626. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.A.A.; Gorbunov, M.V.; Valldor, M.; Hampel, S.; Gräßler, N.; Mikhailova, D. Tuning the electrochemical properties by anionic substitution of Li-rich antiperovskite (Li2Fe)S1−xSexO cathodes for Li-ion batteries. J. Mater. Chem. A 2021, 9, 23095. [Google Scholar] [CrossRef]
- Gorbunov, M.V.; Janson, O.; Stöber, M.; Baran, V.; Mikhailova, D. Correlation between Cationic Composition and Anionic Electrochemical Activity of Li2MSeO Anti-perovskites. J. Mater. Chem. A 2024. submitted. [Google Scholar]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of Lisbwo8 by x-ray powder diffraction. Mat. Res. Bull. 1988, 23, 447. [Google Scholar] [CrossRef]
- Petricek, V.; Dusek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Krist. 2014, 229, 345. [Google Scholar]
- Faraday, M., VI. Experimental researches in electricity. Seventh Series. Philos. Trans. R. Soc. 1834, 124, 77. [Google Scholar]
- Dippel, A.-C.; Liermann, H.-P.; Delitz, J.T.; Walter, P.; Schulte-Schrepping, H.; Seecka, O.H.; Franza, H. Beamline P02.1 at PETRA III for high-resolution and high-energy powder diffraction. J. Synchrotron Radiat. 2015, 22, 675. [Google Scholar] [CrossRef] [PubMed]
- Filik, J.; Ashton, A.W.; Chang, P.C.Y.; Chater, P.A.; Day, S.J.; Drakopoulos, M.; Gerring, M.W.; Hart, M.L.; Magdysyuk, O.V.; Michalik, S.; et al. Processing two-dimensional X-ray diffraction and small-angle scattering data in DAWN 2. J. Appl. Cryst. 2017, 50, 959. [Google Scholar] [CrossRef] [PubMed]
- Herklotz, M.; Weiß, J.; Ahrens, E.; Yavuz, M.; Mereacre, L.; Kiziltas-Yavuz, N.; Dräger, C.; Ehrenberg, H.; Eckert, J.; Fauth, F.; et al. A novel high-throughput setup for in situ powder diffraction on coin cell batteries. J. Appl. Cryst. 2016, 49, 340. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Cryst. 1969, 2, 65. [Google Scholar] [CrossRef]
- Caliebe, W.A.; Murzin, V.; Kalinko, A.; Görlitz, M. High-flux XAFS-beamline P64 at PETRA III. AIP Conf. Proc. 2019, 2054, 060031. [Google Scholar]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, V.M. Die Gesetze der Krystallochemie. Naturwissenschaften 1926, 14, 477. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A Cryst. phys. Diffr. Theor. Gen. Crystallogr. 1976, A32, 751. [Google Scholar] [CrossRef]
- Haynes, W.M.; Lide, D.R.; Bruno, T.J. Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA; London, UK; New York, NY, USA, 2015. [Google Scholar]
- Gorbunov, M.V.; Doert, T.; Mikhailova, D. Na2Fe2Se2O: A double anti-perovskite with prevalence of anionic redox activity in Na-ion batteries. Chem. Commun. 2023, 59, 13763. [Google Scholar] [CrossRef] [PubMed]
- Jahn, H.A.; Teller, E. Stability of Polyatomic Molecules in Degenerate Electronic States. I. Orbital Degeneracy. Proc. R. Soc. A 1937, 161, 220. [Google Scholar]
- Vegard, L. Die Konstitution der Mischkristalle und die Raumfüllung der Atome. Z. Phys. 1921, 5, 17. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Emly, A.; Kioupakis, E.; Van der Ven, A. Phase Stability and Transport Mechanisms in Antiperovskite Li3OCl and Li3OBr Superionic Conductors. Chem. Mater. 2013, 25, 4663. [Google Scholar] [CrossRef]
- Alvin, S.; Cahyadi, H.S.; Hwang, J.; Chang, W.; Kwak, S.K.; Kim, J. Revealing the Intercalation Mechanisms of Lithium, Sodium, and Potassium in Hard Carbon. Adv. Energy Mater. 2020, 10, 2000283. [Google Scholar] [CrossRef]
- Cui, Y.; Abouimrane, A.; Lu, J.; Bolin, T.; Ren, Y.; Weng, W.; Sun, C.; Maroni, V.A.; Heald, S.M.; Amine, K. (De)Lithiation Mechanism of Li/SeSx (x = 0–7) Batteries Determined by in Situ Synchrotron X-ray Diffraction and X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2013, 135, 8047. [Google Scholar] [CrossRef]
- Henthorn, J.T.; Arias, R.J.; Koroidov, S.; Kroll, T.; Sokaras, D.; Bergmann, U.; Rees, D.C.; DeBeer, S. Localized Electronic Structure of Nitrogenase FeMoco Revealed by Selenium K-Edge High Resolution X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2019, 141, 13676. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Newville, M. Fundamentals of XAFS; Rev. 1.7; University of Chicago: Chicago, IL, USA, 2004. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorbunov, M.V.; Mikhailova, D. Structural Behaviour and Charge-Compensation Mechanism in Li2Fe1−xCoxSeO Solid Solutions during Reversible Delithiation. Processes 2024, 12, 756. https://doi.org/10.3390/pr12040756
Gorbunov MV, Mikhailova D. Structural Behaviour and Charge-Compensation Mechanism in Li2Fe1−xCoxSeO Solid Solutions during Reversible Delithiation. Processes. 2024; 12(4):756. https://doi.org/10.3390/pr12040756
Chicago/Turabian StyleGorbunov, Mikhail V., and Daria Mikhailova. 2024. "Structural Behaviour and Charge-Compensation Mechanism in Li2Fe1−xCoxSeO Solid Solutions during Reversible Delithiation" Processes 12, no. 4: 756. https://doi.org/10.3390/pr12040756
APA StyleGorbunov, M. V., & Mikhailova, D. (2024). Structural Behaviour and Charge-Compensation Mechanism in Li2Fe1−xCoxSeO Solid Solutions during Reversible Delithiation. Processes, 12(4), 756. https://doi.org/10.3390/pr12040756