1. Introduction
The CDC estimates that at least 2.5 million individuals experience a TBI annually, resulting in 1.4 million emergency department visits, 235,000 hospitalizations and 52,000 deaths [
1]. Importantly, these numbers do not include TBIs that are treated in outpatient settings or those that go unreported. TBI is particularly common in young adults and children, making it the leading cause of death and disability in the United States among these age groups [
2,
3]. The most recent data from the CDC report that children and adolescents had 16,070 TBI-caused hospitalizations and nearly 3000 TBI-related deaths [
4]. To date, there are no effective pharmacologic treatments approved specifically for those exposed to a TBI. This leaves TBI patients to suffer the debilitating consequences of a head injury, potentially for the rest of their lives [
5].
TBI pathology consists of primary and secondary phases, with primary injury occurring at the time of impact and secondary injury resulting from the biochemical, inflammatory, and structural responses to injury [
6]. Hallmark features of TBI pathology include the disruption of the blood-brain barrier (BBB), increased oxidative stress, and neuroinflammation [
7,
8,
9,
10]. Left unchecked, these injurious processes spread from the focal site of impact into surrounding healthy CNS tissue, increasing the area of damage, similar to the concept of a watershed stroke. The dynamic and debilitating pathophysiology of TBI paired with the extremely limited treatment options make clear the need for therapeutic interventions that can help prevent the spread of injury.
By modulating the pathology of secondary injury, the chronic negative consequences of TBI may be minimized. The BBB regulates access of blood components and immune cells into the brain [
11]. The role of the BBB is to maintain the delicate neuronal environment [
11], and its dysfunction is observed in the pathology of neurotrauma, stroke, and neurological disease [
12]. The injury induced by TBI damages cerebral blood vessels, which circumvents the barrier mechanisms of the BBB [
6]. Once BBB breach occurs, secondary events, such as oxidative stress damage, are triggered that further contribute to injury [
13,
14]. Oxidative stress resulting from neuroinflammation and excitotoxicity may be used as a target for therapeutic intervention [
14]. There are many sources of reactive oxygen species (ROS) in TBI, including the arachidonic acid pathway, mitochondrial damage, xanthine oxidase activity, and the oxidation of heme [
15,
16,
17,
18]. Of particular importance is the prevention of further mitochondrial damage. Dysfunction of this organelle not only decreases total cellular energy output and thus decreases regenerative capacity, but the mitochondria also create more ROS in a self-perpetuating and self-damaging cycle.
Immediately following a TBI, production of superoxide anions is massively increased by electron transport chain enzymes, xanthine oxidase, and NADPH oxidases from injured cells and from the recruited peripheral immune system [
19,
20,
21]. High concentrations of superoxide overwhelm the endogenous antioxidant systems, allowing this anion to react to form additional oxidants that are more stable, such as peroxynitrite or a highly reactive hydroxyl radical via hydrogen peroxide and free heme from damaged red blood cells. These free radicals have deleterious effects on cell physiology, disrupting cell membranes through lipid peroxidation and oxidizing cellular proteins and DNA, which in turn creates more superoxide. This damaging cycle is caused by the cells redox balance, being strongly pro-oxidant and not counteracted by the actions of limited and depleted amounts of endogenous antioxidant enzymes.
In order to correct against the damaging wave of oxidative stress causing imbalance, the notion of targeting two antioxidant enzymes (SOD-1 and catalase) conjugated with an anti-ICAM1 antibody was explored in the studies herein. Early attempts at this strategy have shown strong results. The administration of anti-ICAM-1 antibodies conjugated with catalase following moderate TBI showed brain endothelial cell protection, tissue sparing, reduced inflammatory response, and long-term functional improvement [
22,
23]. Adding SOD-1 to the anti-ICAM-1–catalase conjugate brings two natural antioxidant enzymes directly to the damaged BBB endothelium, where they can dismutate superoxide into hydrogen peroxide (SOD-1) and then immediately convert that hydrogen peroxide into water and oxygen (catalase). Targeted dual antioxidant enzyme therapy could thus help restore the redox balance within damaged cells by decreasing the free radical burden brought on by TBI. The results presented in this report provide proof of concept that the concerted actions of a catalase and superoxide dismutase conjugate cross-linked with an anti-ICAM-1 antibody into nanoparticles greatly lessens the neuroinflammation associated with TBI. Specifically, this study provides evidence that addition of this dual conjugate (1) greatly attenuates the oxidative stress imbalance during neurotrauma (2) preserves BBB integrity and function and (3) reduces inflammatory signatures in the CNS parenchyma.
