Abstract
Post-Translational Modifications (PTMs) are covalent changes to amino acids that occur after protein synthesis, including covalent modifications on side chains and peptide backbones. Many PTMs profoundly impact cellular and molecular functions and structures, and their significance extends to evolutionary studies as well. In light of these implications, we have explored how artificial intelligence (AI) can be utilized in researching PTMs. Initially, rationales for adopting AI and its advantages in understanding the functions of PTMs are discussed. Then, various deep learning architectures and programs, including recent applications of language models, for predicting PTM sites on proteins and the regulatory functions of these PTMs are compared. Finally, our high-throughput PTM-data-generation pipeline, which formats data suitably for AI training and predictions is described. We hope this review illuminates areas where future AI models on PTMs can be improved, thereby contributing to the field of PTM bioengineering.
1. Introduction of Post-Translational Modification
1.1. Definition of Post-Translational Modification
Post-Translational Modifications (PTMs) are covalent alterations to one or more amino acids (AAs) that occur after translation from mRNA to polypeptide chains [1]. These alterations often occur on polar AAs, as well as non-polar residues at the N-termini of proteins [2]. There are more than 400 known types of PTMs, but the most abundant are phosphorylation, glycosylation, acetylation, methylation, and ubiquitin/ubiquitin-like modifications (Figure 1) [1,3,4,5]. In addition, oxidation of methionine and thiol-oxidation of cysteine [6] have been studied as prevalent PTMs in many different organisms.
PTMs come in two forms: reversible/irreversible covalent side chain edits and irreversible peptide backbone cleavage (i.e., proteolytic cleavage) [7]. Changes to side chains (e.g., glycosylation, phosphorylation, and methylation) are either reversible or irreversible, and many residues on a single protein may have side chain modifications, and these side chains can be modified more than once [5].
PTMs may affect the shape and electrostatic properties of the modified residues, therefore having important implications on protein structures and functions, such as expression [8], degradation, protein–protein interaction, catalytic activity, conformational change, and binding to DNA or RNA (Figure 1) [4]. For example, phosphorylation refers to the adding of a phosphoryl group to the side chain of an AA. The phosphoryl group is usually transferred from a donor such as a nucleotide triphosphate or other phosphoryl donor by protein kinases and may be removed by phosphatases [9]. Phosphorylation commonly occurs on serine, threonine, and tyrosine residues. Acetylation can also occur non-specifically, in response to epigenetics regulation, protein damage, aging processes [10]—particularly when there is an excess of acetyl-CoA or acetyl phosphate present. In epigenetic regulation, lysine acetylation on histones influences DNA accessibility and chromatin compaction. Cysteine may undergo PTMs such as nitrosylation, sulfenylation, and glutathionylation. Therefore, along with other physiological factors (e.g., body temperature, infection, inflammation), PTMs contribute to explaining how a limited number of genes can result in a wide range of proteoforms [11] that expand phenotypic diversity [5].
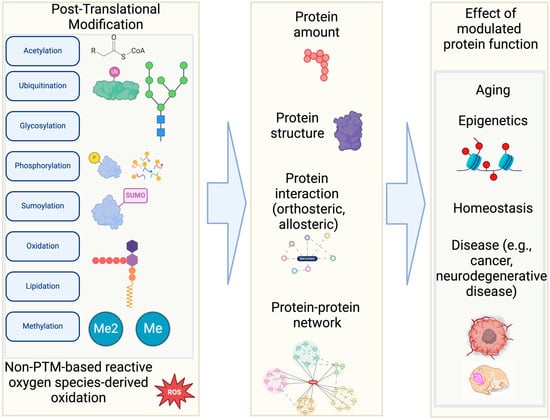
Figure 1.
Various types of Post-Translational Modifications and their effects. This illustration shows several common types of PTMs out of 400 different types. Although these PTMs are often less represented in many computational modeling programs, they control various cellular activities. This figure is generated by BioRender and NIH BIOART [12].
Figure 1.
Various types of Post-Translational Modifications and their effects. This illustration shows several common types of PTMs out of 400 different types. Although these PTMs are often less represented in many computational modeling programs, they control various cellular activities. This figure is generated by BioRender and NIH BIOART [12].
1.2. Importance of Post-Translational Modification
A substantial portion of PTMs can occur incidentally and do not lead to significant functional changes. This is understandable, as most PTMs minimally alter protein structures. For instance, the majority of backbone conformation changes due to phosphorylation are small (median root mean squared deviation-RMSD of 1.1 Å). Only 13% of phosphorylation events cause a structural change greater than 2 Å RMSD; even when focusing on backbone changes, only 28% result in a structural shift exceeding 2 Å RMSD [13].
When PTMs affect protein function, their impact is significant [14], and PTMs that cause larger structural changes tend to correlate more strongly with functional changes [13]. For instance, PTMs are essential for regulating every stage of the nicotinic acetylcholine receptor life cycle, encompassing receptor expression, membrane stability, and function [15]. As a result, PTMs are frequently targeted for disease treatment and detection. For example, cancer therapies have been developed to control the addition or removal of PTMs contributing to the disease [2]. From a metabolic perspective, understanding the role of mitochondria-related PTMs in tumorigenesis is anticipated to guide new approaches for next-generation cancer therapies, as PTMs play a pivotal role in regulating mitochondrial functions in cancer [16].
PTMs can also serve as biomarkers for disease detection. For example, elevated hemoglobin A1C levels are commonly used to diagnose and monitor diabetes management. Hemoglobin A1C is glycosylated when exposed to glucose in the blood, and high blood glucose levels in diabetes result in increased hemoglobin A1C levels [3]. Another example is the use of PTM-related enzymes as biomarkers in glioblastoma multiforme [17].
Given their many uses in clinical and molecular contexts, there has been much effort put towards PTM site discovery and characterizing their respective functions. In particular, irregular phosphorylation is one of the mechanisms underlying the development of many cancers [9,18]. Thus, PTMs are an important subject of study in understanding cellular and molecular regulatory systems.
1.3. Significance of Artificial Intelligence on Post-Translational Modification Research
Computational modeling for PTMs involves multiple complex factors. For instance, whether a sequon is N-glycosylated depends on various factors such as the distance to the next glycosylation site and the surrounding sequences. The need to incorporate complex factors into structural modeling for PTM prediction arises because most PTMs—including phosphorylation—induce only small-scale, stabilizing conformational changes by modulating local residue fluctuations through conformational selection [13]. However, there is a vast number of residues with PTMs, and they are often unstable and low in abundance. Therefore, it has been challenging to conduct large-scale identification and functional characterization using conventional lab methods such as mass spectrometry [19]. Consequently, deep learning (DL) approaches, including protein space embedding large language models, can address these complexities more effectively than traditional machine learning (ML) or shallow neural network models [20]. Thus, DL approaches with large datasets are particularly valuable for extracting meaningful insights related to PTM [20,21]. These methods allow us to use combinations of sequence, structural, and other information to identify residues that may harbor PTMs and their possible implications on function.
