
Fast DLLME-GC-MS Method for Determination of Pesticides in Carmelite Drops and Evaluation of Matrix Effects in Related Medicinal Products


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumental Equipment and Conditions
2.3. Samples and Sample Preparation
2.4. Matrix Effects
2.5. Validation Process
2.6. Real Samples
3. Results and Discussion
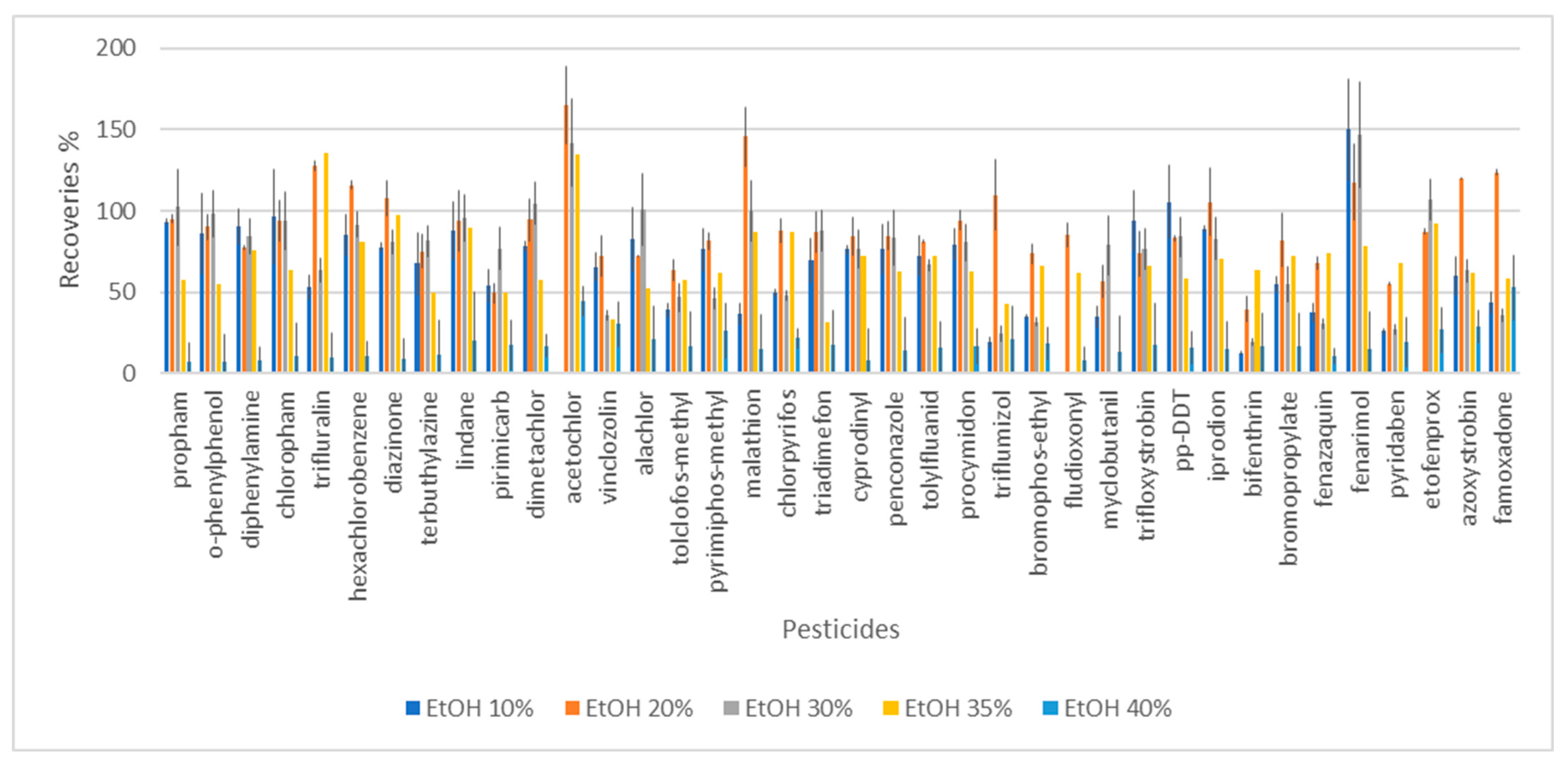
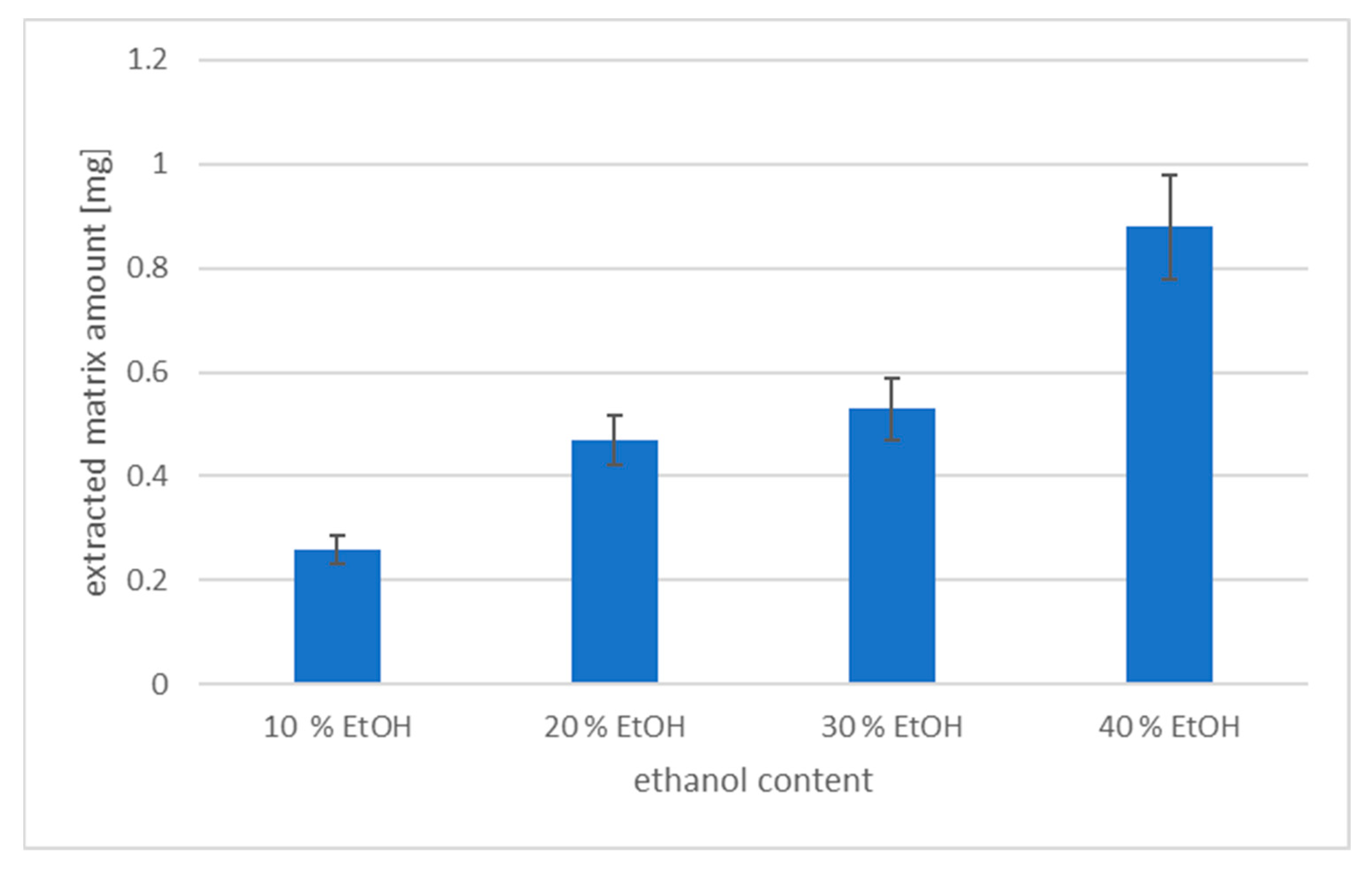
3.1. The Effect of Alcohol Content on DLLME Efficiency
- Alcohol content 10%, RSD in range 2–30%;
- Alcohol content 30%, RSD range 6–23%;
- Alcohol content 40%, RSD range 5–30%.
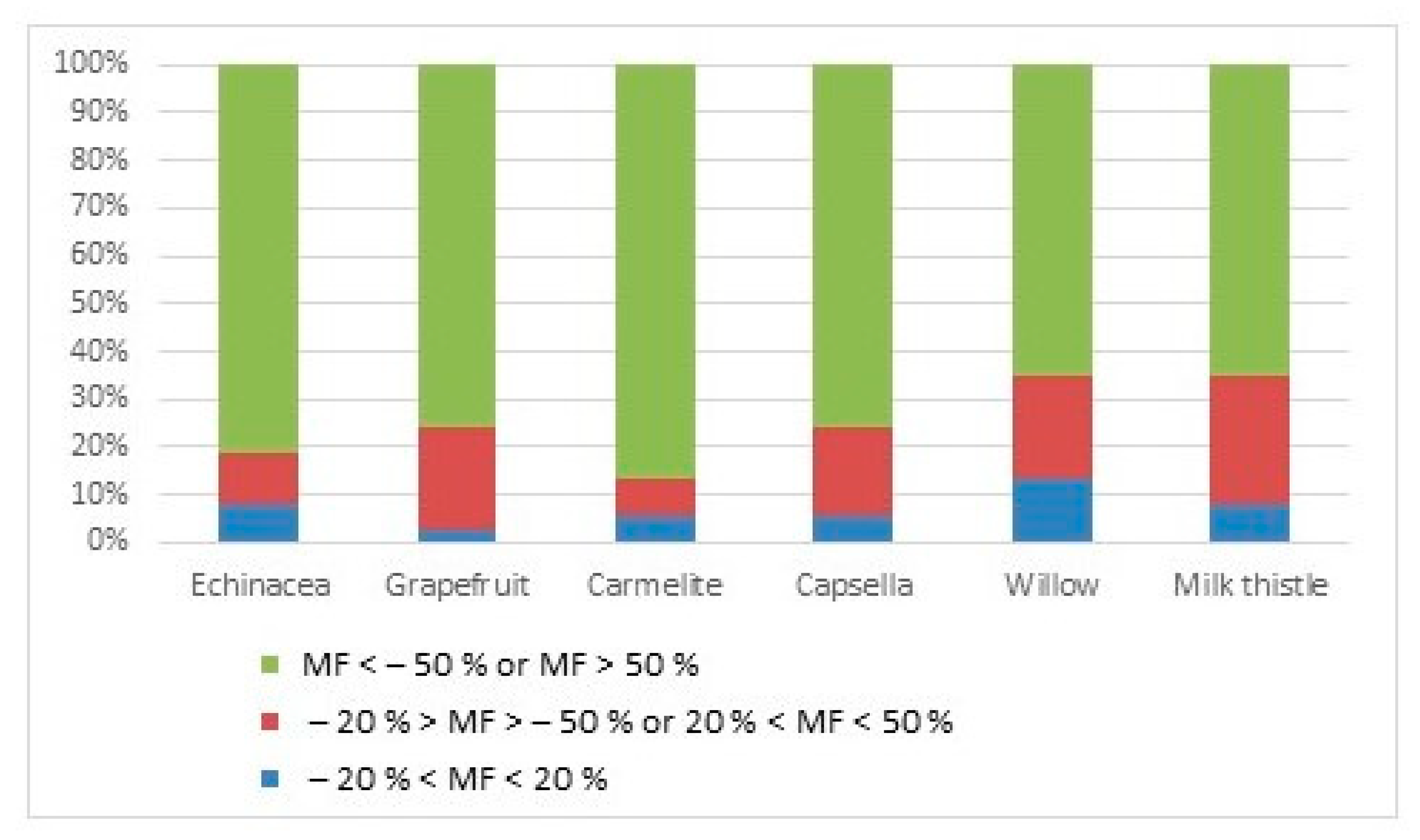
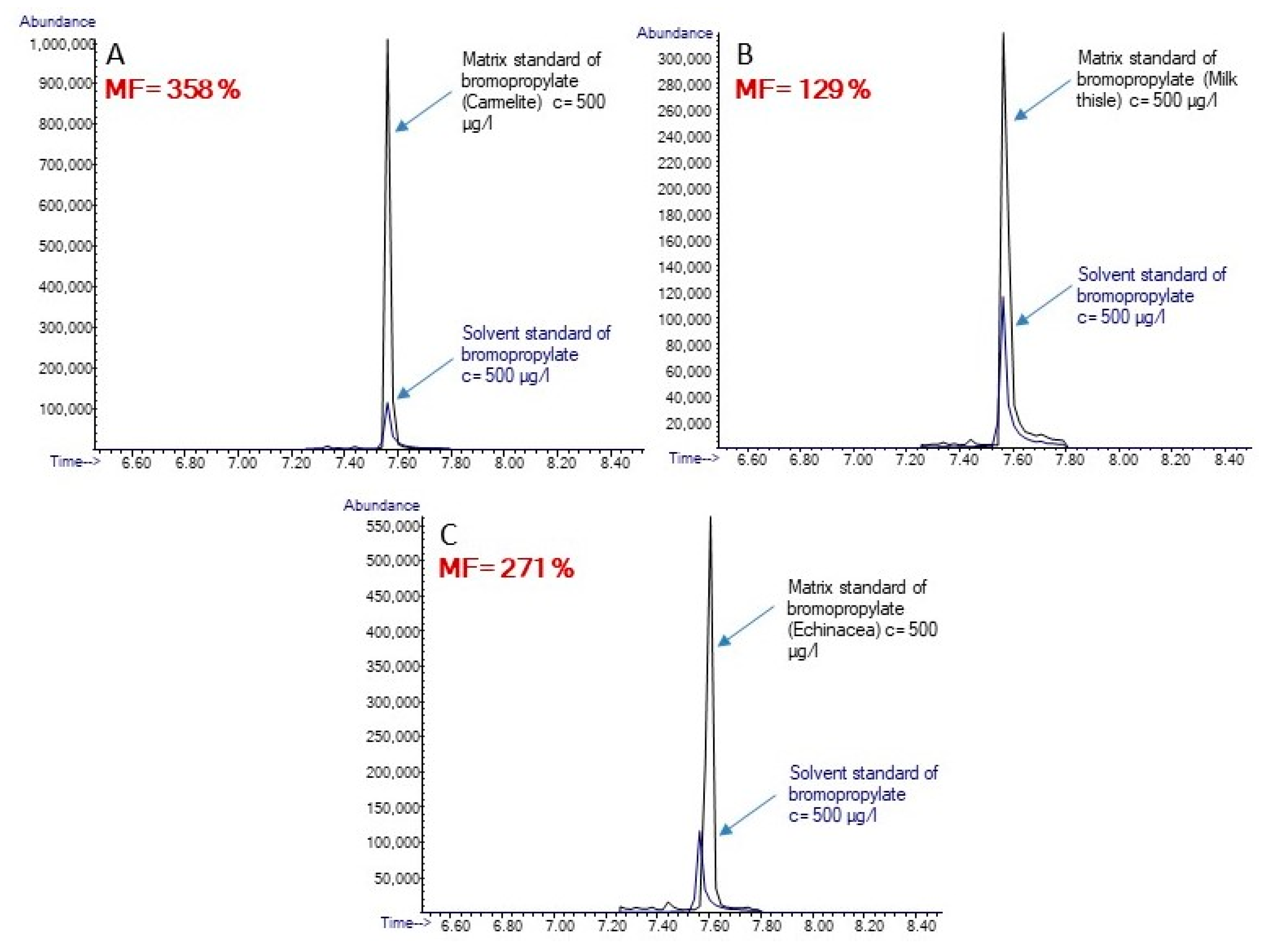
3.2. The Evaluation of the MEs in Related Medicinal Drops
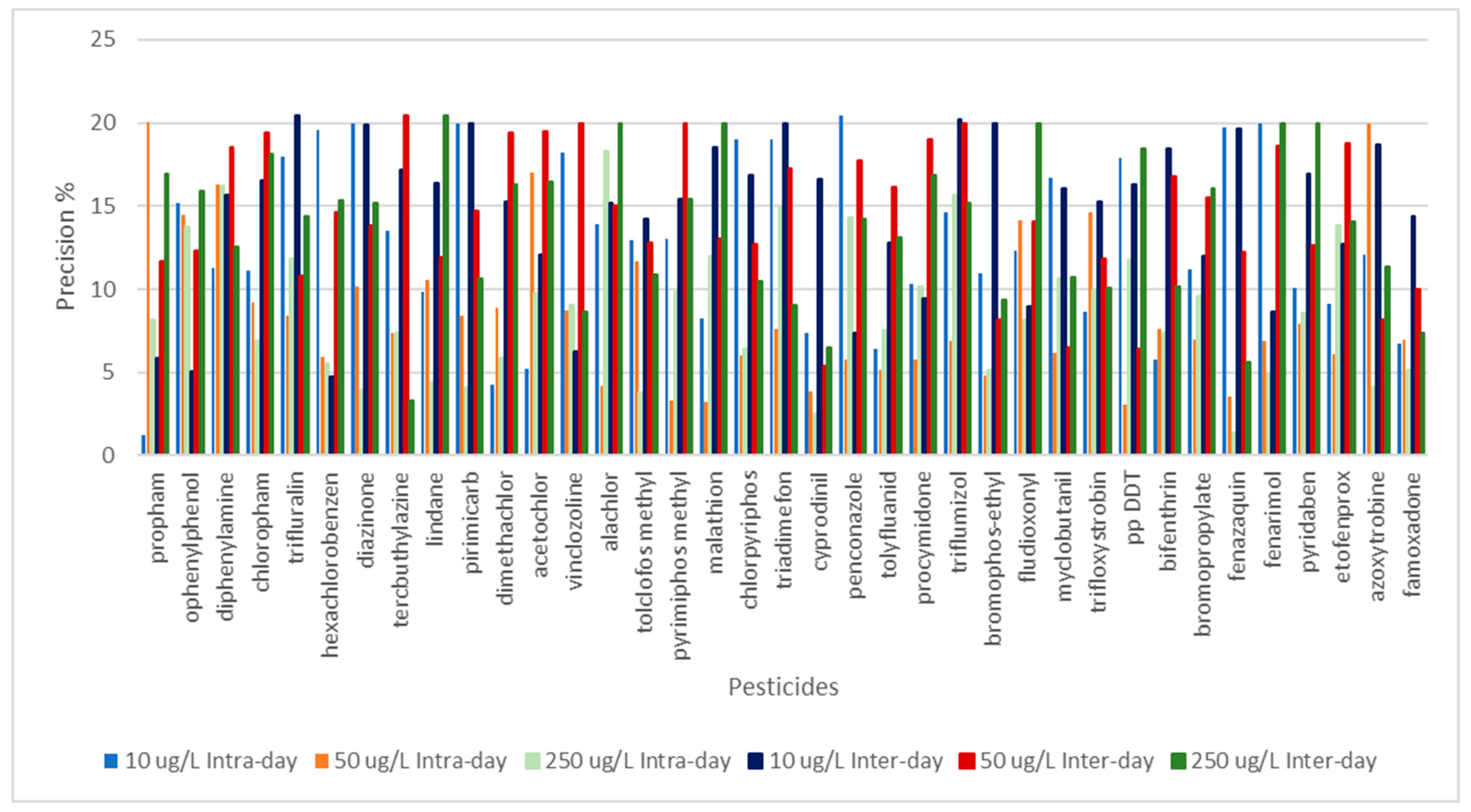
3.3. Validation of the Method