2. Materials and Methods
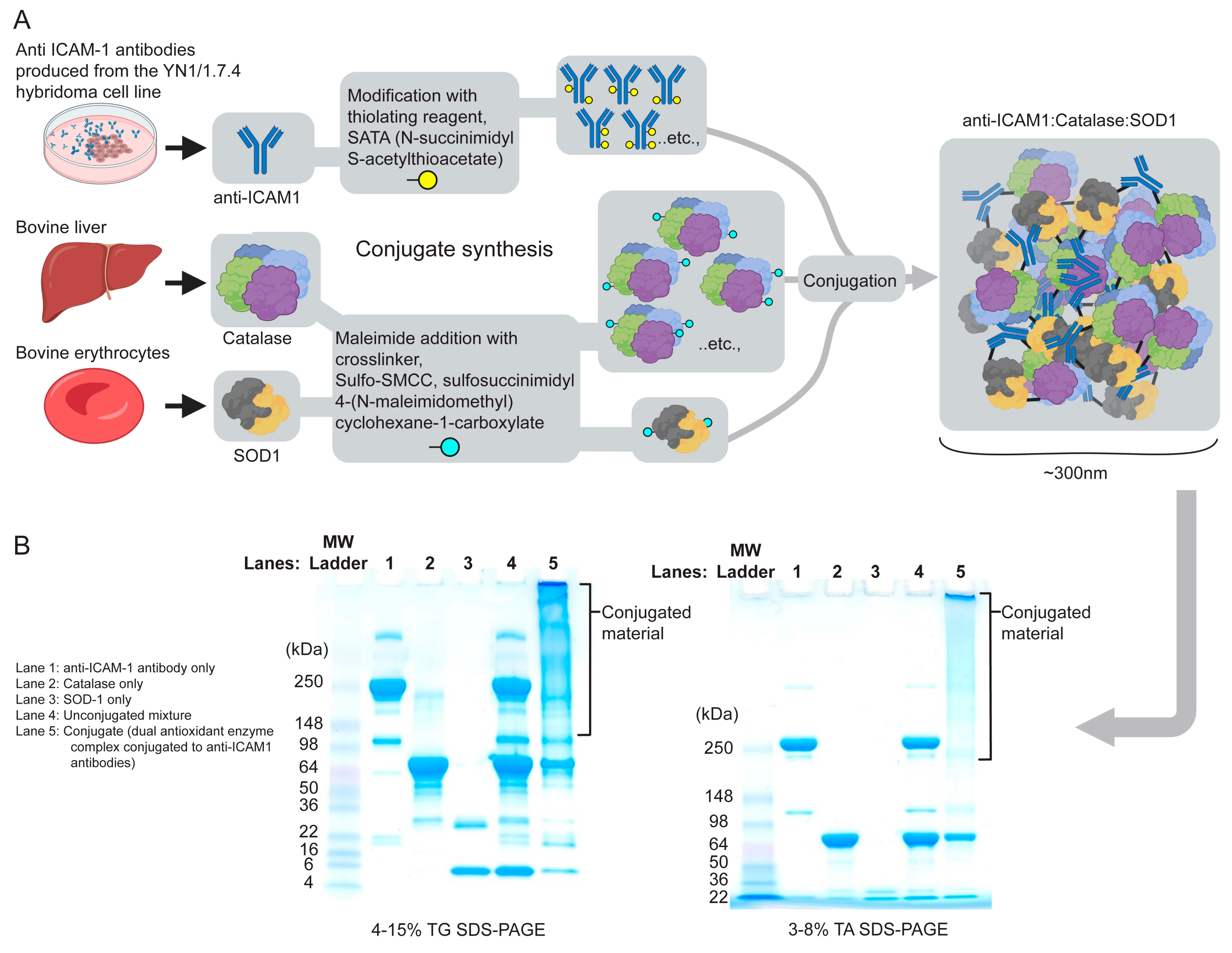
2.1. Dual-Conjugate Preparation
Antioxidant enzymes were acquired from commercial sources. Bovine liver purified catalase was obtained from Calbiochem (San Diego, CA, USA), and superoxide dismutase 1 (SOD-1) from bovine erythrocytes was purchased from EMD Chemicals (San Diego, CA, USA). Succinimidyl-6-[biotinamido]hexanoate (NHS-LC-biotin), 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (SMCC), and N-succinimidyl-S-acetylthioacetate (SATA) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Clone YN1/1.7.4 is a monoclonal antibody of the IgG2b isotype (mAb) against anti-intercellular adhesion molecule 1 (ICAM-1) [
24].
Antibodies were conjugated with catalase and SOD-1 using amino-chemistry fusion methods as previously described [
23]. Protected SH groups were introduced in the molecule of 5.2 mg/mL antibody via primary amines using SATA at an initial molar ratio of 1:10 of antibody:SATA at room temperature for 30 min followed by SH group deprotection with 50 mM hydroxylamine for 2 h. Maleimide groups were introduced into 12 mg/mL catalase and 3.0 mg/mL SOD-1 using SMCC (SOD:SMCC 1:8 and catalase:SMCC 1:20 initial molar ration) for 1 h at room temperature. After protein modification, unreacted reagents were removed by 7K MWCO Zeba Spin Desalting Columns (Thermo Fisher Scientific; Waltham, MA, USA). Conjugation was performed at a 2:1:1 Ab:Catalase:SOD molar ratio on ice and stopped when particle size reached 250–300 nm, as measured by a dynamic light scattering (DLS) Zetasizer Nano ZSP (Malvern Instruments Ltd., Malvern, UK). Conjugates were resuspended with sucrose as a cryoprotector at a final concentration of 20%, then frozen and stored at −80 °C before use. Conjugation was further confirmed by denaturing non-reducing 4–15% SDS-PAGE in a tris-glycine system. The control mixture contained equivalent amounts of unmodified antibody, SOD1 and catalase.
2.2. Cell Culture and Reagents
All experiments used the bEnd.3 cell line, an immortalized brain endothelial cell line derived from mouse cortex and acquired at low passage from the ATCC (Manassas, VA, USA). Cells were grown on rat-tail collagen I-coated flasks in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1% Fungizone, and 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). bEnd.3 cells were grown in an incubator set to 37 °C, 5% CO2, and 95% humidity.
Hydrogen peroxide (H2O2) at 30% w/w solution in water with a stabilizer was purchased from Sigma-Aldrich (St. Louis, MO, USA) and diluted to the desired molarity using serum-free DMEM. For all in vitro experiments, dual conjugates and controls were also diluted in serum-free DMEM prior to use.
2.3. Paracellular Permeability Assays
In order to examine bEnd.3 paracellular permeability, cells were seeded at a density of 16,000 cell per collagen-I coated Transwell inserts (Corning Life Science, Glendale, AZ, USA) on 24-well plates. Pore sizes were 0.4 μm, with a diameter of 0.33 cm2. Cells were seeded in 200 μL of DMEM with 10% FBS, and the basolateral chambers (surrounding the Transwell inserts) were filled with 500 μL of media. Media were changed every 2 days until confluent cell monolayers were formed (~5–6 days). Upon formation of a bEnd.3 cell monolayer, the cells were serum-starved overnight and then treated with 500 μM H2O2, 100 ng/mL dual conjugate, or both in the treatment group for 1 h. Prewarmed and well-mixed FITC–dextran (3 kDa MW, Thermo Fisher Scientific, Waltham, MA, USA) was then added to the inserts at a resulting concentration of 2 mg/mL. After 1 and 3 h, media were taken from the basolateral chambers and the fluorescence of the FITC–dextran permeability tracer was measured at 525 nm using a SpectraMax M5e (Molecular Devices Inc., San Jose, CA, USA).
2.4. Metabolic Flux Assays
Cells were seeded at a density of 16,000 per well onto a Seahorse XFe96 Culture Microplate (Agilent Technologies Inc., Santa Clara, CA, USA) in DMEM with 10% FBS. After seeding, plates were allowed to rest in the culture hood for 30–45 min to reduce edge effects prior to placement in an incubator overnight. The Seahorse XFe96 sensor cartridge was prepared with cell culture-grade sterile water (Corning, Corning, NY, USA) and placed into a 37 °C non-CO2 incubator along with Seahorse Calibrant solution. The following day, the hydrated cartridge was switched to calibrant and placed back into the non-CO2 incubator. Seahorse DMEM assay medium was prepared by supplementing it with pyruvate, glutamine, and glucose to mimic the DMEM the cells were grown in; phenol red and serum were not included in the seahorse assay medium. The complete Seahorse DMEM assay medium was then warmed in a 37 °C water bath until needed. H2O2 was added to wells for a final concentration of 500 μM and left for 1 h in the incubator. After the hour, either dual-conjugate or control groups (unconjugated mixture, free enzymes (SOD-1–catalase) and anti-ICAM-1 antibody) were added to a final concentration of 100 ng/mL in each well.