Therefore, in this review, we discuss recent advancements in structural and sequence-based PTM research leveraging artificial intelligence (AI), explore related non-canonical amino acid studies, and examine experimental data generation as well as the utilization of current PTM databases (DBs).
2. Computational Modeling for PTM Research
2.1. Structural Modeling for PTM Prediction
PTMs often occur on polar residues and non-polar residues near the N-termini. Thus, protein sequence is the most basic input in many computational tools to predict PTM location and function. In addition, some sequons with conserved motifs also give information on where PTMs may be located. For example, the N-X-[S/T] sequon is the site in which glycosylation occurs [20].
However, sequence information is not always sufficient to predict PTM site location or function. Structural information is also crucial for PTM modeling. For example, tools like StructureMap [22] and a specific phosphorylation site prediction program (i.e., PhosAF) [10] suggest that a variety of structural factors (e.g., solvent accessible surface area, dynamic region of structure represented by a predicted local distance difference test—pLDDT) are crucial for understanding the preferred locations and functions of PTMs. These structural requirements are logical, as exposed areas are typically more functionally relevant [13,23] and accessible to “writers” that introduce PTMs. Additionally, pLDDT scores can help identify short intrinsically disordered regions (IDRs), where PTMs are more frequently located. Moreover, conserved sequences alone do not ensure N-linked glycosylation, as many motifs or sequons are buried, making them inaccessible to glycosylation enzymes [20].
In addition to rudimentary sequence and structural information, other advanced annotations have been integrated into PTM models. For example, the FuncPhos-STR tool integrates phosphosite sequence evolution and protein–protein interaction information into structural information from AlphaFold2 [24]. In addition, structural deep network embedding [25] was employed to transform the high-dimensional structural data of a protein–protein interaction network into a more manageable low-dimensional space.
2.1.1. PTM Structural Map
StructureMap analyzed structural trends of extensive lists of PTMs (i.e., phosphorylation, ubiquitination, sumoylation, acetylation, methylation, and glycosylation) [22]. Their analyses have shown that a significant number of phosphorylation sites are located within IDRs, many of which comprise the activation loops of kinases. This suggests a functional relevance to the flexible nature of these regions in protein phosphorylation. However, some analyses of these findings may have been at least slightly biased depending on the choice of datasets used. For example, certain datasets are derived from samples treated under specific conditions. Moreover, PTM sites in physically less accessible regions may have been underrepresented.
2.1.2. Structural Simulation to Study Non-Canonical Amino Acid Effects
PTMs can be considered naturally occurring types of non-canonical amino acids (ncAAs). With much larger chemical space than canonical amino acids (cAA) [26], ncAAs allow more possibilities of protein and peptide engineering [27]. For example, incorporations of ncAAs increased protease resistance [28], membrane permeability [29], and peptide binding affinity [30]. Therefore, various computational approaches have been developed to enhance the utility of ncAAs. For example, in silico screening with the Random Non-standard Peptide Integrated Discovery platform enhances library diversity and enables the discovery of diverse peptide scaffolds containing multiple ncAAs. This approach is particularly useful for identifying mutations possible at position 1 in thioether macrocycle discovery. It also removes the need to create multiple libraries with varying initiators, streamlining the process. Additionally, after the seminal paper of Rosetta-based ncAA rotamer library constructions [31], various computational designs [32,33,34] (e.g., an ncAA probe to study protein–peptide interactions [26] and peptide cylicization [35]) and force field developments [36] have been made. For example, Renfrew et al. have shown that replacing phenylalanine with 4-methyl-phenylalanine at the protein–peptide interface improved binding affinity by 2-fold [31]. Similarly, replacing non-fluorine AAs with fluorine AAs in the core region of helix bundle protein improved its thermal stability [37]. Designing peptides/proteins that are resistant to enzymatic degradation is important for successful biomarker effectiveness and more stable vaccine delivery. Therefore, there have been replacements of some L-AAs with D-AAs to improve peptide stability against protease [38,39,40]. Additionally, mirror images of all protein structures in Protein Data Bank [41] have been generated with the hope of providing potential drug leads [42]. However, D-AA substitutions in the middle of a peptide may disrupt surface topology, secondary structure, and function of the original peptide with L-AAs [40,42].
Force fields (FFs) for ncAAs have not been thoroughly developed for semi-empirical quantum mechanical calculation, and recent extended tight-binding quantum chemistry methods [43] have been tested for cAAs only [44]. However, FFs for empirical methods have been developed. For example, Khoury et al. developed ab initio-derived AMBER FF03 compatible charge parameters for 147 ncAAs including β- and N-methylated AAs [36]. Since more accurate protein FFs have been developed, further FF development for ncAAs is expected. One example of a more accurate protein FF is cross-term map (CMAP); a grid-based correction for the protein φ- and ψ-angular dependence of the energy was added to CHARMM22 FF [45]. The CMAP correction was validated by removing substantial deviations from experimental backbone root-mean-square fluctuations and N-H NMR order parameters [46].
Protein side chains can adopt various conformations influenced by the protein backbone angles (i.e., φ and ψ) and interactions with neighboring residues. These conformations, determined by one or more χ angles, are referred to as rotamers. A collection of such conformations is known as a rotamer library. Sampling expected or favorable rotamers is especially important for the protein core regions and surface regions if they are dominated by electrostatic interactions. In 2012, Rosetta software suite added rotamer libraries of 114 ncAAs [31]. However, more than 200 ncAAs can be incorporated into proteins in prokaryotic and eukaryotic systems [47]. Since the possible combination of these ncAAs can be exponentially large theoretically, scientists have been depositing ncAA rotamers (e.g., residue_types + patches) into Rosetta (1571 as of this writing).