3.4. Real Sample Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Park, J.; Kim, K.; Ryu, D.; Whang, J.H.; Mah, J.H. GC-MS/MS method for determination of polycyclic aromatic hydrocarbons in herbal medicines. Molecules 2023, 28, 3853. [Google Scholar] [CrossRef] [PubMed]
- Dhivya, R.; Manimegalai, K. Preliminary Phytochemical Screening and GC- MS Profiling of Ethanolic Flower Extract of Calotropis gigantea Linn. (Apocynaceae). J. Pharmacogn. Phytochem. 2013, 2, 28–32. [Google Scholar]
- Shendy, A.H.; Eltanany, B.M.; Al-Ghobashy, M.A.; Gadalla, S.A.; Mamdouh, W.; Lotfy, H.M. Coupling of GC-MS/MS to Principal Component Analysis for Assessment of Matrix Effect: Efficient Determination of Ultra-Low Levels of Pesticide Residues in Some Functional Foods. Food Anal. Methods 2019, 12, 2870–2885. [Google Scholar] [CrossRef]
- Sumathi, P.; Parvathi, A. Antimicrobial activity of some traditional medicinal plants. J. Med. Plants Res. 2010, 4, 316–321. [Google Scholar]
- Vaou, N.; Stavropoulou, E.; Voidarou, C.; Tsigalou, C.; Bezirtzoglou, E. Towards Advances in Medicinal Plant Antimicrobial Activity: A Review Study on Challenges and Future Perspectives. Microorganisms 2021, 9, 2041. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Kumar, S. Antioxidant, Lipo-Protective and Antibacterial Activities of Phytoconstituents Present in Solanum. Int. Rev. Biophys. Chem. 2012, 3, 42–47. [Google Scholar]
- Zaidan, M.R.S.; Rain, N.A.; Badrul, A.R. In vitro screening of five local medicinal plants for antibacterial activity using disc diffusion method. Trop. Biomed. 2005, 22, 165–170. [Google Scholar] [PubMed]
- Kumadoh, D.; Ofori-Kwakye, K. Dosage forms of herbal medicinal products and their stability considerations—An overview. J. Crit. Rev. 2017, 4, 1–8. [Google Scholar]
- Bascom, A. Incorporating Herbal Medicine into Clinical Practice; F. A. Davis Company: Philadelphia, PA, USA, 2002. [Google Scholar]
- Hao, W.; Kingston, H.M.; Dillard, A.; Stuff, J.; Pamuku, M. Quantification of Persistent Organic Pollutants in Dietary Supplements Using Stir Bar Sorptive Extraction Coupled with GC-MS/MS and Isotope Dilution Mass Spectrometry. Food Addit. Contam. Part A 2020, 37, 1202–1215. [Google Scholar] [CrossRef]
- Environmental Protection Agency. Electronic Code of Federal Regulations; Title 40: Protection of Environment, PART 180—Tolerances and Exemptions for Pesticide Chemical Residues in Food. 2021. Available online: https://www.ecfr.gov/cgi-bin/retrieveECFR?gp=&SID=07bdbf8dfdc9849ebd84207f283b9289&mc=true&n=pt40.26.180&r=PART&ty=HTML (accessed on 10 July 2023).