The Seahorse XF Cell Mito Stress Test Kit was prepared according to Agilent instructions and reagents were diluted in seahorse DMEM. Final concentrations injected into each well during the assay were as follows: oligomycin at 1.5 μM (mixture of oligomycin A, B, C with A ≥ 60%), carbonyl cyanide-4 [trifluoromethoxy] phenylhydrazone (FCCP) at 0.5 μM, and a mixture of rotenone–antimycin A at 0.5 μM. Oligomycin, FCCP, and rotenone–antimycin were then very carefully loaded in the Seahorse XF cartridge into ports A, B, and C respectively. After loading, the cartridge was placed back into the non-CO2 incubator until the assay was ready to start.
After the 2 h H
2O
2/drug exposure window, standard DMEM without serum was then carefully removed from the cell microplate media and the cells were washed twice with Seahorse assay medium. After the final wash, assay medium was added to the wells and the microplate was placed into the non-CO
2 incubator for 45 min to degas any CO
2 in the wells. The cartridge was added to the Seahorse XFe96 first to allow for calibration and then the microplate with cells was placed into the instrument. The standard Mito Stress Test assay parameters were initiated in Seahorse Wave Controller 2.6.3.8 software (Agilent) and the assay was run as per the manufacturer’s instructions [
25].
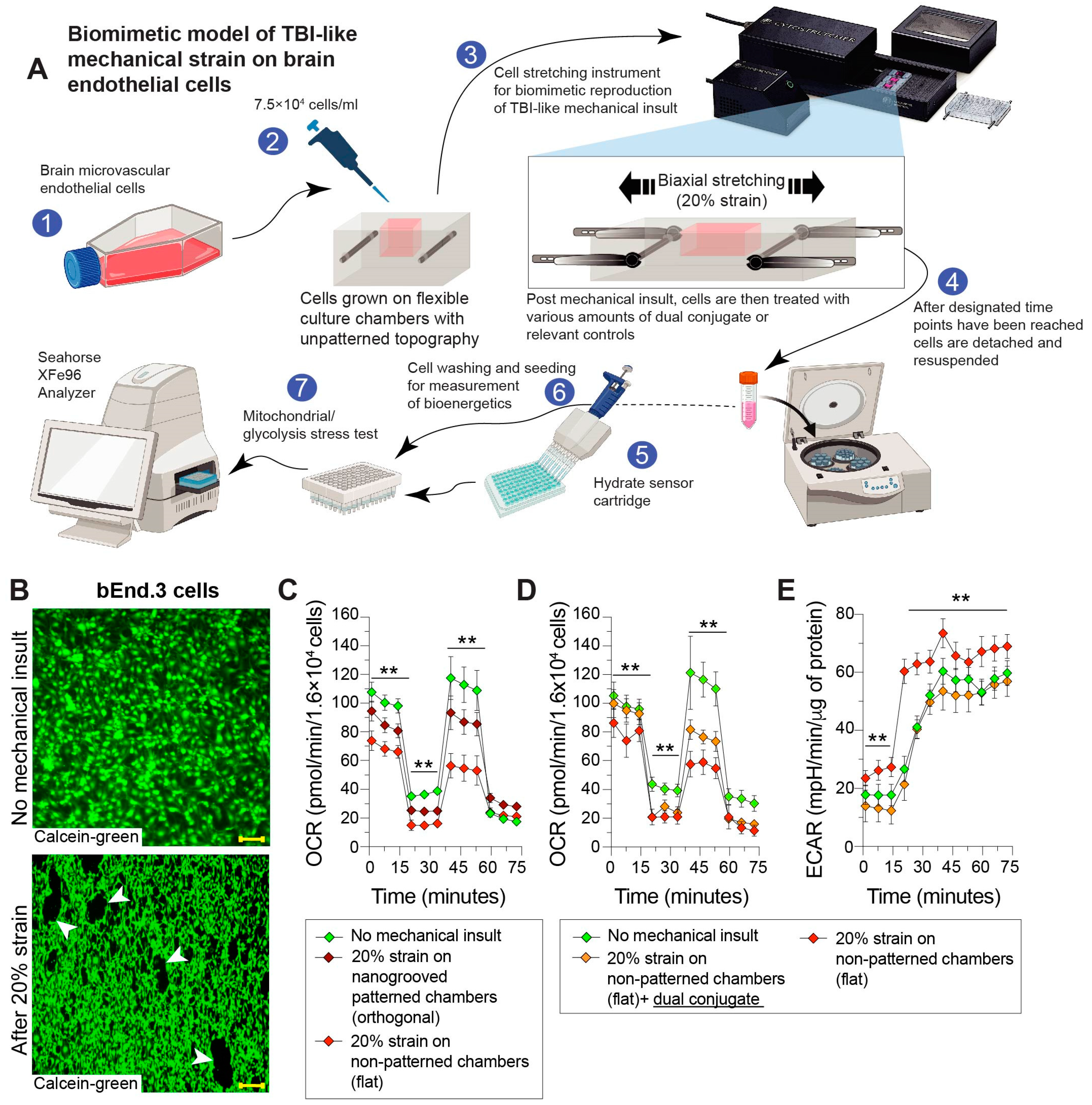
2.5. Mechanical Stretch Injury to Cells (In Vitro TBI Model)
A Cytostretcher mechanical stretch device (Curi Bio Inc., Seattle, WA, USA) with stretch flexible cell culture chambers was used to generate mechanical strain. Membranes were washed with cell culture-grade sterile water, autoclaved and then plasma treated at 100 mTorr for 5 min prior to use. After plasma treatment, the membranes were collagen I-coated overnight. Cells were seeded at a density of 75,000 per Cytostretcher chamber (144 mm2, unpatterned “flat” surface topography membranes) in 10% FBS DMEM media. Medium was changed daily until cells formed a confluent monolayer (~6 days). After monolayers were formed, the cells were switched to serum-free media. The Cytostretcher device was housed within the incubator with the internal region of the device (where the membranes are housed and stretched) and maintained at 37 °C and 95% humidity. Using the NanoSurface Operational Mechanics Interface (NaOMI) computer software (Curi Bio, Seattle, WA, USA Version 4.1.8), stretch parameters were programmed in. All membranes tested were stretched by 4 mm, which is equivalent to a 20% strain on the cell monolayer. Additionally, there was a 0.5 s dwell time after stretching and before compression, thereby mimicking the biomechanical forces commonly observed in TBI acceleration/deacceleration-associated injury. After the cell-containing membrane chambers were stretched, they were immediately exposed to dual-conjugates (100 ng/mL final concentration) and controls (100 ng/mL final concentration), or vehicle.