In addition to the side chain, different backbones can also be modeled. Non-canonical protein backbones, such as oligooxopiperazines, oligopeptoids (peptoids are peptidomimetic oligomers that mimic the motifs of protein secondary structures), β-peptides, hydrogen bond surrogate helices, and oligosaccharides, have been utilized for structure prediction and the design of non-peptidic oligomer scaffolds [48]. For example, the Bonneau group assessed peptoid foldamer conformation as a conventional AA rotamer search [49]. Schneider et al. designed peptoid–peptide macrocycles to inhibit the β-catenin T-cell factor interaction in prostate cancer [50]. These rotamers have been manually parameterized with MakeRotlib [31], which is preceded by quantum mechanical (QM) calculation, OpenBabel [51], and molfile_to_params_polymer.py [52]. However, more automated ncAA rotamer samplings were developed. For example, AutoRotLib generates parameters for an ncAA rotamer library from Simplified Molecular Input Line Entry System code [26]. Unfortunately, license fees associated with using this software can be a barrier for potential users. On the other hand, BioChemical Library [53] automatically generates ncAA rotamer libraries as an open-source program. This highlights a broader impact of open science, as emphasized in other DL review [54]. Developing accurate rotamer libraries is particularly crucial for longer amino acids, such as asparagine, which exhibit a larger number of rotamer conformations [31].
For PTM-specific ncAA simulations, molecular dynamics (MD) simulations have been used. For example, Li et al. used MD simulation and molecular mechanics generalized Born/surface area (MM-GBSA) binding free energy calculations to understand the influence of phosphorylation on death-associated protein kinase 1 activity [55]. Similarly, Mejia-Rodriguez et al. developed a PTM-specific force field with QM calculation and studied the impact of S-nitrosylation of several cysteines using MD and docking simulations [56]. Other docking simulations to model covalent bonding include CovPepDock, which incorporates covalent binding between the peptide and a receptor cysteine [57], covalent labeling-guided protein–protein docking in Rosetta [58], and Meeko-derived [59,60] AutoDock-GPU [61].
2.2. Deep Learning Approaches for PTM
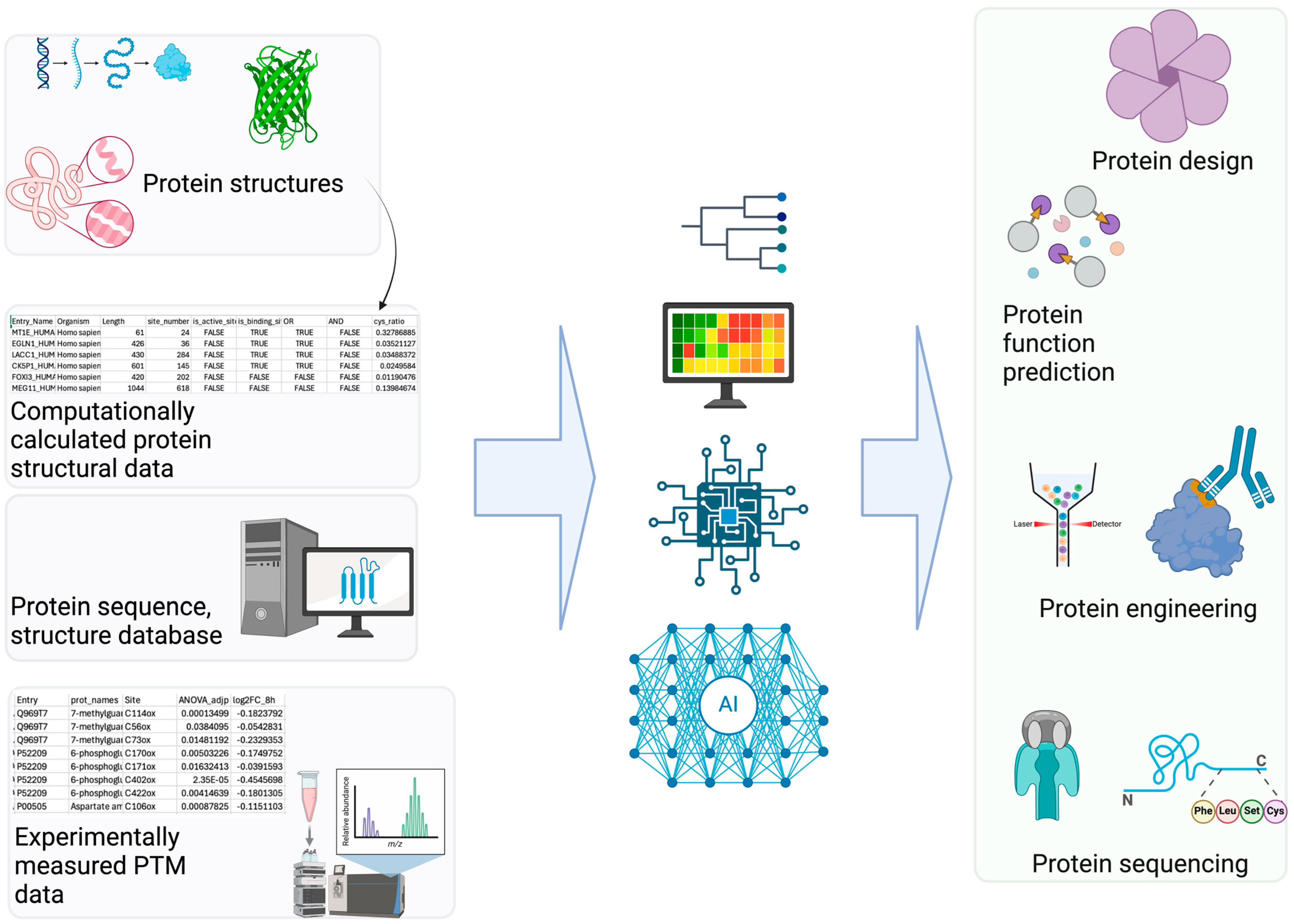
Computational PTM modeling (including site, structure, and function prediction) is a complex problem with numerous contributing features. Additionally, there are many types of data (i.e., sequence, structure, metadata such as species) for PTM modeling. DL is well suited for PTM applications (Figure 2) with its advantage of leveraging large quantities of data effectively [62,63]. DL often performs self-supervised learning more effectively than non-DL ML methods. This advantage of DL stems from its ability to process complex non-linear properties through deeper layers in neural networks compared to other non-DL-based ML methods. Therefore, DL methods often outperform non-DL-based ML methods for PTM modeling [20]. Good examples of DL-based PTM structural modeling includes RosettaFold All-Atom [64] and AlphaFold3 [65]. These programs can predict covalently modified protein structures including PTMs using a denoising diffusion probabilistic model [61]. Therefore, these overcome the limitations of other protein structure prediction models (e.g., OmegaFold [66], Chai-1 [67], ESMFold [68]). Additionally, Meiler and colleagues have incorporated ML into PTM prediction [69]. Specifically, they trained a site prediction model using TensorFlow for 18 of the most abundant PTMs (e.g., glycosylation and phosphorylation) and made it interoperable with existing Rosetta protocols [70]. Other efforts in PTM structural modeling include fine-tuning AlphaFold Multimer to predict phosphopeptide–protein interactions [71].