- European Commission. Regulation (EC) No. 396/2005 on Maximum Residue Levels of Pesticides in or on Food and Feed of Plant and Animal Origin. 2005. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02005R0396-20160513 (accessed on 10 July 2023).
- Narváez, A.; Rodríguez-Carrasco, Y.; Castaldo, L.; Izzo, L.; Ritieni, A. Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Orbitrap High-Resolution Mass Spectrometry for Multi-Residue Analysis of Mycotoxins and Pesticides in Botanical Nutraceuticals. Toxins 2020, 12, 114. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Garrido Frenich, A. Determination of toxic substances, pesticides and mycotoxins, in ginkgo biloba nutraceutical products by liquid chromatography Orbitrap-mass spectrometry. Microchem. J. 2015, 118, 124–130. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Garrido Frenich, A. Multi-class methodology to determine pesticides and mycotoxins in green tea and royal jelly supplements by liquid chromatography coupled with Orbitrap high resolution mass spectrometry. Food Chem. 2016, 197, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Koshy, R.; Yadav, S.; Rajeshkumar, R.; Singh, V.K.; Setty, M.M.; Murali, B.; Agarwal, A. Optimization and Validation of a Multiresidue Method for Screening of 126 Pesticide Residues in Herbal Raw Materials and Extracts Used as Ingredients in Ayurvedic Medicines and Dietary Supplements. J. AOAC Int. 2022, 105, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Z.; Wang, J.H.; Wang, M.L.; Zhang, R.; Lu, X.Y.; Liu, W.H. Dispersive Solid-Phase Extraction Cleanup Combined with Accelerated Solvent Extraction for the Determination of Carbamate Pesticide Residues in Radix Glycyrrhizae Samples by UPLC-MS-MS. J. Chromatogr. Sci. 2011, 49, 702–708. [Google Scholar] [PubMed]
- Chen, Y.; Al-Taher, F.; Juskelis, R.; Wong, J.W.; Zhang, K.; Hayward, D.G.; Zweigenbaum, J.; Stevens, J.; Cappozzo, J. Multiresidue Pesticide Analysis of Dried Botanical Dietary Supplements Using an Automated Dispersive SPE Cleanup for QuEChERS and High-Performance Liquid Chromatography–Tandem Mass Spectrometry. J. Agric. Food Chem. 2012, 60, 9991–9999. [Google Scholar] [CrossRef] [PubMed]
- Szarka, A.; Turková, D.; Hrouzková, S. Dispersive liquid-liquid microextraction followed by gas chromatography–mass spectrometry for the determination of pesticide residues in nutraceutical drops. J. Chromatogr. A 2018, 1570, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, I.; Kanashina, D.; Moskvin, L.; Bulatov, A. Anevaporation-assisteddispersiveliquid–liquid microextractiontechnique as a simpletoolforhighperformanceliquidchromatography tandem–massspectrometrydetermination ofinsecticides in wine. J. Chromatogr. A 2017, 1512, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hrouzková, S.; Brišová, M.; Szarka, A. Development of fast, efficient and ecological method employing vortex-assisted dispersive liquid–liquid microextraction combined with fast gas chromatography–mass spectrometry for pesticide residues analysis in alcohol-content samples. J. Chromatogr. A 2017, 1506, 18–26. [Google Scholar] [CrossRef]
- Panuwet, P.; Hunter, R.E.; D’Souza, P.E.; Chen, X.; Radford, S.A.; Cohen, J.R.; Barr, D.B. Biological matrix effects in quantitative tandem mass spectrometry-based analytical methods: Advancing biomonitoring. Critical Review. Anal. Chem. 2016, 46, 93–105. [Google Scholar] [CrossRef]
- Rahman, M.M.; EI-Aty, A.M.A.; Shim, J.H. Matrix enhancement effect: A blessing or a curse for gas chromatography?—A review. Anal. Chim. Acta 2013, 801, 14–21. [Google Scholar] [CrossRef]
- Wu, X.; Ding, Z. Evaluation of matrix effects for pesticide residue analysis by QuEChERS coupled with UHPLC-MS/MS in complex herbal matrix. Food Chem. 2023, 405, 134755. [Google Scholar] [CrossRef] [PubMed]
