Abstract
Hydrogen peroxide (H2O2) is an important oxidizing molecule that regulates the metabolisms of aerobic organisms. Redox signaling comprises physiological oxidative stress (eustress), while excessive oxidative stress causes damage to molecules. The main enzymatic generators of H2O2 are nicotinamide adenine dinucleotide phosphate oxidases or NADPH oxidases (NOXs) and mitochondrial respiratory chains, as well as various oxidases. The NOX family is constituted of seven enzyme isoforms that produce a superoxide anion (O2−), which can be converted to H2O2 by superoxide dismutase or spontaneously. H2O2 passes through the membranes by some aquaporins (AQPs), known as peroxyporins. It diffuses through cells and tissues to initiate cellular effects, such as proliferation, the recruitment of immune cells, and cell shape changes. Therefore, it has been proposed that H2O2 has the same importance as Ca2+ or adenosine triphosphate (ATP) to act as modulators in signaling and the metabolism. The present overview focuses on the metabolic processes of liver and adipose tissue, regulated by the H2O2 generated by NOXs.
1. Introduction
Living organisms that develop under aerobic conditions generate reactive oxygen species (ROS) by cellular metabolism [1,2,3]. ROS are formed during cellular processes, such as cellular respiration, NADPH oxidase (NOX) activity, and processes related to peroxisomes, nitric oxide synthetase, lipoxygenases, xanthine oxidase, cyclooxygenases, and the cytochrome P450 system, as well as inflammatory processes, mainly in hepatocytes and macrophages [1,2,4,5,6,7].
For a long time, ROS were considered as molecules or entities that could generate damage at different levels, be they cellular, tissue or even at the organ level, or that their production was linked to enzymatic reactions which involve oxygen intermediaries. Its production causes an imbalance in the oxide-reduction (redox) state of the cells. In phagocytes, ROS are produced by the activation of NOX to destroy pathogens during phagocytosis [8]. However, in the last 20 years, it has been shown that the generation of ROS is linked to a variety of essential biological processes, where they participate in the regulation of some metabolic pathways, either negatively or positively. These processes include cell development and differentiation, generation or crosslinking of the extracellular matrix, hormone biosynthesis, cell aging, apoptosis, the responses of oxygen changes (oxygen detection), growth factors, hormones, and cytokines.
2. NOX Family
NOXs represent a family of enzyme complexes present in the membrane which generate ROS. This complex is widely distributed in eukaryotic organisms [9], and recent results indicate that they are also present in bacteria [10]. The first NOX was described in neutrophils and produces a large amount of ROS as a defense system [11]. The catalytic subunit of this enzyme was named gp91phox and is now called NOX2. The development of more of sensitive assays allowed the detection of low amounts of ROS in other cells [12]. NOX1, also known as Mox1, was the first NOX2 homolog to be identified [13]. In mammals, the NOX family is formed by seven members: NOX1, 2, and 3, activated by cytoplasmic subunits; NOX4, which is constitutively active; and NOX5 and DUOX1 and 2, which bind calcium. The product of NOX isoforms is O2−, which rapidly changes to H2O2 [14].
2.1. Hydrogen Peroxide Production
NOX family members have different regulation processes, but they share a catalytic core with two domains: the dehydrogenase and the transmembrane domains [15]. Electrons are transferred from cytosolic NADPH to FAD in the dehydrogenase domain, then to the inner and outer hemes in the transmembrane domain, and finally to the O2 outside the cell, producing O2−, as shown in the following reaction:
NADPH + 2O2 ↔ NADP+ + 2O2− + H+
A superoxide is highly reactive, short-lived, and its charge prevents its diffusion through membranes [16]. However, it can dismutase to H2O2 spontaneously (rate constant = 8 × 104 M−1s−1) or by superoxide dismutase (SOD) (rate constant = 2 × 109 M−1s−1) [17]. H2O2 is diffusible, transverses membranes by aquaporins (AQPs), [18] and initiates cell signaling [19].
2.2. Tissue Distribution of NOX Proteins
NOXs localize in different tissues and membrane compartments, allowing the spatiotemporal organization of ROS signaling. The expression of NOX enzymes in various tissues supports the idea that ROS production is not exclusive to phagocytic cells. The fact that some of these enzymes produce low levels of ROS suggests that they may be participating in other cellular functions than the inflammatory response [20]. NOX2 is referred as phagocyte NADPH oxidase. However, it is also found in other cells such as hepatocytes, endothelial cells, smooth and skeletal muscle, neurons, cardiomyocytes, and hematopoietic stem cells. NOX1 and NOX2 share a 60% amino acid identity. NOX1 is expressed more in the colon epithelium, while NOX3 is almost exclusively expressed in the inner ear. NOX4 expression is strongly observed in the kidney and at moderate levels in vascular endothelial cells, vascular smooth muscle, murine osteoclasts, murine adipocytes, and the pancreas. NOX5 is expressed in the human spleen, testis, and vascular smooth muscle. The dual oxidases DUOX1 and DUOX2 are the H2O2-producing isoforms of the NADPH oxidases (DUOXs) are highly expressed in the thyroid, but they can also be expressed in other cells (see Table 1) [9,21,22,23,24,25].

Table 1.
Tissue distribution and subcellular localization of NOX isoforms.
2.3. Structure and Regulation of NOX
NOX isoform structures were deduced from studies on NOX2 [14]. There are structural properties of NOX2 that are similar to all members of the family. NOX complexes consist of one catalytic subunit and five regulatory subunits (p22phox, p47phox, p67phox, p40phox, and Rac). The catalytic subunit has six transmembrane domains (seven for DUOX1-2) and four hemes in the third and fifth transmembrane domains, as well as an NADPH-binding domain and an FAD-binding domain in the C-terminal in the cytosolic region. NOX5 has additional EF-hands and Ca2+-binding domains at the N-terminus, and DUOX enzymes have EF-hands and a peroxidase-like domain at the N-terminus. NOX2 forms a complex membrane with a stabilizing factor, p22phox [26]. In unstimulated cells, NOX2 and p22phox are found in the plasma membrane, with p67phox associating with the proline-rich region (PRR) of the p47phox in the cytosol. The p47phox has two Src homology 3 (SH3) domains for its association with the PRR of the p22phox [27]. In the resting state, the auto inhibitory region AIR in the p47phox combines with the bis-SH3 domain and the domain phagocyte oxidase (PX) of the p47phox, preventing its binding to p22phox and membrane lipids [28]. The cytoplasmic component p40phox is associated with p67phox. Rac attached to guanosine 5′-diphosphate (GDP) is associated with RhoGDP dissociation inhibitor (RhoGDI) in the cytosol. Rac is a member of the Rho family, and there are three expressed isoforms of Rac: Rac1, Rac2, and Rac3. Rac1 is expressed in non-hematopoietic cells and activates many NOX isoforms. In contrast, Rac2 is expressed in hematopoietic cells, and Rac3 is predominantly found in the central nervous system [29,30]. The phagocytosis of bacteria induces phosphorylation in p47phox and conformational change, allowing its interaction with p22phox [14,27]. Subunit p47phox, attached to the membrane, transports the activator subunits p67phox and p40phox to NOX2 [31]. Phagocytosis also causes the generation of Rac2-GTP, which is released from the RhoGDI. Rac-GTP goes toward the membrane and interacts with NOX2 and p67phox [32,33]. The assembled complex generates superoxide in the extracellular space (Figure 1).