After the treatment period, media were removed and cells washed with 1× PBS twice. Following the second wash, Accutase (Sigma Aldrich, St. Louis, MO, USA) was added to each membrane to lift the cells for further analysis. Cells were spun at 1000 RPM for 5 min and then resuspended with 10% FBS DMEM. Cells were then plated onto Seahorse microplates at 16,000 cells per well and incubated for 1–2 days for later bioenergetic assays, as outlined above.
2.6. Vertebrate Animals and Controlled Cortical Impact (CCI) Traumatic Brain Injury Model
The Institutional Animal Care and Use Committee (IACUC) at Temple University (Philadelphia, PA, USA) approved all procedures detailed in this study that required the use of vertebrate animals prior to initiating any experimental objectives. Additionally, all methods were performed in full compliance with Temple University’s IACUC policies and the National Institutes of Health (NIH) ethical guidelines. Six-week-old male C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Animals were housed in cages and allowed to acclimate for two weeks in the Temple University Central Animal Facility. The animals were provided standard environmental enrichment conditions and were fed a commercial pellet diet and water ad libitum. Animals were weighed daily before and after surgery to ensure a stable body weight prior to euthanasia. The mice were anesthetized using 5% (induction) and 2% (maintenance) isoflurane in oxygen; consciousness was checked by hind paw pinching throughout the surgery. Surgical procedures for the experimental TBI model were followed as previously described [
23].
Briefly, a 4 mm craniectomy was performed over the right somatosensory cortex between bregma and lambda suture lines. Artificial cerebrospinal fluid was dropped onto the surgical area as needed to prevent drying out and overheating from drilling. A moderate controlled cortical impact (CCI) was delivered at a speed of 3.5 m/s with a compression depth of 1 mm using an Impact One™ Stereotaxic CCI Instrument (Leica Microsystems; Buffalo Grove, IL, USA) outfitted with a 2 mm-diameter piston. The dwell time was 0.5 s. After impact, the craniectomy was sealed with a 5 mm glass coverslip (Electron Microscopy Sciences; Hatfield, PA, USA) using VetBond to allow for monitoring of the impact site.
All animals were individually housed following the CCI-TBI procedure. Naïve animals did not undergo any surgical intervention. Dual conjugates (1.0 mg/kg of anti-ICAM-1–SOD-1–catalase) or an unconjugated mixture of anti-ICAM-1, SOD-1, and catalase (1.0 mg/kg) were administered by retro-orbital injection 4 h following impact. Experimental groups were as follows: non-craniotomized controls (naïve), CCI-TBI only, CCI-TBI + dual conjugates, or CCI-TBI + unconjugated mixture.
2.7. Immunohistochemistry
Immunohistochemistry was performed on brain tissue segments to evaluate the extent of neuroinflammation and neuropathology in all experimental groups listed above. At 48 h after CCI-TBI, mice were anesthetized and then transcardially perfused with PBS followed by Poly/LEM fixative (Polysciences, Inc., Warrington, PA, USA). Perfused brains were removed from the skull and placed in Poly/LEM fixative for 24 h at 4 °C. Brains were then dissected into 2 mm segments using a stainless-steel brain matrix (CellPoint Scientific, Inc.; Gaithersburg, MD, USA). Segments were post-fixed in Poly/LEM fixative at 4 °C for an additional 24 h. Next, segments were washed with PBS, processed using a Tissue-Tek VIP6 (Sakura Finetek USA, Inc., Torrance, CA, USA), paraffin-embedded using a TN-1500 Embedding Console System (Tanner Scientific, Inc., Sarasota, FL, USA), and sectioned using a rotary microtome (Leica Microsystems, Inc., Buffalo Grove, IL, USA). Paraffin-embedded sections (5 μm) from each experimental group were cleared, rehydrated and stained with antibodies against NeuN, GFAP or Iba-1 to determine neuronal survival, extent of astrogliosis, or microglial activation. respectively. in the region of impact. Prior to primary antibody incubation. Sections stained for GFAP and Iba-1 were HIER-pretreated with 10 mM citric acid buffer (pH 6.0). Sections stained for NeuN received no pretreatment. All sections were incubated in primary antibody prepared in Dako Antibody Diluent either for 1 h at RT (NeuN, Iba1) or overnight at 4 °C (GFAP) at the following dilutions: NeuN (1:500, Abnova Corp., Walnut, CA, USA), Iba-1 (1:400, (Fujifilm Wako Chemicals Inc., Richmond, VA, USA), GFAP (1:2000, Cell Signaling Technologies Inc., Danvers MA, USA). Positive antibody staining was detected using an HRP- or AP-conjugated labeled polymer system (ImmPRESS Staining Kits, Vector Laboratories, Newark, CA, USA) and subsequently visualized using Sigma DAB (NeuN and Iba1) or Vector Blue (GFAP). Sections were then dehydrated, and cover-slipped in preparation for imaging.