- Raposo, F.; Barceló, D. Challenges and strategies of matrix effects using chroma-tography-mass spectrometry: An overview from research versus regulatory viewpoints. Trends Anal. Chem. 2021, 134, 116068. [Google Scholar] [CrossRef]
- Van Eeckhaut, A.; Lanckmans, K.; Sarre, S.; Smolders, I.; Michotte, Y. Validation of bioanalytical LC-MS/MS assays: Evaluation of matrix effects. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Hewavitharana, A.K.; Abu Kassim, N.S.; Shaw, P.N. Standard addition with internal standardisation as an alternative to using stable isotope labelled internal standards to correct for matrix effects—Comparison and validation using liquid chromatography-tandem mass spectrometric assay of vitamin D. J. Chromatogr. A 2018, 1553, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pérez, S.M.L.; Plaza-Bolaños, P.; Romero-González, R.; Martínez Vidal, J.L.; Garrido Frenich, A. Comprehensive qualitative and quantitative determination of pesticides and veterinary drugs in honey using liquid chromatography—Orbitrap high-resolution mass spectrometry. J. Chromatogr. A 2010, 1217, 5872–5885. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, H.; Kittlaus, S.; Kempe, G.; Alder, L. Reduction of matrix effects in liquid chromatography–electrospray ionization–mass spectrometry by dilution of the sample extracts: How much dilution is needed? Anal. Chim. Acta 2012, 721, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Bolzan, C.M.; Caldas, S.S.; Guimarães, B.S.; Primel, E.G. Dispersiveliquid-liquid microextraction with liquid chromatography-tandem mass spectrometry for the determination of triazine, neonicotinoid, triazole and imidazoline pesticides in mineral water samples. J. Braz. Chem. Soc. 2015, 26, 1902–1913. [Google Scholar]
- National Library of Medicine, PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/#query=log%20kow (accessed on 15 March 2024).
- Mukherjee, P.K. Evidence-Based Validation of Herbal Medicine, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Campillo, N.; Vinas, P.; Cacho, J.I.; Penalver, R.; Hernández-Córdoba, M. Evaluation of dispersive liquid–liquid microextraction for the simultaneous determination of chlorophenols and haloanisoles in wines and cork stoppers using gas chromatography-mass spectrometry. J. Chromatogr. A 2010, 1217, 7323–7330. [Google Scholar] [CrossRef]
- Montevecchi, G.; Masino, F.; Zanasi, L.; Antonelli, A. Determination of phthalate esters in distillates by ultrasound-vortex-assisted dispersive liquid-liquid micro-extraction (USVADLLME) coupled with gas chromatography/mass spectrometry. Food Chem. 2017, 221, 1354–1360. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, H.; Huang, X.; Hang, F.; Luo, H. Rapid analysis of short- and medium-chain chlorinated paraffins in wine by dispersive liquid–liquid micro-extraction coupled with high performance liquid chromatography-electrospray ionization quadrupole time-of-flight mass spectrometry. Food Chem. 2020, 319, 126583. [Google Scholar] [CrossRef] [PubMed]
- SANTE/11813/2017; Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. European Commission Directorate General for Health and Food Safety, Safety of the Food Chain Pesticides and Biocides: Bruxelles/Brussel, Belgium, 2017. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf (accessed on 17 January 2024).
- Łozowicka, B.; Rutkowska, E.; Jankowska, M. Influence of QuEChERS modifica-tions on recovery and matrix effect during the multi-residue pesticide analysis in soil by GC/MS/MS and GC/ECD/NPD. Environ. Sci. Pollut. Res. 2017, 24, 7124–7138. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Peer review of the pesticide risk assessment of the active substance tolclofos-methyl. EFSA J. 2018, 16, e05130. [Google Scholar]
- European Food Safety Authority (EFSA). The 2018 European Union report on pesticide residues in food. EFSA J. 2020, 18, e06057. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Pesticide | log Kow | Retention Time (min) | Monitored Ions (m/z) |