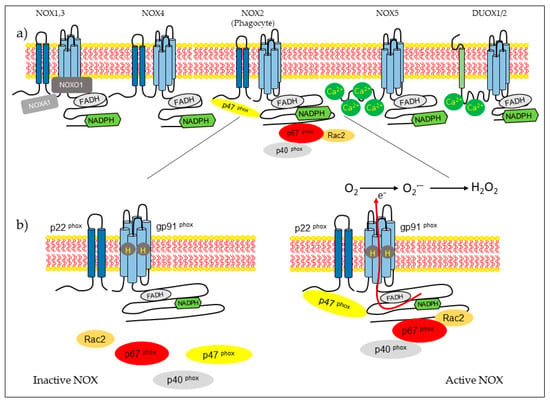
Figure 1.
NADPH oxidase (NOX) types and activation of the NOX2 macrophage for the formation of reactive oxygen species. (a) Structure of the isoforms that are part of the NOX family. (b) Activation of the NOX2 system, which occurs by the assembly of cytosolic regulatory proteins (p40phox, p47phox, p67phox, and Rac2) with the catalytic subunit gp91phox and p22phox in the membrane, to activate the enzyme complex and generate superoxide anions (O2−) and hydrogen peroxide (H2O2).
The other isoforms share some characteristics of NOX2 activation. The NOX1 isoform exhibits 60% of its identity with NOX2. Its activity depends on cytosolic subunits. NOX1 requires p22phox, Rac GTPase, the first organizer (NOXO1), which is a homolog of p47phox, and the first activator (NOXA1), a homolog of p67phox [34]. NOX3 shares 56% of its amino acid identity with NOX2 [35]. This NOX requires NOXO1 and NOXA1 for activation, but p47phox, p67phox, and Rac have not been studied [36]. NOX4 shares approximately 39% of it sequence homology with NOX2. This isoform is constitutively active, and its activity depends on p22phox [37]. Recently, it has been described that polymerase δ-interacting protein 2 (POLDIP2) regulates NOX4 [38]. NOX5 comprises five isoforms: a, b, c, d, and e or S [39,40]. NOX5 a–d isoforms have an intracellular domain that contains a Ca+2 -binding EF-hand region [41]. NOX5 a–d isoforms do not require p22phox or cytosolic subunits for activation, but they need cytosolic calcium. Heat shock protein 90 (HSP90) has been described as a regulatory protein for NOX1–3 and NOX5 [42]. The NOX5 e isoform lacks a Ca2+-binding domain and depends on the cAMP response element-binding protein for activity [43]. DUOX1 and DUOX2 were first identified in the thyroid gland. These enzymes share 50% of their amino acid identity with NOX2 [44]. They have a NOX domain similar to that of NOX1-4, an EF-hand region similar to NOX5, and a seventh transmembrane peroxidase-like domain. Both dual oxidases have been identified in the plasma membrane and endoplasmic reticulum [45]. The permanence of DUOX proteins in the endoplasmic reticulum depends on the maturation factors of DUOXA1 and DUOXA2. The DUOX2 immature form generates O2−, whereas the mature form generates H2O2 [46]. DUOX proteins do not need a cytosolic activator or organizer subunits for activation, but they do require calcium [47].
2.4. The Role of NOXs in Human Physiology and Pathophysiology
NOX proteins regulate physiological processes such as host defense, cell growth, differentiation, metabolism, and apoptosis [9,21,24]. The deregulation or absence of some NOXs has been linked to a variety of diseases. The earliest discovery was chronic granulomatous disease (CGD), caused by the absence of active NOX2. Patients with CGD exhibit chronic infections, impaired wound healing, and cognitive deficits [48,49].
NOX1 is strongly expressed in the colon epithelium, where its physiological role is controversial, and its participation in immune system defense and cell proliferation has been proposed. It has also been shown to be present in vascular smooth muscle cells, where it regulates its growth and migration [24]. NOX1 can be also found in cells of the central nervous system (CNS). NOX1-deficient mice exhibit a reduction in pain sensitivity accompanied by inflammation (hyperalgesia). It has been suggested to have a role in the defense of the CNS in microglia, while it has been implicated in the growth of neurites [50]. More recently, NOX1 has been associated with processes related to cancer through participation in the control of the cell cycle, in particular of the cyclin D1. NOX1 overexpression in the epithelial cells of the lung induces an increase in the expression and proliferation of cyclin D1. On the other hand, a correlation between Ras mutations and overexpressed NOX1 was shown in studies of human colon cancer [24].
NOX2 is known to be essential in innate defense of the guest, as well as being protective against colon inflammation, as CGD patients exhibit non-infective colitis [51]. Contrarily, NOX2 regulates the normal renal blood flow and enhances skeletal muscle metabolism and insulin sensitivity [52,53]. Overactivation of NOX1 and NOX2 is involved in developing hypertension [54,55] and inflammation [56].
NOX3 participates in the formation of the inner ear [57]. NOX3 and NOX5 induce the differentiation of human oligodendrocytes [58]. Moreover, NOX3 mutant mice showed motor, coordination, orientation, and behavior disorders. The degree of specialization of NOX3 and its importance in physiological processes of the nervous system stand out. It has also been shown that NOX3 is of functional importance in the endothelial cells of the lung. In mice, where NOX3 activity increases, emphysema develops, though so far the involved mechanism is unknown. Another line of study has demonstrated a new role for NOX3 in insulin resistance. In a diabetic mouse model, an increase of NOX3 expression was shown, as well as the generation of ROS in the liver [24].
NOX4, after NOX2, constitutes the homolog with the highest expression, compared with other NOX homologues. It has been associated with the expression of oxygen-dependent genes in the kidney. NOX4 has also been involved in other physiological processes, including cell senescence, apoptosis, survival, insulin signaling, migration, and differentiation [9]. It has also been shown that NOX4 is responsible for the production of O2− in osteoclasts, participating in the process of bone resorption. Additionally, it was recently found that NOX4 participates in adipocyte differentiation [59], insulin sensitivity in the liver and adipose tissue [60], and glucose-stimulated insulin secretion [61]. This homologue has also been found in smooth muscle cells, endothelial cells, fibroblasts, keratinocytes, neurons, and hepatocytes. In vascular smooth muscle cells (VSMCs), insulin-like growth factor I (IGF-I) induces NOX4-mediated migration by an unknown mechanism. NOX4 regulates growth and survival in VSMCs treated with urokinase, plasminogen activators, or transforming growth factor beta (TGF-β). Studies conducted with VSMCs, fibroblasts, adipocytes, and embryonic stem cells showed that ROS production by NOX4 promotes differentiation [24], angiogenesis, vasodilation [62], and apoptosis [63], and it protects the vasculature from ischemic and inflammatory stress [64].