FuncPhos-STR makes predictions about phosphosite function. This tool uses phosphosite sequence evolution data with protein–protein interaction information, which allows them to integrate information about the structure and dynamics of the protein into their DL model [24]. Other tools investigate how mutations affect PTM sites in protein structure. For example, MIND-S does this using a graph neural network with multi-head attention [72]. Cao et al. applied DL methods to advance nanopore-based protein sequencing, extending its capabilities to the PTM level [73]. In their approach, a peptide is passed through a nanopore, causing changes in the electrical current that correspond to the different AA residues traversing it. They utilized a long short-term memory (LSTM) recurrent neural network to interpret these electrical signals and a multi-layer perceptron (MLP) to predict the peptide sequences. This methodology allows them to associate specific current patterns with PTM predictions, as modified residues exhibit distinct electrical signatures compared to their non-modified counterparts.
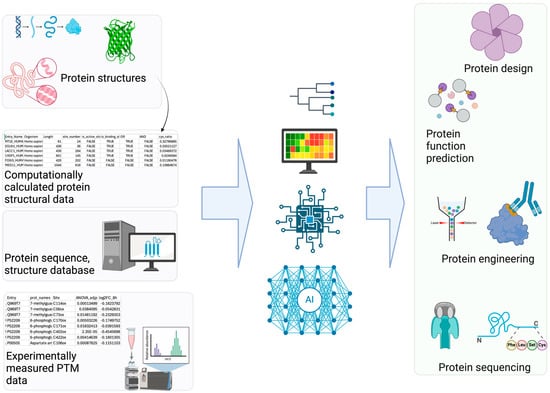
Figure 2.
AI Applications in PTM Research. (Left): Various PTM-related datasets serve as the foundation for building AI/ML models. (Right): These models enable diverse applications utilizing accurate PTM information, such as predicting PTM sites and their associated functions [74].
Figure 2.
AI Applications in PTM Research. (Left): Various PTM-related datasets serve as the foundation for building AI/ML models. (Right): These models enable diverse applications utilizing accurate PTM information, such as predicting PTM sites and their associated functions [74].
2.2.1. Language Models for PTM
Other recently developed PTM DL tools include the use of pre-trained protein language models (pLMs). PLMs use ML techniques typically developed to analyze human language to make context-specific predictions about protein structure and function [75,76]. Therefore, diverse protein-related language models have been developed [68,77,78,79]. For PTM research, LMPhosSite makes predictions on phosphosite locations. This tool integrates contextualized embeddings from a pLM to improve performance [80]. LMNglyPred also utilizes pLMs by repurposing embeddings from a pre-trained pLM to predict N-linked glycosylation sites. For this, the authors used an MLP for feature extraction with pre-trained per residue pLMs [20]. PTM-Mamba includes information from its previous pLMs in their model but also incorporates PTM tokens into training a pLM, improving its model’s accuracy for PTM-specific tasks [81]. Supervised word embeddings from a pLM (i.e., ProtT5) were also used to predict protein succinylation sites [82]. Recently, prompt-based fine-tuning of a GPT-2 model (i.e., PTMGPT2) was reported as an interpretable PTM prediction [83]. It identifies sequence motifs crucial for molecular recognition and analyzes the effects of mutations occurring at or near PTM sites, providing better insights into protein functionality.
2.2.2. Comparison of Deep Learning Approaches for PTM
Meng et al. presented a comprehensive compilation of studies employing DL techniques for PTM in early 2022 [84]. Apart from the datasets they introduced, primary distinctions among these studies were evident: the methods employed for handling PTM data and the subsequent encoding or embedding strategies used to feed this data into the DL models, as well as the diverse DL model architectures employed. While some studies had already incorporated attention mechanisms, the adoption of transformer models for PTM tasks was still not widespread.
The types of input data are closely related to the encoding strategies employed in PTM research (mostly PTM site prediction) using DL models. Most studies utilize protein sequences as input, as seen in tools like MusiteDeep (Table 1) [85]. Some studies incorporate protein structural information, e.g., models such as MIND-S [72]. Others also include annotation information, like PTM-Mamba [81].

Table 1.
Publicly accessible PTM modeling programs.
Encoding is the process of converting biological information into a numerical format that can be processed by DL models. For protein sequences, major encoding methods include one-hot encoding (or one-of-k encoding), where each AA is represented as a binary vector with a length equal to the number of possible AAs. Only one position in the vector is set to 1, corresponding to the specific AA, while all other positions are set to 0. Various embedding methods are also widely used, including embedding layers integrated within DL models that transform AA sequences into continuous vector spaces, capturing contextual relationships. Word embedding techniques, such as Word2Vec or FastText, treat AAs or k-mers as words and generate embeddings based on their contextual similarity. Pre-trained protein models like ProtT5 provide embeddings that capture rich contextual information about protein sequences, learned from large-scale protein datasets [76]. Although BLOSUM62, a substitution matrix that scores alignments between protein sequences, has been used historically, its popularity has declined. When the data includes evolutionary information, Position-Specific Scoring Matrices (PSSMs) can be employed [86]. PSSMs are generated from multiple sequence alignments and reflect the evolutionary conservation of AA residues at each position. This encoding method has been utilized by PTM models (i.e., DeepAcet) and a general protein structure prediction and design program (i.e., transform-restrained Rosetta, which is abbreviated as trRosetta) [87,88].
MLPs have historically been the initial approach in the field of PTM prediction, serving as a reliable baseline method, e.g., DL-based protein lysine acetylation modification prediction (Deep-PLA) and Histone-Net [89,90]. However, more complex neural network architectures, such as CNNs and RNNs, specifically LSTMs, have gained prominence, such as models like DeepPhos, MusiteDeep, and LMPhosSite [80,85,91]. CNNs excel at detecting local patterns within sequences, which is particularly useful for identifying conserved motifs or sequence motifs around PTM sites. Their ability to learn features through convolutional layers allows them to detect motifs regardless of their position in the input sequence, making CNNs robust to slight shifts in sequence position. The layered architecture of CNNs enables them to build hierarchical representations of the input data, where lower layers capture simple patterns such as small motifs, and higher layers capture more complex patterns like secondary structure elements. LSTMs, on the other hand, are designed to handle sequential data and can capture long-range dependencies within protein sequences. This capability is crucial for PTM prediction, where the modification site might depend on residues that are far apart in the sequence. Additionally, bidirectional LSTMs can consider information from both former and latter residues, providing a more comprehensive understanding of the sequence context. When protein structural information is available, Graph Neural Networks (GNNs) also represent a highly effective choice, such as MIND-S [72]. GNNs can model the complex relationships between residues in a protein structure, capturing the three-dimensional spatial arrangements and interactions that are crucial for understanding protein function and PTMs.