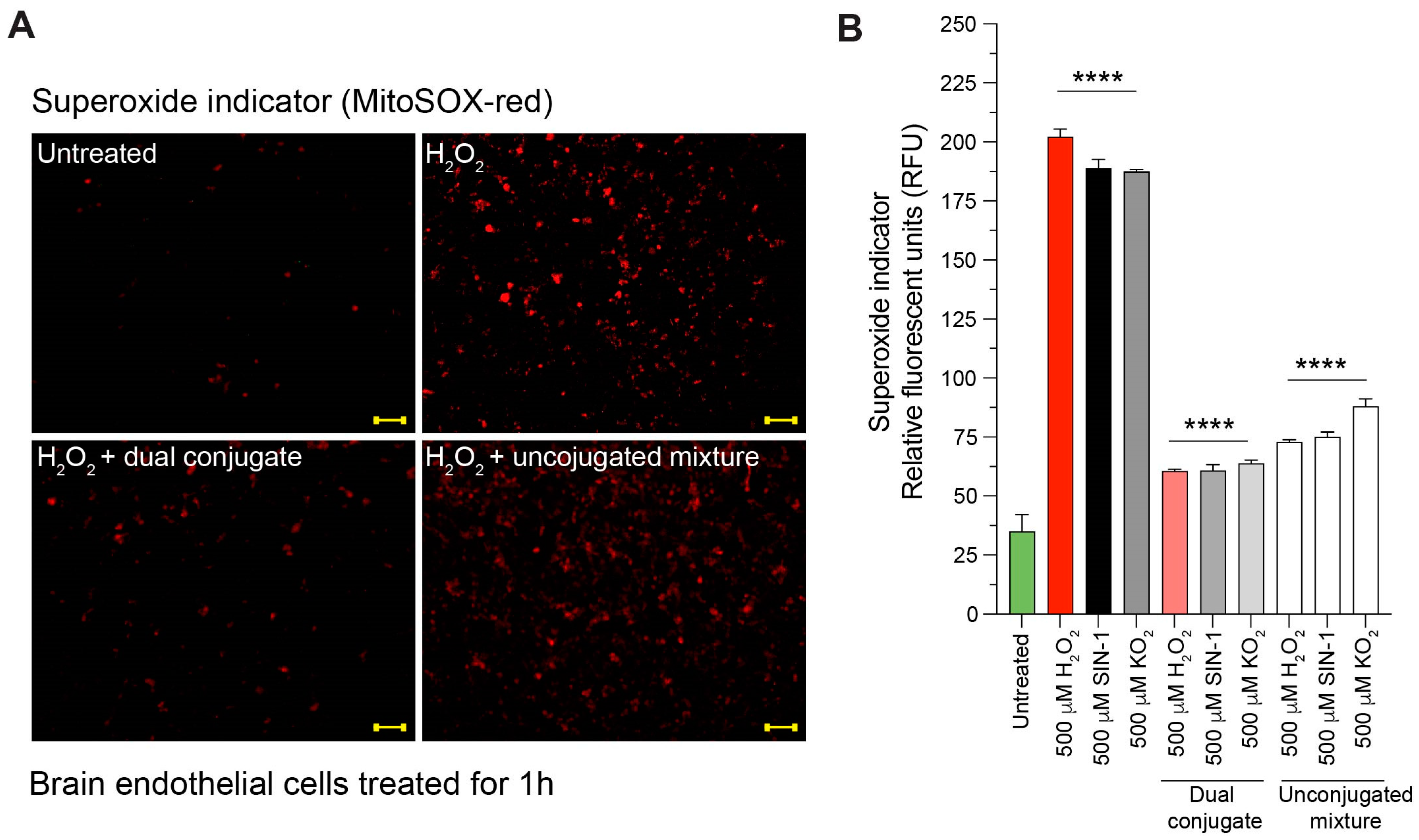
2.8. Reactive Oxygen Species (ROS) Detection Assay
Cells were seeded at a density of 55,000 per well on a 24-well cell culture plate and allowed to grow until confluent monolayers were formed. Prior to exposure, wells were changed to 0% FBS DMEM media and cells were given time to adjust. MitoSOX red (Thermo Fisher Scientific, Waltham, MA, USA) was prepared in sterile, anhydrous DMSO to make a stock solution of 5 mM as per manufacturer instructions. A working solution was then prepared in DPBS with calcium and magnesium to yield a 500 nM working solution (protected from light at all times). Cells were exposed to either H
2O
2, SIN-1, and/or 1 mM KO
2 (superoxide anion solution), as indicated in the Results. The superoxide anion solution was prepared as described previously [
26,
27]: 60 mg of 18-crown-6 ether was made into a fine powder and immediately dissolved in 10 mL of sterile, anhydrous DMSO, and 7 mg of powdered KO
2 was quickly added to the solution to prevent any reactions with water vapor in the air. This mixture was then heated in a water bath for 10 min followed by vigorous mixing on a shaker for 1 h until a pale-yellow solution was formed with no remaining crystals of KO
2. Appropriate concentrations of the mixtures were added to their respective wells and incubated for 1 h. Next, 100 ng/mL of dual conjugate or the unconjugated mixture were added, and the plates were once again incubated for 1 h. Cells were then washed with media to remove reagents and drugs, and then 1 mL of the MitoSOX red working solution was added to each well. Plates were then covered and incubated for 45 min, after which they were washed 3×with warm DPBS with calcium and magnesium. Cells were imaged using an EVOS FL Cell Imaging System (Thermo Fisher Scientific, Waltham, MA, USA). In order to quantify the fluorescent data from the above experiments, 96-well black/clear-bottom plates were used and the wells were read on a plate reader (SpectraMax M5e, Molecular Devices, San Jose, CA, USA) using the spectral properties provided by the manufacturer.
2.9. Immunofluorescence Assay
Immunofluorescence detection of ZO-1 was performed as previously described [
28]. Briefly, cell monolayers were washed with 1× PBS and fixed with 4% paraformaldehyde for 10 min. Cells were then washed with 1× PBS, blocked and permeabilized with 5% donkey serum and 0.3% Triton-X for 20 min. To visualize the tight-junction protein zonula occludens 1 (ZO-1), cells were stained anti-ZO-1 (BD Biosciences, Franklin Lakes, NJ, USA) and diluted at 1:100 for one hour at RT. Secondary antibody anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) was used at a dilution of 1:500 for 1 h at RT. The cell nuclei were counterstained with DAPI and the monolayer was mounted with ProLong antifade reagent. Images were acquired using a Nikon A1R inverted confocal microscope (Nikon Instruments, Tokyo, Japan).
2.10. Imaging and Image Analysis
Analysis of chromogen immunostaining of GFAP, Iba-1, and NeuN was performed with FUJI/ImageJ analysis software (Version 1.54g; NIH, Bethesda, MD, USA) using manual counting or particle counting on high-resolution images taken with a Nikon 80i microscope (Nikon instruments, Tokyo, Japan; 5/sample, 3/group, taken at 20× objective magnification) on immunolabeled cells. For manual counting, DAB+ nuclear staining in sections immunolabeled for NeuN were counted in an area of 2.5 × 105 microns2. The hematoxylin counterstains nuclei to aid classification of DAB+ NeuN live or dead neurons. GFAP manual counting for activated astrocytes was performed based on density/color intensity above twice the threshold of that found in the naïve controls. DAB+ IHC for IBA-1 was counted as positive based on resting or activated microglia based on classical morphological presentation of their cellular status. For automated particle counting, the images were calibrated and then sequentially processed for background subtraction, color threshold segmentation, and binary conversion (with application of the watershed function) based on the above indicated parameters for manual counting. Cells identified from the above parameters were counted in the entire area of the image by use of the “analyze particles” function (selected by particle area and circularity).