NOX5 regulates cell signaling and functions, and it contributes to sperm motility and viability [65], as well as vascular smooth muscle cell contraction [66]. Several authors have proposed that NOX5 plays a role in cell proliferation. For example, in humans, VSMC platelet derived growth factor (PDGF)-induced proliferation is mediated by NOX5. This coincides with the fact that NOX5 is expressed in various cancer cell lines. In this sense, NOX5 has been implicated in the regulation of prostate cancer and in Barrett’s esophagus adenocarcinoma. Moreover, it has been proposed that platelet-activating factor (PAF) induces the expression of NOX5 in these cells. In this context, it has been suggested that NOX5 is involved in the malignancy of malignant mature Β cells, since this complex is not expressed in normal B cells [24].
DUOX enzymes were isolated originally in thyroid thyrocytes, and they are associated with the production of H2O2 used for the oxidation of iodide during the synthesis of thyroid hormones. In the lungs and salivary glands, DUOX provides H2O2 to lactoperoxidase that converts thiocyanate anions into a hypothiocyanate oxidant with microbicidal functions, which gives it an essential role in defending the guest [9]. DUOX1 and 2 deregulations have been associated with hypothyroidism [67]. DUOXs also play a role in host defense in the gastrointestinal tract and the lung epithelium [68].
Most of the functions of NOXs in humans are not yet understood. However, NOXs have also been associated with the pathogenesis of rheumatoid arthritis [69], diabetes, atherosclerosis [70], and several neurological diseases including Alzheimer’s [71] and Parkinson’s diseases [72].
2.5. Effect of NOX-Derived H2O2 on the Tissues
2.5.1. Liver
NOXs are widely distributed in the whole liver. The main isoforms in this organ are NOX1, NOX2, and NOX4 [73]. NOX2 expression has been suggested for a long time in this gland, in murine Kupffer cells [74], rat hepatocytes [75], endothelial cells [76], and in hepatic stellate cells (HSCs) [77]. NOX1 is expressed in sinusoidal endothelial cells, hepatocytes, and HSCs. While NOX4 is expressed in hepatocytes, Kupffer cells, and culture-activated rat HSCs [77]. Finally, DUOX1 and DUOX2 are expressed in rat hepatocytes, Kupffer cells, and either activated or non-activated HSCs [78]. The functions of NOXs in the liver are not well studied. The role of NOX2 in Kupffer cells should namely be the killing of microorganisms. ROS have been suggested to play an essential role in clearing pathogens derived from the gut [79], regulating expression of the CD95 ligand [80], and NFκB-dependent production of tumor necrosis factor alpha (TNF-α) [81]. In addition, ROS may intervene in hepatic fibrosis and may directly intoxicate hepatocytes [82]. Hepatic stellate cells are involved in liver development and regeneration, the production of cytokines, retinoid metabolism, and secretion of lipoproteins and growth factors, but the physiological role of NOXs has not been studied [77]. Hepatocytes contain many ROS-generating systems, including cytochromes, mitochondrial respiration, arachidonic acid oxidation, and NOXs. The generation of ROS through an NOX has been described in response to many stimuli like adrenaline [83,84,85], CD95 ligand [78], TGF-β [86], and bile production in hepatocytes [87]. Adrenaline activates the NOX2 enzyme, localized in hepatocyte plasma membranes through the α1-adrenoreceptors (α1-AR). This activation requires GTP, but does not require adenosine triphosphate (ATP). The NOX2 complex present in the hepatocyte plasma membrane contains gp91phox, p22phox, p40phox, p47phox, p67phox, and Rac1 proteins. The Gα13 protein links α1-AR and the NOX complex in this cell. α1-AR stimulation induces Rac1-GTP generation, which is necessary for H2O2 synthesis. Negative cross-talk between α1-ARs and β-ARs for H2O2 synthesis, gluconeogenesis, and ureagenesis was observed in these cells [83,84]. AQP8 is necessary for H2O2 to enter the hepatocyte and carry out its function [85]. Based on previous work [88], H2O2 generated after NOX2 activation by α1-AR in hepatocytes may react with cAMP-dependent protein kinase A (PKA) and inhibit gluconeogenesis and ureagenesis processes (Figure 2).
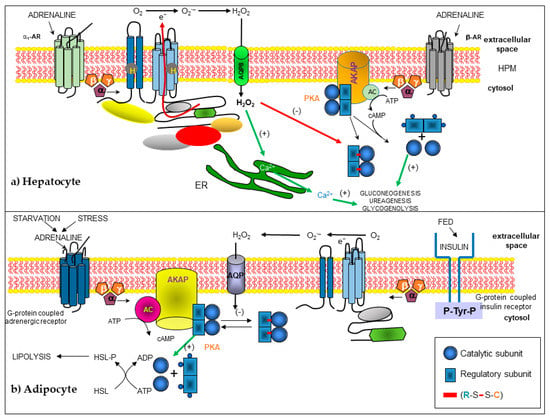
Figure 2.
Modified schemes from [85] and [89], in which the signaling and cross-linking pathways are presented and where H2O2 participates as a second messenger, generated by the activation of an NOX. H2O2 enters the cell through an aquaporin that modifies the redox state of protein kinase A (PKA), producing a disulfide (-) bond between the regulatory and catalytic subunits that alters the activation and function of the holoenzyme PKA by cAMP. (a) In the hepatocyte, the activation of α1 adrenergic receptors (α1-AR) inhibits the activation of PKA. Reproduced with permission from Magdalena Vilchis-Landeros, Raquel Guinzberg, Héctor Riveros-Rosas et al., and FEBS Letters, and published by John Wiley and Sons, 2020. (b) In the adipocyte, the lipolysis pathway is inhibited by the oxidation of PKA, due to the generation of H2O2, stimulated by insulin.
Hepatocyte-derived ROS might lead to apoptosis, but ROS generated by neighboring cells might cause hepatocyte proliferation [78]. The CD95 ligand, hyperosmotic exposure, or hydrophobic bile salts in primary cultured hepatocytes induce apoptosis by the activation of NOX isoforms [78,90]. Furthermore, in FaO hepatoma cells, apoptosis promoted by TGF-β increases NOX1 and NOX4 expression [91,92]. In most recent studies, authors reported the role of NOX1 as a controller of the autocrine cell growth of FaO hepatoma cells through regulation of the epidermal growth factor receptor (EGFR) pathway [93].