Recently, transformers have gained popularity in this field, such as TransPTM [92]. The advantages of transformers in this context include their ability to capture longer-range dependencies than LSTM, their parallel processing capabilities, and their scalability with large datasets. Transformers excel at capturing long-range dependencies within protein sequences using the self-attention mechanism. This mechanism allows each position in the sequence to directly attend to all other positions, effectively capturing relationships between distant residues. Hence, it allows the modeling of even proteins with high contact order. The parallel processing capability of transformers is another significant advantage. Traditional LSTMs can be computationally intensive and slow, especially for long sequences. In contrast, transformers process all positions in the sequence simultaneously, leading to more efficient training and inference. Pre-training transformer models (like ProtT5) on large protein datasets allows them to learn rich, generalizable representations of protein sequences. These pre-trained models can then be fine-tuned on specific PTM prediction tasks.
3. Experimental Data for PTM ML
3.1. Mass Spectrometry-Based PTM Proteomics for ML
Mass spectrometry-based proteomics is the preferred technology for providing large-scale and unbiased PTM measurements suitable for ML. Due to the unique chemistries and usually low-abundance nature of many PTMs, specific sample processing workflows and MS data acquisition methods have been developed for each PTM of interest. In general, these approaches aim to provide high coverage of the PTMome, precise localization of the modification site, and accurate quantification of the level of modification. To reach high PTMome coverage, selective enrichment and fractionation are often used prior to MS analysis. For example, peptides were fractionated by basic reversed-phase liquid chromatography (bRPLC), and each fraction was enriched by immobilized metal affinity chromatography for phosphoproteomics [93]. In the case of redox modifications, our group developed resin-assisted capture to enrich oxidized cysteines first, followed by bRPLC to achieve in-depth profiling of the redox proteome [94]. These approaches usually employ data-dependent acquisition to collect MS data and require a long instrument time due to extensive fractionation. Recently, data-independent acquisition (DIA) methods have been evaluated for PTM works, with some promising results showing the identification of >30,000 phosphorylation sites using a short single-shot LC–MS run [95]. In addition, precise localization of PTM sites is important because PTM events are site-specific, and knowledge of the modification site is critical for subsequent PTM-based engineering. MS data contains sequence-level information (e.g., b and y fragment ions in MS/MS spectra) and thus can be used to infer the site localization of modified AAs. Many algorithms have been developed for PTM site localization [96,97], and an interesting recent trend is to use DL-based framework to control the false localization rate [98]. Lastly, high-quality quantitative PTMomics data is required to enable ML to identify PTM signatures contributing a particular biological phenotype. Both isobaric tagging-based methods and label-free approaches are widely used in quantitative proteomics. Since a large sample size is beneficial for ML, isobaric tagging-based methods such as tandem mass tags are advantageous because of the sample multiplexing power. However, a shorter LC–MS run time coupled with advanced DIA approaches also provides an alternative for high-throughput analysis of hundreds of PTMomes for future ML studies.
3.2. Public PTM Databases
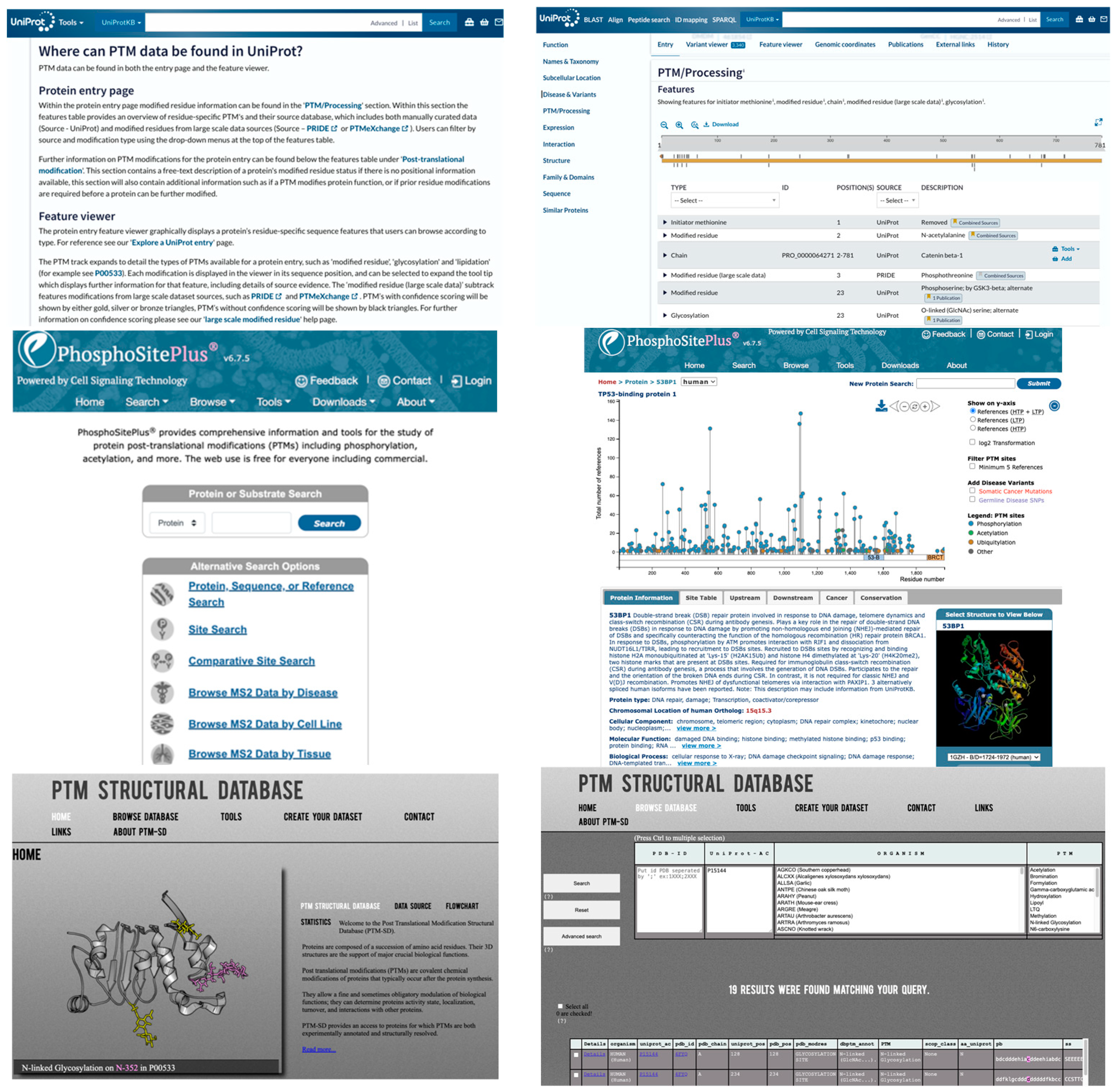
To accelerate the application of DL in PTM research, leveraging publicly available PTM DBs, as previously summarized (Table 1 in Meng et al. [84]), is essential. Additional PTM DBs include GlycoEP datasets, which were compiled using sequence, evolutionary, and structural information [20]. Moreover, advancements in high-throughput proteomics have led to the development of extensive quantitative PTM proteome datasets, such as qPTM [99]. These datasets can also be integrated with other chemoproteomics DBs (e.g., CysDB) [100], further enhancing PTM research capabilities. Figure 3 shows three popular DBs (i.e., PhosphoSitePlus [101], UniProt [102], and PTM-Structural Database [103]). They offer various search options, including protein or substrate name, specific sites, disease, cell line, and tissue, and provide details such as the number of references for each PTM, along with the corresponding residue number, PDB ID, and UniProt ID.