Assessment of dual-conjugate cellular binding and internalization was performed by indirect immunofluorescence and confocal imaging. bEnd.3 cells were grown to confluence and then stimulated with 50 ng/mL of LPS (Sigma, St. Louis, MO, USA) overnight. Cells were then exposed to 100 ng/mL of dual conjugate. Cells were fixed/permeabilized in 4% formaldehyde for 15 min. The fixative was rinsed and permeabilization solution consisting of 0.1% Triton X-100 in 1× PBS was added to the cells for 10 min. Cells were placed in blocking solution (1% donkey serum in 1× PBS) for 1 h and then rinsed 3× with 1× PBS. The primary antibodies anti-ICAM-1 (1:200, Abcam, Waltham, MA, USA), anti-SOD-1 (clone 2F5, 1:200, Thermo Fisher Scientific, Waltham, MA, USA) were diluted in blocking solution. Primary antibodies were incubated with the cells overnight at 4 °C. Cells were then rinsed 3× in 1× PBS and secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA) were added for 2 h at room temperature. Secondary antibodies used were anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 594. Cells were then rinsed 3× with 1× PBS and incubated with the nuclear stain counterstain DAPI (4′,6-diamidino-2-phenylindole, Invitrogen/Thermo Fischer Waltham, MA, USA), then rinsed with 1× PBS, and mounted using ProLong antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA). Immunofluorescence was visualized using a Nikon A1R confocal microscope (Nikon Instruments, Tokyo, Japan). Images were acquired with Nikon NIS Elements AR imaging software (Version AR 2.2; Nikon, Tokyo, Japan).
2.11. Statistical Analysis
For in vivo experiments, all animals were randomly assigned to an injury group and endpoint time (n = 3). Tissue was isolated from both the contralateral and ipsilateral hemispheres to the craniectomy and the corresponding anatomical location in naïve mice. Data from image analysis were compared using one-way ANOVA and Dunnett’s post hoc test for multiple comparisons against the naïve control. All data are expressed as means ± S.E.M. For all in vitro cellular and biochemical assays, the experiments were conducted at least three times (three to four replicates per experiment) and compared using one-way ANOVA with post hoc Tukey’s or Dunnett’s test, as indicated in the Results section. Results are expressed as means ± S.E.M. All data were analyzed with GraphPad Prism software v9.5.1 (GraphPad, San Diego, CA, USA).
4. Discussion
According to the CDC’s latest reports from 2021, there were 69,413 TBI-related deaths, and nearly 2500 of those were children or adolescents. Children and teenagers are particularly vulnerable to head injuries, as the sequelae of TBI disrupt normal neurocognitive and behavioral development [
43]. While public awareness of concussions, TBI, and their dangers has grown significantly, there are still no effective clinical options to treat patients of all ages [
44]. Our group’s work is a step in the direction of filling this therapeutic gap for patients suffering from TBI.
It is known that a TBI will cause damage to the BBB, leading to endothelial cell activation, inflammatory responses and potentially a BBB breach, allowing toxic substances to enter the CNS parenchyma [
45,
46]. Previously, we explored attenuating the oxidative stress response following TBI in a mouse model using a novel targeted single antioxidant enzyme therapy [
23]. Also, our team pioneered the strategy of vascular immunotargeting of drugs and drug carriers to surface determinants of the endothelial cells [
47,
48]. Studies of targeting antioxidant agents to PECAM-1, stable and highly expressed pan-endothelial cell adhesion molecule, provided the proof of concept for precise antioxidant interventions detoxifying endothelial ROS [
49]. More recently, we expanded the arsenal of the targeting of SOD or catalase to ICAM-1, providing advantages of enhanced delivery to pathologically activated endothelial cells and relatively well-controlled intracellular delivery of the antioxidant cargoes [
50,
51,
52,
53]. The current study builds on this work by now using two antioxidant enzymes (rather than a single one, previously tested) conjugated with ICAM-1. Here, we demonstrate that this novel dual-conjugate construct (anti-ICAM-1–SOD-1–catalase) protects brain endothelial cells from the deleterious effects of TBI by counteracting BBB leakiness and the increased levels of free radicals while promoting mitochondrial health.
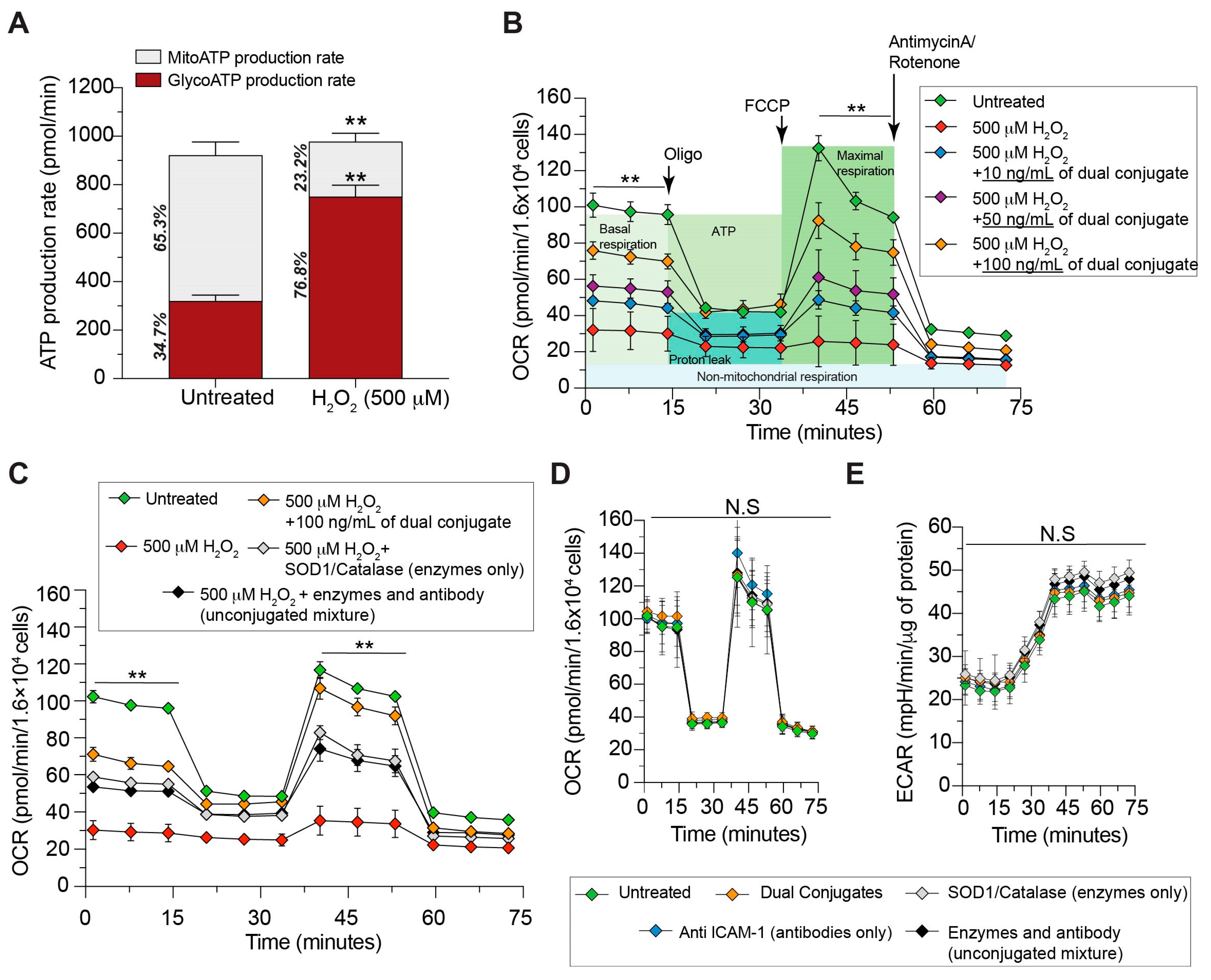