Regardless of their physiological function, NOXs in the liver may participate in different pathological processes by chronic production of high ROS levels in hyperinsulinemia and hyperglycemia [94]. More recently, it has been reported that a critical role of NOX3-derived ROS is palmitate-induced insulin resistance in hepatocytes [95]. NOX also plays a role in hepatic cancer in many rodent models [96,97]. Increased NOX activity has been implicated in nonalcoholic fatty liver disease [98]. In addition, NOX2 might be necessary for liver ischemia–reperfusion injury because deficient mice in this isoform show reduced hepatic reperfusion injury [99,100]. On the other hand, much evidence supports the role of NOX in tissue inflammation and fibrosis. The role of increased oxidative stress in the pathogenesis of liver fibrosis is based on studies using clinical samples and experimental animal models of liver fibrosis [101,102]. NOX4 expression is elevated in a murine model of steatosis-induced fibrosis. In this model, hepatocyte-specific deletion of NOX4 results in lower oxidative stress, reduced lipid peroxidation, and the attenuation of liver fibrosis [103]. NOX4-derived ROS has also been implicated in the transdifferentiation and proliferation of HSCs [102]. NOX2-/- mice are protected against CCl4−-induced liver fibrosis [101]. These studies support the role of NOX2 in regulating both the expression of matrix metalloproteinase-2 and -9 (MMP-2 and MMP-9) and the expression of the MMP tissue inhibitors (TIMP-1 and TIMP-2). NOX2 has been shown to mediate the activation of HSCs into myofibroblasts after the phagocytosis of dying hepatocytes [104]. NOX1 is also expressed and activated in HSCs, and NOX1 knockout mice are also protected against CCl4−induced liver fibrosis [105]. NOX1 has also been involved in the transdifferentiation and proliferation of HSCs, similar to NOX2 and NOX4. In vitro studies using a genetic approach to delete NOX5 in HSCs have also suggested that NOX5 may be involved in the activation of HSCs through the modulation of p38 mitogen activated protein kinase (MAPK) [106].
2.5.2. Adipose Tissue and Other Tissues
ROS have been increasingly recognized for their role in the regulation of intracellular signaling pathways [107,108,109]. H2O2 is particularly suitable for redox signaling because it has a longer half-life than other ROS, which allows it to diffuse longer distances in cells [110]. In the international literature, there are a large number of reports that verify the effect of H2O2 on the regulation of enzymatic activity and how it affects the response of one or more metabolic pathways [111,112,113,114,115,116]. For this process to be carried out, it is necessary that the structural conditions exist within the proteins or enzymes, in addition to a temporary increase in the concentration of H2O2, so that this ROS can act and modify the redox state of the enzymes [117,118]. H2O2 is a weak oxidant that reacts primarily with the thiol groups of the cysteines of various proteins. Four oxidation states of cysteines can be generated: disulfide (-S-S), sulfenic acid (-SOH), sulfinic acid (-O2H), and sulfonic acid (-SO3H). Very strong oxidants, such as vanadate, are required for the generation of the last two oxidized states of thiols, and their formation is irreversible under physiological conditions. In contrast, H2O2 oxidizes the SH groups of cysteines to sulfenic acid, which is rapidly reduced by various intracellular reducing agents, including glutathione (GSH) and thioredoxin (Trx). However, cysteine residues are only a good target for the oxidizing action of H2O2 if they are deprotonated and exist as a thiolate anion (-S-) [119].
There are several examples where the activity of different enzymes is regulated or affected by the presence of H2O2. A clear example is the protein tyrosine phosphatases (PTPs), including PTP1B, which counteract the phosphorylation of protein tyrosine and, therefore, act together with the tyrosine kinase receptor (TKR) to regulate cell signaling. PTP activity is dependent on a cysteine (Cys) residue in the conserved active site, which renders these enzymes susceptible to oxidative inactivation. During stimulation of the platelet-derived growth factor receptor and epidermal growth factor (EGF) pathways, the catalytic Cys residue of PTP1B is reversibly oxidized and thus inhibited. The product of the initial oxidation is a sulfenic acid, which can be condensed to an internal sulfenilamide or undergo glutathionylation. Reversibly oxidized PTP1B can be reactivated by the thioredoxin (Trx) system, allowing the Trx system to modulate cellular TKR signaling [120].
Another relevant example is the one observed in the activation of the transcription factor NF-κB by H2O2, which is produced endogenously by the activation of protein kinase C [121]. On the other hand, it was demonstrated that the increase in tyrosine phosphorylation of many proteins and the activation of the extracellular signal-regulated kinase (ERK) pathway by the activators of respiratory bursts in macrophages depended on H2O2 [122].
In 2006, Brennan et al. [123] showed that the regulatory subunits of PKA type I (RI) have thiol groups that detect the cellular redox state, since high concentrations of H2O2 cause the 2 RI subunits to form disulfide bridges, which had been previously described to be constitutively formed, even when purified, but may be formed by the oxidizing conditions of the process. The formation of disulfide bridges is associated with the translocation of PKA from the cytoplasm to the nucleus or to the myofilament compartment. This redistribution is accompanied by the phosphorylation of PKA substrates, which include troponin I, myocin-linked protein C, and phospholamban. This phosphorylation is sensitive to H89, an inhibitor of PKA, demonstrating that phosphorylation is mediated by PKA. Lower concentrations of H2O2 are required to activate type I PKA than those necessary to activate type II PKA.
Increased H2O2 contraction is mediated by an adrenergic discharge that increases cAMP levels, activating PKA. This leads us to think that a treatment with H2O2 can increase contraction, although previous studies showed that H2O2 decreases contraction. However, these studies were carried out at very high concentrations of H2O2 or for very long periods of time [123].
An emblematic example that has been studied since the late 1970s is that an increase in the intracellular H2O2 concentration can mimic the effects of insulin in adipose tissue, including the inhibition of epinephrine-stimulated lipolysis [118,124,125]. Another described effect related to insulin is that it activates NOX in adipose tissue, which transiently increases the cellular H2O2 concentration [126].
Years later, new reports expanded the information on how this pathway is regulated; that is, H2O2 is produced by NOX4, which is characteristic of adipocytes, during the physiological transduction of insulin in adipose cells [127]. In 2001, Mahadev et al. demonstrated that insulin causes the rapid formation of H2O2 in 3T3-L1 adipocytes, a redox signal that enhances the early cascade of insulin-stimulated tyrosine phosphorylation through reversible oxidative inactivation of the protein tyrosine phosphatase thiol dependent (PTPase) 1B and other enzymes [128,129], which leads to an understanding of the regulatory mechanism that complements the first steps in insulin amplification signaling. A more recent report on insulin signaling through H2O2 during lipolysis showed that H2O2, whether generated by insulin or added, reversibly inhibited lipolysis rates activated by epinephrine or Bt2cAMP [88]. This effect was produced through H2O2-mediated oxidation of two sulfhydryl groups of the PKA holoenzyme: Cys 97 of the regulatory IIα or IIβ subunits and Cys 199 of the catalytic α subunit, which formed a disulfide bond that inhibited holoenzyme activation by cAMP, thus avoiding PKA-stimulated lipolysis (Figure 2b). This information, together with the inhibition of lipolysis stimulated by non-steroidal anti-inflammatory drugs (NSAIDs), led us to propose that H2O2, generated by the activation of NOX4 and located in the plasma membrane of adipocytes by NSAIDs, was the molecule that acted as an agent that mimicked the activity of insulin. This effect is reflected in the modification and inhibition of PKA type II in rat adipocytes [89].