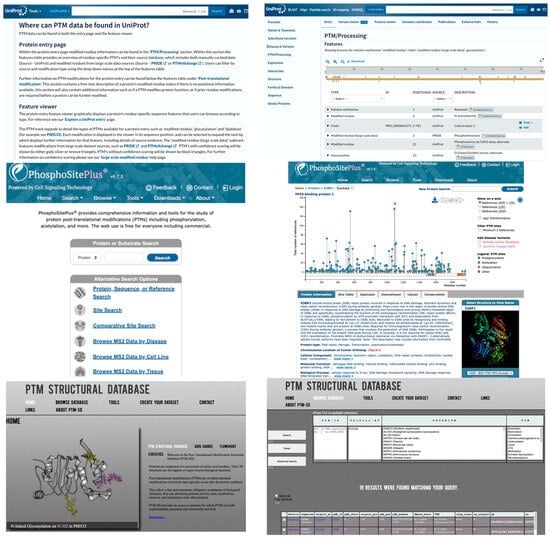
Figure 3.
Representative PTM Databases. Upper: UniProt. Middle: PhosphoSitePlus. Lower: PTM-Structural Database. Left: input pages. Right: data retrieval pages.
4. Discussion
It is expected that AI-accelerated PTM study will propagate to other fields including more clinically relevant therapeutics and evolution research [104,105]. However, most current applications of ML and DL in PTM research have predominantly centered on predicting PTM sites [106]. This trend has been found for both mass spectrometry-derived proteomics data [96,97,98] and general protein structure and sequence data [20,24]. Predicting PTM sites holds significant values, as specific PTMs can bring about orthosteric or allosteric regulation in signal transduction [13,107]. For example, phosphorylation often exerts allosteric effects that extend well beyond the immediate vicinity of the phosphorylation site [13]. However, other aspects of ML/DL-assisted PTM research—such as nanopore-based PTM detection [73] and direct functional prediction [24,108]—remain relatively underexplored and require further investigation. Predicting PTM-induced functions will naturally involve structural studies, as larger conformational changes are generally more functionally relevant, particularly for many phosphorylation events [13].
Additionally, more structural biology data from methods like NMR [109,110] would be beneficial in these new research directions. PTM data is currently underrepresented in structural biology databases [13]. Moreover, advanced structural modeling that incorporates a conformational selection approach—often elucidated by techniques like NMR or cryo-EM single particle analysis—will provide a more accurate explanation for PTMs such as phosphorylation, which aligns more with conformational selection than with induced-fit structural rearrangement [13]. Expanding these efforts will enhance the application of PTM research, especially considering that, for phosphorylation alone, over 100,000 PTM sites have been identified, yet the enzymes regulating these sites are known for only a small fraction, and even fewer functions have been elucidated [111].
5. Conclusions
PTM studies are contributing significantly to the development of therapeutic and diagnostic tools for diseases such as cancer, metabolic disorders, and neurodegenerative conditions. As the field progresses, AI-driven PTM research is expected to drive innovations in clinical applications and evolutionary biology, transforming understanding of protein regulation and cellular systems. Therefore, we have discussed the significant role of AI in advancing PTM research, and the benefits of generating high-throughput PTM data for AI training. Additionally, advanced machine learning architectures, such as language models and graph neural networks, will continue uncovering novel aspects of PTM biology, while public DBs and collaborative efforts continue to expand the accessibility and scope of this endeavor. We expect that the combination of AI-driven predictions and more experimental structural biology data will further elucidate the molecular mechanisms underlying PTM-induced conformational changes and functional impacts.
Funding
Pacific Northwest National Laboratory is a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under Contract No. DE-AC05-76RL01830.
Acknowledgments
The research described in this paper is part of the Predictive Phenomics Initiative at Pacific Northwest National Laboratory and conducted under the Laboratory Directed Research and Development Program. Additional funding was received from Defense Threat Reduction Agency. Douglas Renfrew and Richard Overstreet advised several ncAA modeling programs. We appreciate reviewers for their invaluable comments and suggestions, which have enhanced the clarity and readability of the manuscript. Pacific Northwest National Laboratory is a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under Contract No. DE-AC05-76RL01830.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database J. Biol. Databases Curation 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liu, J.; Inuzuka, H.; Wei, W. Targeted protein posttranslational modifications by chemically induced proximity for cancer therapy. J. Biol. Chem. 2023, 299, 104572. [Google Scholar] [CrossRef]
- Marx, V. Inside the chase after those elusive proteoforms. Nat. Methods 2024, 21, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Leutert, M.; Entwisle, S.W.; Villén, J. Decoding Post-Translational Modification Crosstalk With Proteomics. Mol. Cell. Proteom. MCP 2021, 20, 100129. [Google Scholar] [CrossRef]
- Bobalova, J.; Strouhalova, D.; Bobal, P. Common Post-translational Modifications (PTMs) of Proteins: Analysis by Up-to-Date Analytical Techniques with an Emphasis on Barley. J. Agric. Food Chem. 2023, 71, 14825–14837. [Google Scholar] [CrossRef]
- Chung, H.S.; Wang, S.-B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine Oxidative Posttranslational Modifications. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef]
- ThermoFisher Scientitic Overview of Post-Translational Modifications (PTMs). Available online: https://www.thermofisher.com/ie/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-post-translational-modification.html (accessed on 28 December 2024).