Mitochondria are damaged in the primary injury of TBI through excessive stretching forces as well as the secondary injury phase by both nonmitochondrial and intramitochondrial biochemical cascades. Outside the mitochondria, excitotoxicity leads to increased intracellular calcium ion levels, which in turn activates pro-oxidative enzymes such as xanthine oxidase and nitric oxide synthase. Within the mitochondria, there are numerous biochemical pathways and enzymes, including at the electron transport chain or ETC, which generate large amounts of ROS. These systems produce free radicals such as superoxide and ROS like H2O2 that under physiological conditions are reduced by endogenous antioxidant enzymes. Following TBI, however, the natural antioxidative protective systems quickly become overwhelmed by the massive increase in ROS, which subsequently leads to intracellular damage through cell membrane lipid peroxidation, DNA damage, and inhibition of the ETC function. Two major imbalances then follow: one is in the redox status of the cell, while the other corresponds to the energetics within the cell that can ultimately lead to cell death unless corrected. The cell has a high energetic demand to repair the damage caused by TBI that is hampered, as damaged mitochondria are unable to provide the ATP needed for repair mechanisms to fully function. Therefore, we investigated the effects of direct ROS exposure to monolayers of brain endothelial cells along with the anti-ICAM-1–SOD-1–catalase dual conjugates. We sought to determine the level of mitochondrial damage induced by H2O2 as measured by ATP assays and the cellular bioenergetics measuring platform of Agilent’s Seahorse Mito Stress assays.
H
2O
2 causes the cells to enter a highly glycolytic state (
Figure 2A), shifting ATP production away from the more efficient process of aerobic respiration. Additionally, the H
2O
2 resulted in a drastically reduced ability of cells to respond to mitochondrial stress in the form of an uncoupling agent during the Mito Stress Test (
Figure 2B). A metabolic shift towards glycolytic ATP production as well as an inability to accommodate the increased energy demands are pathological hallmarks following TBI.
Figure 2B shows that the dual conjugate was able to rescue these deficits in a dose-dependent fashion, suggesting that reducing H
2O
2 following TBI via targeted antioxidant delivery (
Figure 2C) is advantageous against untargeted antioxidant enzyme delivery to brain endothelial cells.
Next, the ability for the dual conjugates to provide protection in the context of a mechanical stretch injury applied to the cells was evaluated. Of note, the strain was intended to simulate similar stretching and shear forces experienced by cells during a TBI [
54]. To our knowledge, this is the first example of combining the use of a stretch device to mimic TBI-associated forces with the Seahorse technological platform to determine cellular bioenergetics (
Figure 4A).
Figure 4D shows that the stretch-injured cells follow the abovementioned patterns of TBI-damaged cells: an increased reliance on glycolysis for ATP production as well as the inability to properly respond to stress. These cells display little to no metabolic reserve postinsult, posing a key limitation for brain endothelial cell recovery from injury. Administration of the dual conjugate provided the cells with greater resilience (reserve) following stretch injury. As such, they had increased metabolic reserve, as observed by OCR readings of patterning closer to measurements seen in the sham condition. ECAR in cells treated with the dual conjugate also followed the sham condition, whereas the stretch-injured cells showed a rapid increase in the rate of glycolysis (
Figure 4D). These results suggest that the targeted delivery of the dual conjugates to injured brain endothelial cells conveys near-immediate metabolic benefits, as measured by mitochondrial health, allowing faster cellular recovery.
Another hallmark of TBI is disruption of BBB function and stability at the level of brain microvascular endothelial cells [
55,
56,
57]. Mechanical or chemical injury can cause a breakdown of the BBB and the tightness between cells of the monolayer [
58,
59]. Therefore, we set forth to determine how H
2O
2 can alter the permeability of the BBB and if the dual conjugates could have a healing effect in the context of such an injury. Low-molecular-weight FITC–dextran (4 kD) was used to measure BBB permeability after injury at both early (1 h) and late (3 h) time points in comparison to groups treated with the dual conjugates. Our results suggest that H
2O
2 transiently increases the leakiness of the endothelial cell barrier, whereas treatment with the dual conjugates prevents the increase in paracellular permeability to a level approaching control groups. The results point to a direct benefit on barrier integrity when free radical damage is minimized following the use of this targeted intervention.