In addition, the production of ROS in adipose tissue, especially H2O2, is physiologically relevant in the response to insulin not only at lipolysis level, but also by exerting a protective antihyperglycemic action through adipocyte differentiation and the uptake of glucose by differentiated adipocytes. However, if there is a sustained excess of glucose in the body’s metabolic balance, NOX activity will contribute to the development of obesity [130].
3. Conclusions
Cellular processes are regulated by different factors, both external and internal. ROS generated by changes in the environment and cell metabolism itself, along with antioxidant defenses, directly influence cell homeostasis. An important component within this regulation is NOX, which generates the production of ROS in a controlled manner and participates in the regulation of different cellular components.
NOX2 is the most studied enzyme, since it is found in phagocytes and involved in the elimination of invading agents such as bacteria. However, the other components of the NOX family are of great interest because they regulate different metabolic pathways in tissues and organs, activating or inhibiting key enzymes to regulate metabolic pathways that have importance within organisms and enhance the effects of hormones like insulin. On the other hand, it is important to mention that NOX, in turn, is regulated by different processes such as phosphorylation (NOX2) or the presence of calcium in the medium. For this reason, they can be activated (NOX5, DUOX1 or 2). NOX4 can be also found which, apparently, is constitutively active, so it does not need cytosolic subunits and is ready to generate ROS when activated by some hormone.
All of this means that the study of these enzymes is increasing, since many metabolic processes are regulated, either positively or negatively, by the metabolites generated by NOXs. As can be seen in this review, there are many cellular processes that are regulated by the ROS generated by NOX in the liver and adipose tissue. However, the target proteins of this redox mechanism have yet to be defined in many of them. We have shown that, in the case of some metabolic pathways in the liver such as gluconeogenesis, glycogenolysis, ureogenesis, and lipolysis in the adipocyte, the target protein of H2O2, generated by NOX2 and NOX4, can be PKA, which is an important enzyme in all of these metabolic pathways, thus indicating them as possible pharmacological targets to regulate different pathological processes in which they participate (e.g., inflammation, cell signaling, and immune response, among others). There is still a long way to go to understand the several roles that ROS perform in different cellular settings, such as signaling mechanisms, the regulation of major metabolic pathways and the cell cycle, immune defense, and both programmed and non-programmed death processes, as well as the target proteins of ROS in these processes. However, the prospects in this regard are encouraging, thanks to the large number of new techniques involved in the study of these byproducts of the aerobic metabolism and the effort and investment of different research groups at an international level. Therefore, the study of the regulation of metabolic processes by H2O2, generated by NOX, represents a very broad field of study to understand and realize the importance of these enzymes within cells.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Osorio Alves, J.; Matta Pereira, L.; Cabral Coutinho do Rego Monteiro, I.; Pontes Dos Santos, L.H.; Soares Marreiros Ferraz, A.; Carneiro Loureiro, A.C.; Calado Lima, C.; Leal-Cardoso, J.H.; Pires Carvalho, D.; Soares Fortunato, R.; et al. Strenuous acute exercise induces slow and fast twitch-dependent NADPH oxidase expression in rat skeletal muscle. Antioxidants (Basel) 2020, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Breton-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Sulciner, D.J.; Gutkind, J.S.; Irani, K.; Goldschmidt-Clermont, P.J.; Finkel, T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 1996, 318 Pt 2, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, C.; Cherrier, M.V.; Dias Mirandela, G.; Petit-Hartlein, I.; Stasia, M.J.; Fontecilla-Camps, J.C.; Fieschi, F.; Dupuy, J. The NOX family of proteins is also present in bacteria. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Lambeth, J.D.; Nauseef, W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Cheng, G.; Arnold, R.S.; Edens, W.A. Novel homologs of gp91phox. Trends Biochem. Sci. 2000, 25, 459–461. [Google Scholar] [CrossRef]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Magnani, F.; Nenci, S.; Millana Fananas, E.; Ceccon, M.; Romero, E.; Fraaije, M.W.; Mattevi, A. Crystal structures and atomic model of NADPH oxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 6764–6769. [Google Scholar] [CrossRef]
- Pryor, W.A. Oxy-radicals and related species: Their formation, lifetimes, and reactions. Annu. Rev. Physiol. 1986, 48, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE 2000, 2000, pe1. [Google Scholar] [CrossRef]
- Krause, K.H. Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn. J. Infect. Dis. 2004, 57, S28–S29. [Google Scholar]
- Bedard, K.; Lardy, B.; Krause, K.H. NOX family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Buvelot, H.; Jaquet, V.; Krause, K.H. Mammalian NADPH oxidases. Methods Mol. Biol. 2019, 1982, 17–36. [Google Scholar] [PubMed]
- Katsuyama, M.; Matsuno, K.; Yabe-Nishimura, C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J. Clin. Biochem. Nutr. 2012, 50, 9–22. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef]
- Al Ghouleh, I.; Khoo, N.K.; Knaus, U.G.; Griendling, K.K.; Touyz, R.M.; Thannickal, V.J.; Barchowsky, A.; Nauseef, W.M.; Kelley, E.E.; Bauer, P.M.; et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011, 51, 1271–1288. [Google Scholar] [CrossRef]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef]
- Groemping, Y.; Lapouge, K.; Smerdon, S.J.; Rittinger, K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 2003, 113, 343–355. [Google Scholar] [CrossRef]
- Hiroaki, H.; Ago, T.; Ito, T.; Sumimoto, H.; Kohda, D. Solution structure of the PX domain, a target of the SH3 domain. Nat. Struct. Biol. 2001, 8, 526–530. [Google Scholar] [CrossRef]