- Qian, M.; Yan, F.; Yuan, T.; Yang, B.; He, Q.; Zhu, H. Targeting post-translational modification of transcription factors as cancer therapy. Drug Discov. Today 2020, 25, 1502–1512. [Google Scholar] [CrossRef]
- Dunphy, K.; Dowling, P.; Bazou, D.; O’Gorman, P. Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers. Cancers 2021, 13, 1930. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.; Lindner, A.B. Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxid. Med. Cell. Longev. 2017, 2017, 5716409. [Google Scholar] [CrossRef]
- Leonard, B.; Danna, V.; Gorham, L.; Davison, M.; Chrisler, W.; Kim, D.N.; Gerbasi, V.R. Shaping Nanobodies and Intrabodies against Proteoforms. Anal. Chem. 2023, 95, 8747–8751. [Google Scholar] [CrossRef] [PubMed]
- hamster prion disease with brain. Illustration from NIH BIOART Source.
- Correa Marrero, M.; Mello, V.H.; Sartori, P.; Beltrao, P. Global comparative structural analysis of responses to protein phosphorylation. bioRxiv 2024. bioRxiv:2024.10.18.617420. [Google Scholar] [CrossRef]
- Pieroni, S.; Castelli, M.; Piobbico, D.; Ferracchiato, S.; Scopetti, D.; Di-Iacovo, N.; Della-Fazia, M.A.; Servillo, G. The Four Homeostasis Knights: In Balance upon Post-Translational Modifications. Int. J. Mol. Sci. 2022, 23, 14480. [Google Scholar] [CrossRef] [PubMed]
- Chrestia, J.F.; Turani, O.; Araujo, N.R.; Hernando, G.; Esandi, M.d.C.; Bouzat, C. Regulation of nicotinic acetylcholine receptors by post-translational modifications. Pharmacol. Res. 2023, 190, 106712. [Google Scholar] [CrossRef]
- Peng, Y.; Liu, H.; Liu, J.; Long, J. Post-translational modifications on mitochondrial metabolic enzymes in cancer. Free Radic. Biol. Med. 2022, 179, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Gupta, R.; Ambasta, R.K.; Kumar, P. Emerging trends in post-translational modification: Shedding light on Glioblastoma multiforme. Biochim. Biophys. Acta BBA—Rev. Cancer 2023, 1878, 188999. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Smith, L.E.; Rogowska-Wrzesinska, A. The challenge of detecting modifications on proteins. Essays Biochem. 2020, 64, 135–153. [Google Scholar] [CrossRef]
- Pakhrin, S.C.; Pokharel, S.; Aoki-Kinoshita, K.F.; Beck, M.R.; Dam, T.K.; Caragea, D.; KC, D.B. LMNglyPred: Prediction of human N-linked glycosylation sites using embeddings from a pre-trained protein language model. Glycobiology 2023, 33, 411–422. [Google Scholar] [CrossRef]
- Yu, Z.; Yu, J.; Wang, H.; Zhang, S.; Zhao, L.; Shi, S. PhosAF: An integrated deep learning architecture for predicting protein phosphorylation sites with AlphaFold2 predicted structures. Anal. Biochem. 2024, 690, 115510. [Google Scholar] [CrossRef]
- Bludau, I.; Willems, S.; Zeng, W.-F.; Strauss, M.T.; Hansen, F.M.; Tanzer, M.C.; Karayel, O.; Schulman, B.A.; Mann, M. The structural context of posttranslational modifications at a proteome-wide scale. PLOS Biol. 2022, 20, e3001636. [Google Scholar] [CrossRef] [PubMed]
- Kamacioglu, A.; Tuncbag, N.; Ozlu, N. Structural analysis of mammalian protein phosphorylation at a proteome level. Structure 2021, 29, 1219–1229.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, C.; Cai, M.; Luo, C.; Zhu, F.; Liang, Z. FuncPhos-STR: An integrated deep neural network for functional phosphosite prediction based on AlphaFold protein structure and dynamics. Int. J. Biol. Macromol. 2024, 266, 131180. [Google Scholar] [CrossRef]
- Wang, D.; Cui, P.; Zhu, W. Structural Deep Network Embedding. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; Association for Computing Machinery, New York, NY, USA, 13–17 August 2016; pp. 1225–1234. [Google Scholar]
- Holden, J.K.; Pavlovicz, R.; Gobbi, A.; Song, Y.; Cunningham, C.N. Computational Site Saturation Mutagenesis of Canonical and Non-Canonical Amino Acids to Probe Protein-Peptide Interactions. Front. Mol. Biosci. 2022, 9, 848689. [Google Scholar] [CrossRef] [PubMed]
- Aphicho, K.; Kittipanukul, N.; Uttamapinant, C. Visualizing the complexity of proteins in living cells with genetic code expansion. Curr. Opin. Chem. Biol. 2022, 66, 102108. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Nickling, J.H.; Bartholomae, M.; Buivydas, A.; Kuipers, O.P.; Budisa, N. Prospects of In vivo Incorporation of Non-canonical Amino Acids for the Chemical Diversification of Antimicrobial Peptides. Front. Microbiol. 2017, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Walport, L.J.; Obexer, R.; Suga, H. Strategies for transitioning macrocyclic peptides to cell-pxermeable drug leads. Curr. Opin. Biotechnol. 2017, 48, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, Z.; Zhou, Z.; Shen, H.C.; Yan, S.F.; Mayweg, A.V.; Xu, Z.; Qin, N.; Wong, J.C.; Zhang, Z.; et al. Structure-Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP-TEAD Protein-Protein Interaction. ACS Med. Chem. Lett. 2014, 5, 993–998. [Google Scholar] [CrossRef]
- Renfrew, P.D.; Choi, E.J.; Bonneau, R.; Kuhlman, B. Incorporation of Noncanonical Amino Acids into Rosetta and Use in Computational Protein-Peptide Interface Design. PLoS ONE 2012, 7, e32637. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, V.K.; Kang, C.S.; Sawaya, M.R.; Rettie, S.; Li, X.; Antselovich, I.; Craven, T.W.; Watkins, A.M.; Labonte, J.W.; DiMaio, F.; et al. Computational design of mixed chirality peptide macrocycles with internal symmetry. Protein Sci. 2020, 29, 2433–2445. [Google Scholar] [CrossRef]