TBI induces an immediate production of superoxide, which begins a cascade of damaging oxidative stress, starting with the breakdown of superoxide into oxygen and H
2O
2. Additionally, superoxide can react with nitric oxide (NO), forming the highly oxidative peroxynitrite molecule, which can further cellular damage. Since our previous experiments explored the direct influence of H
2O
2 on dual conjugates or the indirect influence of the full superoxide cascade through mechanical stretch injury, we conducted experiments to directly measure the ROS reduction of the dual conjugates when exposed to superoxide, H
2O
2, or SIN-1, which can form both superoxide and peroxynitrite [
60]. Administration of H
2O
2, superoxide via KO
2 or SIN-1 resulted in clearly evident increases in ROS measured by superoxide MitoSOX red fluorescence in this particular set of experiments. Adding the anti-ICAM-1–SOD-1–catalase dual conjugate after administering the above chemical insults greatly reduced the ROS signal, whereas administration of the unconjugated mixture control group did not have as drastic an effect (
Figure 5). These results suggest that the targeted dual conjugates can have a beneficial effect on brain endothelial cells experiencing oxidative stress, such as that seen in TBI. It stands to reason that the anti-ICAM-1 antibody allows for the SOD-1 and catalase enzymes to be brought in close proximity to the cell, which helps accelerate the breakdown of superoxide and subsequently H
2O
2.
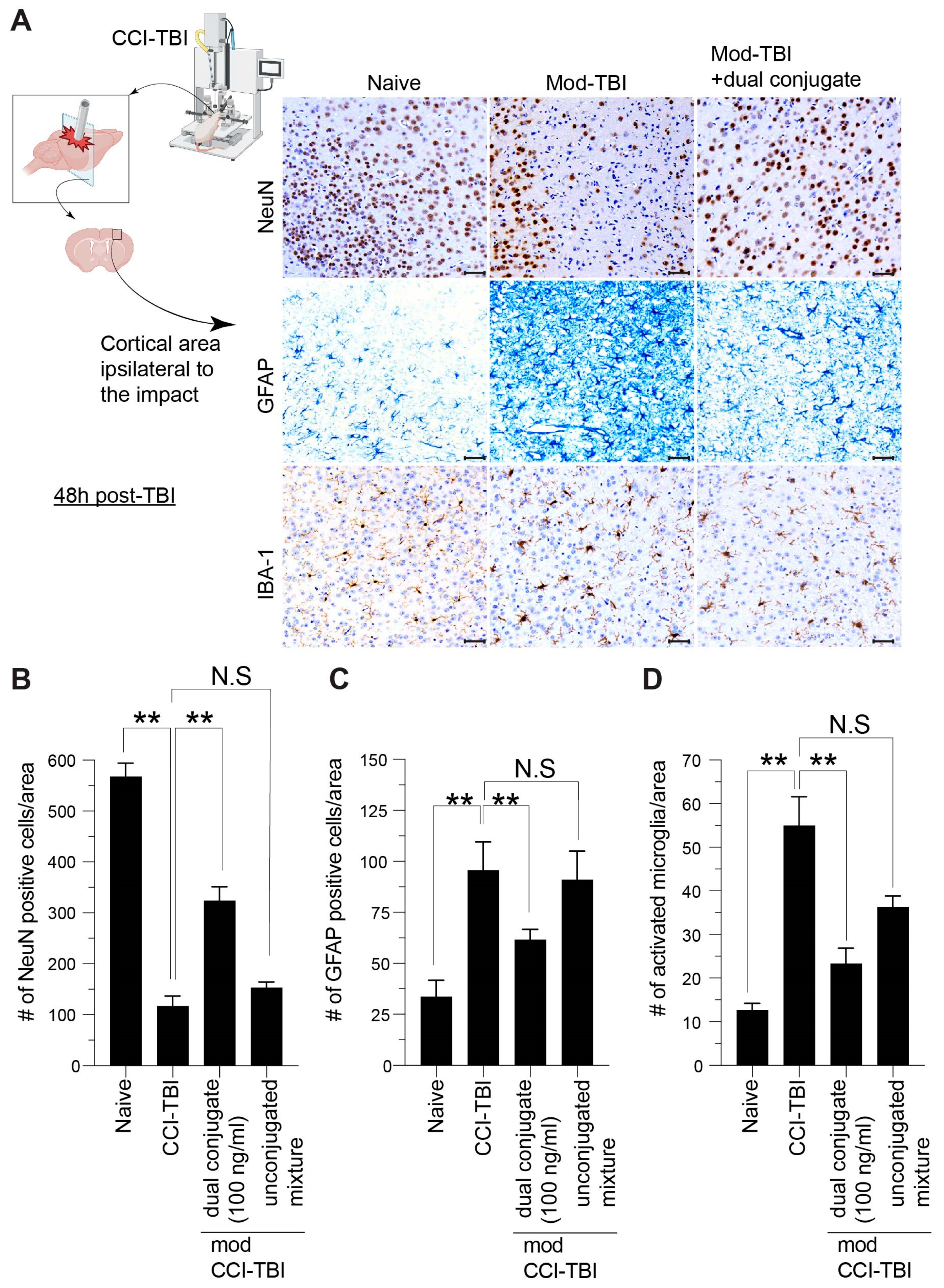
Although the abovementioned studies provided a better understanding of the benefits conveyed by the dual conjugates at the cellular level, we wondered whether similar outcomes would be observed in vivo. Therefore, the studies here also attempted to gather information as to the therapeutic effectiveness of the dual conjugate in vivo via a controlled cortical impact mouse model of TBI. Established neuropathological indices of TBI were used to conduct this study, NeuN to monitor cortical neuron loss after injury along with GFAP and Iba-1 for astrocytic and microglial activation, respectively. These markers provide similar tissue pathology in both mouse models of TBI and in humans who have experienced neurotrauma [
61]. Consistent with our previous published work along with other groups, in moderate CCI-TBI, affected mice displayed decreased NeuN levels concurrent with increased markers of neuroinflammation—GFAP and Iba-1. In TBI patients, the resulting chronic neuroinflammation can be extremely debilitating, leading to cognitive and motor deficits along with other comorbidities, such as substance abuse [
62]. Targeting neuroinflammation in the acute phase of TBI through reducing the overabundance of ROS could help reduce persistent activation of astrocytes and microglia (
Figure 6). Significantly, administration of anti-ICAM-1–SOD-1–catalase 4 h after CCI-TBI resulted in increased neuronal survival and decrease in glial activation, whereas the unconjugated mixture of anti-ICAM-1, SOD-1, and catalase did not convey the same beneficial effect (
Figure 6). These results provide strong evidence that targeted antioxidant enzyme therapy to the brain vasculature is highly effective for rendering neuroprotection following a brain injury.
The results presented in this report provide strong proof of concept that two potent antioxidant enzymes can be covalently fused to a targeting antibody in a single construct that is both biologically active and targets inflamed brain endothelial cells. It remains to be tested, but it is thought that such an approach of minimizing trauma-associated neuroinflammation could also extend to other pathological conditions in which the cerebral vasculature is activated and the BBB is compromised.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}