- Bokoch, G.M. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005, 15, 163–171. [Google Scholar] [CrossRef]
- Mizuno, T.; Kaibuchi, K.; Ando, S.; Musha, T.; Hiraoka, K.; Takaishi, K.; Asada, M.; Nunoi, H.; Matsuda, I.; Takai, Y. Regulation of the superoxide-generating NADPH oxidase by a small GTP-binding protein and its stimulatory and inhibitory GDP/GTP exchange proteins. J. Biol. Chem. 1992, 267, 10215–10218. [Google Scholar]
- Han, C.H.; Freeman, J.L.; Lee, T.; Motalebi, S.A.; Lambeth, J.D. Regulation of the neutrophil respiratory burst oxidase. Identification of an activation domain in p67(phox). J. Biol. Chem. 1998, 273, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- Diebold, B.A.; Bokoch, G.M. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2001, 2, 211–215. [Google Scholar] [CrossRef]
- Lapouge, K.; Smith, S.J.; Walker, P.A.; Gamblin, S.J.; Smerdon, S.J.; Rittinger, K. Structure of the TPR domain of p67phox in complex with Rac.GTP. Mol. Cell 2000, 6, 899–907. [Google Scholar] [CrossRef]
- Geiszt, M.; Lekstrom, K.; Witta, J.; Leto, T.L. Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J. Biol. Chem. 2003, 278, 20006–20012. [Google Scholar] [CrossRef]
- Ueno, N.; Takeya, R.; Miyano, K.; Kikuchi, H.; Sumimoto, H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: Its regulation by oxidase organizers and activators. J. Biol. Chem. 2005, 280, 23328–23339. [Google Scholar] [CrossRef]
- Ueyama, T.; Geiszt, M.; Leto, T.L. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol. Cell Biol. 2006, 26, 2160–2174. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassegue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef]
- Banfi, B.; Molnar, G.; Maturana, A.; Steger, K.; Hegedus, B.; Demaurex, N.; Krause, K.H. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Banfi, B.; Tirone, F.; Durussel, I.; Knisz, J.; Moskwa, P.; Molnar, G.Z.; Krause, K.H.; Cox, J.A. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). J. Biol. Chem. 2004, 279, 18583–18591. [Google Scholar] [CrossRef]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal 2011, 14, 2107–2119. [Google Scholar] [CrossRef]
- Fu, X.; Beer, D.G.; Behar, J.; Wands, J.; Lambeth, D.; Cao, W. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J. Biol. Chem. 2006, 281, 20368–20382. [Google Scholar] [CrossRef]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noel-Hudson, M.S.; Deme, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef]
- De Deken, X.; Wang, D.; Dumont, J.E.; Miot, F. Characterization of ThOX proteins as components of the thyroid H(2)O(2)-generating system. Exp. Cell Res. 2002, 273, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Sugawara, Y.; Wen, K.; Giulivi, C. Generation of oxygen free radicals in thyroid cells and inhibition of thyroid peroxidase. Exp. Biol Med. (Maywood) 2002, 227, 141–146. [Google Scholar] [CrossRef]
- Ameziane-El-Hassani, R.; Morand, S.; Boucher, J.L.; Frapart, Y.M.; Apostolou, D.; Agnandji, D.; Gnidehou, S.; Ohayon, R.; Noel-Hudson, M.S.; Francon, J.; et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J. Biol. Chem. 2005, 280, 30046–30054. [Google Scholar] [CrossRef]
- Kishida, K.T.; Hoeffer, C.A.; Hu, D.; Pao, M.; Holland, S.M.; Klann, E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell Biol. 2006, 26, 5908–5920. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef]
- Arimura, Y.; Goto, A.; Yamashita, K.; Endo, T.; Ikeda, H.; Tanaka, K.; Tsutsumi, H.; Shinomura, Y.; Imai, K. Intractable colitis associated with chronic granulomatous disease. J. Med. Microbiol. 2006, 55, 1587–1590. [Google Scholar] [CrossRef]
- Haque, M.Z.; Majid, D.S. Assessment of renal functional phenotype in mice lacking gp91PHOX subunit of NAD(P)H oxidase. Hypertension 2004, 43, 335–340. [Google Scholar] [CrossRef]
- Henriquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 2019, 10, 4623. [Google Scholar] [CrossRef]
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef]
- Fukui, T.; Ishizaka, N.; Rajagopalan, S.; Laursen, J.B.; Capers, Q.t.; Taylor, W.R.; Harrison, D.G.; de Leon, H.; Wilcox, J.N.; Griendling, K.K. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 1997, 80, 45–51. [Google Scholar] [CrossRef]
- Rokutan, K.; Kawahara, T.; Kuwano, Y.; Tominaga, K.; Nishida, K.; Teshima-Kondo, S. Nox enzymes and oxidative stress in the immunopathology of the gastrointestinal tract. Semin. Immunopathol. 2008, 30, 315–327. [Google Scholar] [CrossRef]
- Paffenholz, R.; Bergstrom, R.A.; Pasutto, F.; Wabnitz, P.; Munroe, R.J.; Jagla, W.; Heinzmann, U.; Marquardt, A.; Bareiss, A.; Laufs, J.; et al. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004, 18, 486–491. [Google Scholar] [CrossRef]
- Accetta, R.; Damiano, S.; Morano, A.; Mondola, P.; Paterno, R.; Avvedimento, E.V.; Santillo, M. Reactive oxygen species derived from NOX3 and NOX5 drive differentiation of human oligodendrocytes. Front. Cell Neurosci. 2016, 10, 146. [Google Scholar] [CrossRef]
- Schroder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 239–245. [Google Scholar] [CrossRef]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef]
- Plecita-Hlavata, L.; Jaburek, M.; Holendova, B.; Tauber, J.; Pavluch, V.; Berkova, Z.; Cahova, M.; Schroder, K.; Brandes, R.P.; Siemen, D.; et al. Glucose-stimulated insulin secretion fundamentally requires H2O2 signaling by NADPH Oxidase 4. Diabetes 2020, 69, 1341–1354. [Google Scholar] [CrossRef]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target. 2015, 23, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Kennedy, K.; Chew, P.; Okabe, J.; El-Osta, A.; Calkin, A.C.; Biessen, E.A.; Touyz, R.M.; Cooper, M.E.; et al. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 295–307. [Google Scholar] [CrossRef]
- Ghanbari, H.; Keshtgar, S.; Kazeroni, M. Inhibition of the CatSper channel and NOX5 enzyme activity affects the functions of the progesterone-stimulated human sperm. Iran. J. Med. Sci. 2018, 43, 18–25. [Google Scholar]
- Montezano, A.C.; De Lucca Camargo, L.; Persson, P.; Rios, F.J.; Harvey, A.P.; Anagnostopoulou, A.; Palacios, R.; Gandara, A.C.P.; Alves-Lopes, R.; Neves, K.B.; et al. NADPH Oxidase 5 is a pro-contractile nox isoform and a point of cross-talk for calcium and redox signaling-implications in vascular function. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.; van Trotsenburg, A.S.; Baas, F.; de Vijlder, J.J.; Vulsma, T.; Ris-Stalpers, C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef]