|---|---|---|---|---|
| 1 | Propham | 2.60 | 3.811 | 179, 93, 120 |
| 2 | o-Phenylphenol | 3.18 | 4.119 | 170, 169, 141 |
| 3 | Diphenylamine | 3.82 | 4.553 | 169, 168, 167 |
| 4 | Trifluralin | 5.27 | 4.578 | 306, 264, 307 |
| 5 | Chlorpropham | 3.4 | 4.673 | 127, 171, 213 |
| 6 | Hexachlorobenzene | 3.93 | 4.864 | 284, 286, 249 |
| 7 | Terbuthylazine | 3.70 | 5.085 | 214, 229, 173 |
| 8 | Diazinon | 3.69 | 5.103 | 304, 179, 107 |
| 9 | Lindane | 3.50 | 5.151 | 181, 219, 183 |
| 10 | Pirimicarb | 1.70 | 5.380 | 166, 72, 238 |
| 11 | Acetochlor | 4.14 | 5.420 | 149, 146, 223 |
| 12 | Vinclozolin | 3.02 | 5.545 | 212, 285, 178 |
| 13 | Dimethachlor | 2.17 | 5.552 | 197, 134, 149 |
| 14 | Alachlor | 3.09 | 5.620 | 160, 188, 146 |
| 15 | Tolclofos-methyl | 5.631 | 265, 267 | |
| 16 | Parathion-methyl | 3.00 | 5.650 | 263, 125, 109 |
| 17 | Pirimiphos-methyl | 3.90 | 5.772 | 290, 305, 276 |
| 18 | Malathion | 2.75 | 5.790 | 173, 127, 93 |
| 19 | Chlorpyrifos | 4.70 | 5.945 | 314, 197, 199 |
| 20 | Triadimefon | 2.77 | 6.099 | 208, 181, 210 |
| 21 | Cyprodinil | 4.00 | 6.291 | 224, 225, 86 |
| 22 | Tolylfluanid | 3.90 | 6.327 | 137, 238, 101 |
| 23 | Penconazole | 4.64 | 6.356 | 248, 159, 161 |
| 24 | Triflumizole | 3.67 | 6.446 | 278, 206, 179 |
| 25 | Bromophos-ethyl | 6.15 | 6.450 | 359, 303, 242 |
| 26 | Procymidone | 3.30 | 6.450 | 283, 285, 96 |
| 27 | Fludioxonil | 4.12 | 6.640 | 248, 249, 86 |
| 28 | Myclobutanil | 2.89 | 6.954 | 179, 245, 288 |
| 29 | Trifloxystrobin | 4.50 | 7.161 | 172, 116, 187 |
| 30 | p,p-DDT | 7.00 | 7.173 | 235, 165, 237 |
| 31 | Iprodione | 3.00 | 7.285 | 314, 184, 316 |
| 32 | Bifenthrin | 6.60 | 7.428 | 181, 165, 166 |
| 33 | Bromopropylate | 5.40 | 7.538 | 341, 339, 343 |
| 34 | Fenazaquin | 5.51 | 7.672 | 145, 160, 146 |
| 35 | Pyridaben | 6.37 | 7.985 | 147, 309, 207 |
| 36 | Fenarimol | 3.69 | 8.005 | 139, 251, 207 |
| 37 | Etofenprox | 6.80 | 8.413 | 161, 86, 135 |
| 38 | Difenoconazole | 4.40 | 8.752 | 323, 207, 281 |
| 39 | Azoxystrobin | 2.50 | 9.055 | 344, 207, 224 |
| 40 | Famoxadone | 4.65 | 9.233 | 330, 207, 224 |
| Type of Tinctures | Alcohol Content (%) | Description |
|---|---|---|
| Carmelite drops | 40 | 10% extract from a mixture of herbs (lemon balm leaves, lemon perch, nutmeg, cinnamon bark, and clove flower) in 40% alcohol |
| Milk thistle drops | 40 | 10% extract of milk thistle fruit in 40% alcohol |
| Willow flower drops | 40 | 10% extract from the stem of a small-flowered willow in 40% alcohol |
| Echinacea drops | 24.5 | The drops contain the juice from the flowers of purple coneflower (Echinacea purpurea) |
| Grapefruit drops | 66 | Extract of grapefruit seeds and peel |
| Capsella drops | 40 | 10% coconut stem extract in 40% alcohol |
| Pesticides | MF/% Echinacea | MF/% Grapefruit | MF/% Carmelite | MF/% Capsella | MF/% Willow | MF/% Milk Thistle |
|---|---|---|---|---|---|---|
| Propham | 3 | 23 | 35 | 8 | 8 | 74 |
| o-Phenylphenol | 45 | 75 | −10 | −46 | 26 | 60 |
| Diphenylamine | 236 | 106 | 85 | 124 | 111 | 109 |
| Chlorpropham | 409 | 97 | 134 | 69 | 89 | 125 |
| Trifluralin | 21 | 29 | 67 | 46 | 30 | 61 |
| Hexachlorobenzene | 11 | 14 | −40 | 9 | 8 | 36 |
| Diazinone | 233 | 84 | 125 | 52 | 33 | 115 |
| Tercbuthylazine | −38 | 65 | −88 | −56 | −26 | 13 |
| Lindane | 1012 | 50 | 274 | 614 | 103 | 438 |
| Pirimicarb | 99 | 77 | 131 | 56 | 39 | 30 |
| Dimethachlor | 78 | 43 | 77 | 45 | 25 | 44 |
| Acetochlor | 199 | 39 | 80 | 47 | 34 | 35 |
| Vinclozoline | 70 | 69 | 62 | 44 | 41 | 37 |
| Alalchlor | 75 | 36 | 88 | 36 | 19 | 30 |
| Tolclophos methyl | 113 | 105 | 106 | 87 | 74 | 83 |
| Pirmiphos methyl | 129 | 110 | 131 | 108 | 74 | 71 |
| Malathion | 320 | 326 | 263 | 435 | 237 | 214 |