- Knaus, U.G. Oxidants in physiological processes. Handb. Exp. Pharmacol. 2020. [CrossRef]
- Liu, E.; Perl, A. Pathogenesis and treatment of autoimmune rheumatic diseases. Curr. Opin. Rheumatol. 2019, 31, 307–315. [Google Scholar] [CrossRef]
- Laddha, A.P.; Kulkarni, Y.A. NADPH oxidase: A membrane-bound enzyme and its inhibitors in diabetic complications. Eur. J. Pharmacol. 2020, 881, 173206. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9 (Suppl. 2), S8. [Google Scholar] [CrossRef]
- Wu, D.C.; Teismann, P.; Tieu, K.; Vila, M.; Jackson-Lewis, V.; Ischiropoulos, H.; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 6145–6150. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Torok, N.J. NADPH oxidases in chronic liver diseases. Adv. Hepatol. 2014, 2014. [Google Scholar] [CrossRef]
- Ding, A.; Nathan, C. Analysis of the nonfunctional respiratory burst in murine Kupffer cells. J. Exp. Med. 1988, 167, 1154–1170. [Google Scholar] [CrossRef]
- Mizukami, Y.; Matsubara, F.; Matsukawa, S.; Izumi, R. Cytochemical localization of glutaraldehyde-resistant NAD(P)H-oxidase in rat hepatocytes. Histochemistry 1983, 79, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; O’Donnell, V.B.; Wood, J.D.; Broughton, J.P.; Hughes, E.J.; Jones, O.T. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am. J. Physiol. 1996, 271, H1626–H1634. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Reinehr, R.; Becker, S.; Eberle, A.; Grether-Beck, S.; Haussinger, D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J. Biol. Chem. 2005, 280, 27179–27194. [Google Scholar] [CrossRef]
- LaCourse, R.; Ryan, L.; North, R.J. Expression of NADPH oxidase-dependent resistance to listeriosis in mice occurs during the first 6 to 12 h of liver infection. Infect. Immun. 2002, 70, 7179–7181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uchikura, K.; Wada, T.; Hoshino, S.; Nagakawa, Y.; Aiko, T.; Bulkley, G.B.; Klein, A.S.; Sun, Z. Lipopolysaccharides induced increases in Fas ligand expression by Kupffer cells via mechanisms dependent on reactive oxygen species. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G620–G626. [Google Scholar] [CrossRef]
- Rose, M.L.; Rusyn, I.; Bojes, H.K.; Belyea, J.; Cattley, R.C.; Thurman, R.G. Role of Kupffer cells and oxidants in signaling peroxisome proliferator-induced hepatocyte proliferation. Mutat. Res. 2000, 448, 179–192. [Google Scholar] [CrossRef]
- McKim, S.E.; Gabele, E.; Isayama, F.; Lambert, J.C.; Tucker, L.M.; Wheeler, M.D.; Connor, H.D.; Mason, R.P.; Doll, M.A.; Hein, D.W.; et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: Studies with knockout mice. Gastroenterology 2003, 125, 1834–1844. [Google Scholar] [CrossRef]
- Diaz-Cruz, A.; Guinzberg, R.; Guerra, R.; Vilchis, M.; Carrasco, D.; Garcia-Vazquez, F.J.; Pina, E. Adrenaline stimulates H2O2 generation in liver via NADPH oxidase. Free Radic. Res. 2007, 41, 663–672. [Google Scholar] [CrossRef]
- Diaz-Cruz, A.; Vilchis-Landeros, M.M.; Guinzberg, R.; Villalobos-Molina, R.; Pina, E. NOX2 activated by alpha1-adrenoceptors modulates hepatic metabolic routes stimulated by beta-adrenoceptors. Free Radic. Res. 2011, 45, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Vilchis-Landeros, M.; Guinzberg, R.; Riveros-Rosas, H.; Villalobos-Molina, R.; Pina, E. Aquaporin 8 is involved in H2O2-mediated differential regulation of metabolic signaling by alpha1- and beta-adrenoceptors in hepatocytes. FEBS Lett. 2020, 594, 1564–1576. [Google Scholar] [CrossRef]
- Herrera, B.; Murillo, M.M.; Alvarez-Barrientos, A.; Beltran, J.; Fernandez, M.; Fabregat, I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic. Biol. Med. 2004, 36, 16–26. [Google Scholar] [CrossRef]
- Roma, M.G.; Sanchez Pozzi, E.J. Oxidative stress: A radical way to stop making bile. Ann. Hepatol. 2008, 7, 16–33. [Google Scholar] [CrossRef]
- de Pina, M.Z.; Vazquez-Meza, H.; Pardo, J.P.; Rendon, J.L.; Villalobos-Molina, R.; Riveros-Rosas, H.; Pina, E. Signaling the signal, cyclic AMP-dependent protein kinase inhibition by insulin-formed H2O2 and reactivation by thioredoxin. J. Biol. Chem. 2008, 283, 12373–12386. [Google Scholar] [CrossRef]
- Vazquez-Meza, H.; de Pina, M.Z.; Pardo, J.P.; Riveros-Rosas, H.; Villalobos-Molina, R.; Pina, E. Non-steroidal anti-inflammatory drugs activate NADPH oxidase in adipocytes and raise the H2O2 pool to prevent cAMP-stimulated protein kinase a activation and inhibit lipolysis. BMC Biochem. 2013, 14, 13. [Google Scholar] [CrossRef]
- Becker, S.; Reinehr, R.; Graf, D.; vom Dahl, S.; Haussinger, D. Hydrophobic bile salts induce hepatocyte shrinkage via NADPH oxidase activation. Cell Physiol. Biochem. 2007, 19, 89–98. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Herrera, B.; Ventura, J.J.; Roncero, C.; Fernandez, M.; Fabregat, I. EGF blocks NADPH oxidase activation by TGF-beta in fetal rat hepatocytes, impairing oxidative stress, and cell death. J. Cell Physiol. 2006, 207, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Bertran, E.; Caja, L.; Carmona-Cuenca, I.; Murillo, M.M.; Fabregat, I. The inhibition of the epidermal growth factor (EGF) pathway enhances TGF-beta-induced apoptosis in rat hepatoma cells through inducing oxidative stress coincident with a change in the expression pattern of the NADPH oxidases (NOX) isoforms. Biochim. Biophys. Acta 2009, 1793, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Fabregat, I. NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J. Biol. Chem. 2010, 285, 24815–24824. [Google Scholar] [CrossRef]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The effects of palmitate on hepatic insulin resistance are mediated by NADPH oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef]
- Rao, M.S.; Reddy, J.K. The relevance of peroxisome proliferation and cell proliferation in peroxisome proliferator-induced hepatocarcinogenesis. Drug Metab. Rev. 1989, 21, 103–110. [Google Scholar]
- Teufelhofer, O.; Parzefall, W.; Kainzbauer, E.; Ferk, F.; Freiler, C.; Knasmuller, S.; Elbling, L.; Thurman, R.; Schulte-Hermann, R. Superoxide generation from Kupffer cells contributes to hepatocarcinogenesis: Studies on NADPH oxidase knockout mice. Carcinogenesis 2005, 26, 319–329. [Google Scholar] [CrossRef]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005, 19, 136–138. [Google Scholar] [CrossRef]
- Harada, H.; Hines, I.N.; Flores, S.; Gao, B.; McCord, J.; Scheerens, H.; Grisham, M.B. Role of NADPH oxidase-derived superoxide in reduced size liver ischemia and reperfusion injury. Arch. Biochem. Biophys. 2004, 423, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Hines, I.N.; Hoffman, J.M.; Scheerens, H.; Day, B.J.; Harada, H.; Pavlick, K.P.; Bharwani, S.; Wolf, R.; Gao, B.; Flores, S.; et al. Regulation of postischemic liver injury following different durations of ischemia. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G536–G545. [Google Scholar] [CrossRef]
- Aram, G.; Potter, J.J.; Liu, X.; Wang, L.; Torbenson, M.S.; Mezey, E. Deficiency of nicotinamide adenine dinucleotide phosphate, reduced form oxidase enhances hepatocellular injury but attenuates fibrosis after chronic carbon tetrachloride administration. Hepatology 2009, 49, 911–919. [Google Scholar] [CrossRef]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernandez-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology 2015, 149, 468–480 e410. [Google Scholar] [CrossRef]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef] [PubMed]
- Andueza, A.; Garde, N.; Garcia-Garzon, A.; Ansorena, E.; Lopez-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martinez-Irujo, J.J. NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Gladyshev, V.N. Role of reactive oxygen species-mediated signaling in aging. Antioxid. Redox Signal. 2013, 19, 1362–1372. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.J.; Bolduc, J.A.; Wu, H.; Collins, J.A.; Burke, E.A.; Reisz, J.A.; Klomsiri, C.; Wood, S.T.; Yammani, R.R.; Poole, L.B.; et al. H2O2 oxidation of cysteine residues in c-Jun N-terminal kinase 2 (JNK2) contributes to redox regulation in human articular chondrocytes. J. Biol. Chem. 2018, 293, 16376–16389. [Google Scholar] [CrossRef]
- Haval, G.A.; Pekhale, K.D.; Perween, N.A.; Ghaskadbi, S.M.; Ghaskadbi, S.S. Excess hydrogen peroxide inhibits head and foot regeneration in hydra by affecting DNA repair and expression of essential genes. J. Biochem. Mol. Toxicol. 2020. [CrossRef]
- Tsubata, T. Involvement of reactive oxygen species (ROS) in BCR signaling as a second messenger. Adv. Exp. Med. Biol. 2020, 1254, 37–46. [Google Scholar]
- Wang, G.; Yang, Q.; Zheng, C.; Li, D.; Li, J.; Zhang, F. Physiological Concentration of H2O2 supports dopamine neuronal survival via activation of Nrf2 signaling in glial cells. Cell Mol. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rashdan, N.A.; Pattillo, C.B. Hydrogen peroxide in the ER: A tale of triage. Redox Biol. 2020, 28, 101358. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medrano-Fernandez, I. Human aquaporin-11 guarantees efficient transport of H2O2 across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef]
- Lismont, C.; Koster, J.; Provost, S.; Baes, M.; Van Veldhoven, P.P.; Waterham, H.R.; Fransen, M. Deciphering the potential involvement of PXMP2 and PEX11B in hydrogen peroxide permeation across the peroxisomal membrane reveals a role for PEX11B in protein sorting. Biochim. Biophys. Acta Biomembr. 2019, 1861, 182991. [Google Scholar] [CrossRef]
- Mukherjee, S.P. Mediation of the antilipolytic and lipogenic effects of insulin in adipocytes by intracellular accumulation of hydrogen peroxide. Biochem. Pharmacol. 1980, 29, 1239–1246. [Google Scholar] [CrossRef]
- Mukherjee, S.P.; Lynn, W.S. Reduced nicotinamide adenine dinucleotide phosphate oxidase in adipocyte plasma membrane and its activation by insulin. Possible role in the hormone’s effects on adenylate cyclase and the hexose monophosphate shunt. Arch. Biochem. Biophys. 1977, 184, 69–76. [Google Scholar] [CrossRef]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arner, E.S.J. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019, 294, 12330–12338. [Google Scholar] [CrossRef]
- Kaul, N.; Gopalakrishna, R.; Gundimeda, U.; Choi, J.; Forman, H.J. Role of protein kinase C in basal and hydrogen peroxide-stimulated NF-kappa B activation in the murine macrophage J774A.1 cell line. Arch. Biochem. Biophys. 1998, 350, 79–86. [Google Scholar] [CrossRef]
- Torres, M.; Forman, H.J. Activation of several MAP kinases upon stimulation of rat alveolar macrophages: Role of the NADPH oxidase. Arch. Biochem. Biophys. 1999, 366, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schroder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef]
- Little, S.A.; de Haen, C. Effects of hydrogen peroxide on basal and hormone-stimulated lipolysis in perifused rat fat cells in relation to the mechanism of action of insulin. J. Biol. Chem. 1980, 255, 10888–10895. [Google Scholar]
- Mukherjee, S.P.; Lane, R.H.; Lynn, W.S. Endogenous hydrogen peroxide and peroxidative metabolism in adipocytes in response to insulin and sulfhydryl reagents. Biochem. Pharmacol. 1978, 27, 2589–2594. [Google Scholar] [CrossRef]
- Lawrence, J.C., Jr.; Larner, J. Activation of glycogen synthase in rat adipocytes by insulin and glucose involves increased glucose transport and phosphorylation. J. Biol. Chem. 1978, 253, 2104–2113. [Google Scholar]
- Krieger-Brauer, H.I.; Medda, P.K.; Kather, H. Insulin-induced activation of NADPH-dependent H2O2 generation in human adipocyte plasma membranes is mediated by Galphai2. J. Biol. Chem. 1997, 272, 10135–10143. [Google Scholar] [CrossRef]
- Mahadev, K.; Zilbering, A.; Zhu, L.; Goldstein, B.J. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J. Biol. Chem. 2001, 276, 21938–21942. [Google Scholar] [CrossRef]
- Mahadev, K.; Wu, X.; Motoshima, H.; Goldstein, B.J. Integration of multiple downstream signals determines the net effect of insulin on MAP kinase vs. PI 3’-kinase activation: Potential role of insulin-stimulated H(2)O(2). Cell Signal. 2004, 16, 323–331. [Google Scholar] [CrossRef]
- Porras, A.; Zuluaga, S.; Valladares, A.; Alvarez, A.M.; Herrera, B.; Fabregat, I.; Benito, M. Long-term treatment with insulin induces apoptosis in brown adipocytes: Role of oxidative stress. Endocrinology 2003, 144, 5390–5401. [Google Scholar] [CrossRef][Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).