| Chlorpyriphos | 115 | 141 | 158 | 96 | 58 | 63 |
| Triadimefon | 269 | 117 | −10 | −22 | 349 | −3 |
| Cyprodinyl | 1138 | 253 | 98 | 96 | 645 | 599 |
| Penconazole | 371 | 814 | 155 | 123 | 307 | 245 |
| Tolylfluanid | 201 | 198 | 170 | 144 | 163 | 119 |
| Procymidone | 104 | 113 | 52 | 57 | 62 | 40 |
| Triflumizol | 513 | 500 | 2379 | 1002 | 597 | 51 |
| Bromophos ethane | 129 | 179 | 94 | 105 | 100 | 45 |
| Fludioxonyl | −42 | −25 | 32 | 52 | −15 | −26 |
| Myclobutanyl | 635 | 234 | 126 | 325 | 115 | 50 |
| Trifloxystrobin | 1350 | 1540 | 322 | 532 | 437 | 689 |
| p,p-DDT | 181 | 182 | 59 | 65 | 144 | 139 |
| Bifenthrin | −12 | 277 | 135 | 93 | 9 | 2 |
| Bhromopropylate | 271 | 527 | 358 | 420 | 253 | 129 |
| Fenazaquin | 637 | 1814 | 649 | 1329 | 1004 | 634 |
| Fenarimol | 135 | 142 | 98 | 235 | 88 | 87 |
| Pyridaben | 365 | 850 | 656 | 379 | 457 | 357 |
| Etophenprox | 532 | 616 | 687 | 432 | 521 | 357 |
| Azoxystrobine | 718 | 1772 | 1795 | 1192 | 1259 | 768 |
| Famoxadone | 68 | 49 | 86 | 98 | 56 | 60 |
| Pesticides | R2 | LOD µg/L | LOQ µg/L | LCL µg/L |
|---|---|---|---|---|
| Propham | 0.9941 | 0.082 | 0.271 | 0.5 |
| o-Phenylphenol | 0.9736 | 0.009 | 0.030 | 0.05 |
| Diphenylamine | 0.9845 | 0.057 | 0.188 | 0.5 |
| Chlorpropham | 0.9984 | 0.225 | 0.743 | 1 |
| Trifluralin | 0.9750 | 0.002 | 0.007 | 0.01 |
| Hexachlorobenzene | 0.9764 | 0.011 | 0.036 | 0.05 |
| Diazinone | 09872 | 0.003 | 0.010 | 0.01 |
| Tercbuthylazine | 0.9725 | 0.111 | 0.366 | 0.5 |
| Lindane | 0.9799 | 0.416 | 1.373 | 5 |
| Pirimicarb | 0.9864 | 0.250 | 0.825 | 1 |
| Dimethachlor | 0.9882 | 0.028 | 0.092 | 0.1 |
| Acetochlor | 0.9875 | 0.123 | 0.406 | 0.5 |
| Vinclozoline | 0.9849 | 0.078 | 0.257 | 0.5 |
| Alachlor | 0.9997 | 0.094 | 0.310 | 0.5 |
| Tolclofos-methyl | 0.9947 | 0.018 | 0.059 | 0.1 |
| Pirimiphos-methyl | 0.9926 | 0.054 | 0.178 | 0.5 |
| Malathion | 0.9871 | 0.054 | 0.178 | 0.5 |
| Chlorpyriphos | 0.9778 | 0.005 | 0.017 | 0.05 |
| Triadimefon | 0.9890 | 0.039 | 0.129 | 0.5 |
| Cyprodinil | 0.9760 | 0.159 | 0.525 | 1 |
| Penconazole | 0.9877 | 0.011 | 0.036 | 0.05 |
| Tolylfluanid | 0.9947 | 0.089 | 0.294 | 0.5 |
| Procymidone | 0.9983 | 0.022 | 0.073 | 0.1 |
| Triflumizol | 0.9967 | 0.106 | 0.350 | 0.5 |
| Bromophos-ethyl | 0.9774 | 0.002 | 0.007 | 0.01 |
| Fludioxonyl | 0.9973 | 0.055 | 0.182 | 0.5 |
| Myclobutanil | 0.9764 | 0.040 | 0.132 | 0.5 |
| Trifloxystrobin | 0.9735 | 0.277 | 0.914 | 1 |
| p,p-DDT | 0.9836 | 0.098 | 0.323 | 0.5 |
| Bifenthrin | 0.9938 | 0.050 | 0.165 | 0.5 |
| Bromopropylate | 0.9741 | 0.011 | 0.036 | 0.05 |
| Fenazaquin | 0.9966 | 0.014 | 0.046 | 0.05 |
| Fenarimol | 0.9976 | 0.127 | 0.419 | 0.5 |
| Pyridaben | 0.9837 | 0.137 | 0.452 | 0.5 |
| Etofenprox | 0.9866 | 0.007 | 0.023 | 0.05 |
| Azoxystrobine | 0.9755 | 0.063 | 0.208 | 0.5 |
| Famoxadone | 0.9748 | 0.057 | 0.188 | 0.5 |
| Type of Tinctures | Alcohol Content (%) | Concentration of Pesticides | RSD% * |
|---|---|---|---|
| Carmelite drops | 40 | o-Phenylphenol 16.47 µg/L Bromopropylate 0.67 µg/L | 2 5 |
| Echinacea drops | 24.5 | Tolclofos-methyl 2.24 µg/L | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szarka, A.; Hrouzková, S. Fast DLLME-GC-MS Method for Determination of Pesticides in Carmelite Drops and Evaluation of Matrix Effects in Related Medicinal Products. Foods 2024, 13, 1745. https://doi.org/10.3390/foods13111745
Szarka A, Hrouzková S. Fast DLLME-GC-MS Method for Determination of Pesticides in Carmelite Drops and Evaluation of Matrix Effects in Related Medicinal Products. Foods. 2024; 13(11):1745. https://doi.org/10.3390/foods13111745
Chicago/Turabian StyleSzarka, Agneša, and Svetlana Hrouzková. 2024. "Fast DLLME-GC-MS Method for Determination of Pesticides in Carmelite Drops and Evaluation of Matrix Effects in Related Medicinal Products" Foods 13, no. 11: 1745. https://doi.org/10.3390/foods13111745