- Beyer, J.N.; Hosseinzadeh, P.; Gottfried-Lee, I.; Van Fossen, E.M.; Zhu, P.; Bednar, R.M.; Karplus, P.A.; Mehl, R.A.; Cooley, R.B. Overcoming Near-Cognate Suppression in a Release Factor 1-Deficient Host with an Improved Nitro-Tyrosine tRNA Synthetase. J. Mol. Biol. 2020, 432, 4690–4704. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Hauf, M.; Richter, F.; Albers, S.; Möglich, A.; Ignatova, Z.; Budisa, N. Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine. Int. J. Mol. Sci. 2019, 20, 2343. [Google Scholar] [CrossRef] [PubMed]
- Karami, Y.; Murail, S.; Giribaldi, J.; Lefranc, B.; Leprince, J.; de Vries, S.J.; Tufféry, P. A novel computational method for head-to-tail peptide cyclization: Application to urotensin II. bioRxiv 2022. [Google Scholar] [CrossRef]
- Khoury, G.A.; Smadbeck, J.; Tamamis, P.; Vandris, A.C.; Kieslich, C.A.; Floudas, C.A. Forcefield_NCAA: Ab Initio Charge Parameters to Aid in the Discovery and Design of Therapeutic Proteins and Peptides with Unnatural Amino Acids and Their Application to Complement Inhibitors of the Compstatin Family. ACS Synth. Biol. 2014, 3, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Buer, B.C.; Meagher, J.L.; Stuckey, J.A.; Marsh, E.N. Structural basis for the enhanced stability of highly fluorinated proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 4810–4815. [Google Scholar] [CrossRef]
- Jia, F.; Wang, J.; Peng, J.; Zhao, P.; Kong, Z.; Wang, K.; Yan, W.; Wang, R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim. Biophys. Sin. 2017, 49, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Regina, T.; Katalin, U.; Dóra, I.; Erzsébet, F.; Alan, P.; Ferenc, H. Partial d-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar] [CrossRef]
- Hong, S.Y.; Oh, J.E.; Lee, K.-H. Effect of d-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Garton, M.; Nim, S.; Stone, T.A.; Wang, K.E.; Deber, C.M.; Kim, P.M. Method to generate highly stable D-amino acid analogs of bioactive helical peptides using a mirror image of the entire PDB. Proc. Natl. Acad. Sci. USA 2018, 115, 1505–1510. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Kesharwani, M.K.; Karton, A.; Martin, J.M.L. Benchmark ab Initio Conformational Energies for the Proteinogenic Amino Acids through Explicitly Correlated Methods. Assessment of Density Functional Methods. J. Chem. Theory Comput. 2016, 12, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Freedberg, D.I.; Venable, R.M.; Rossi, A.; Bull, T.E.; Pastor, R.W. Discriminating the Helical Forms of Peptides by NMR and Molecular Dynamics Simulation. J. Am. Chem. Soc. 2004, 126, 10478–10484. [Google Scholar] [CrossRef]
- Buck, M.; Bouguet-Bonnet, S.; Pastor, R.W.; MacKerell Jr, A.D. Importance of the CMAP correction to the CHARMM22 protein force field: Dynamics of hen lysozyme. Biophys. J. 2006, 90, L36–L38. [Google Scholar] [CrossRef] [PubMed]
- Pagar, A.D.; Patil, M.D.; Flood, D.T.; Yoo, T.H.; Dawson, P.E.; Yun, H. Recent Advances in Biocatalysis with Chemical Modification and Expanded Amino Acid Alphabet. Chem. Rev. 2021, 121, 6173–6245. [Google Scholar] [CrossRef]
- Drew, K.; Renfrew, P.D.; Craven, T.W.; Butterfoss, G.L.; Chou, F.-C.; Lyskov, S.; Bullock, B.N.; Watkins, A.; Labonte, J.W.; Pacella, M.; et al. Adding Diverse Noncanonical Backbones to Rosetta: Enabling Peptidomimetic Design. PLoS ONE 2013, 8, e67051. [Google Scholar] [CrossRef] [PubMed]
- Renfrew, P.D.; Craven, T.W.; Butterfoss, G.L.; Kirshenbaum, K.; Bonneau, R. A Rotamer Library to Enable Modeling and Design of Peptoid Foldamers. J. Am. Chem. Soc. 2014, 136, 8772–8782. [Google Scholar] [CrossRef]
- Schneider, J.A.; Craven, T.W.; Kasper, A.C.; Yun, C.; Haugbro, M.; Briggs, E.M.; Svetlov, V.; Nudler, E.; Knaut, H.; Bonneau, R.; et al. Design of Peptoid-peptide Macrocycles to Inhibit the β-catenin TCF Interaction in Prostate Cancer. Nat. Commun. 2018, 9, 4396. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Watkins, A.; Renfrew, D. Working with Noncanonical Amino Acids in Rosetta. Available online: https://new.rosettacommons.org/docs/latest/rosetta_basics/non_protein_residues/Noncanonical-Amino-Acids (accessed on 5 May 2022).
- Brown, B.P.; Vu, O.; Geanes, A.R.; Kothiwale, S.; Butkiewicz, M.; Lowe, E.W.; Mueller, R.; Pape, R.; Mendenhall, J.; Meiler, J. Introduction to the BioChemical Library (BCL): An Application-Based Open-Source Toolkit for Integrated Cheminformatics and Machine Learning in Computer-Aided Drug Discovery. Front. Pharmacol. 2022, 13, 833099. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.N.; McNaughton, A.D.; Kumar, N. Leveraging Artificial Intelligence to Expedite Antibody Design and Enhance Antibody–Antigen Interactions. Bioengineering 2024, 11, 185. [Google Scholar] [CrossRef]
- Li, X.; Hou, C.; Yang, M.; Luo, B.; Mao, N.; Chen, K.; Chen, Z.; Bai, Y. The effect of phosphorylation on the conformational dynamics and allostery of the association of death-associated protein kinase with calmodulin. J. Biomol. Struct. Dyn. 2024. [Google Scholar] [CrossRef]
- Mejia-Rodriguez, D.; Kim, H.; Sadler, N.; Li, X.; Bohutskyi, P.; Valiev, M.; Qian, W.-J.; Cheung, M.S. PTM-Psi: A python package to facilitate the computational investigation of ost-ranslational odification on rotein tructures and their mpacts on dynamics and functions. Protein Sci. 2023, 32, e4822. [Google Scholar] [CrossRef] [PubMed]
- Tivon, B.; Gabizon, R.; Somsen, B.A.; Cossar, P.J.; Ottmann, C.; London, N. Covalent flexible peptide docking in Rosetta. Chem. Sci. 2021, 12, 10836–10847. [Google Scholar] [CrossRef] [PubMed]
- Drake, Z.C.; Seffernick, J.T.; Lindert, S. Protein complex prediction using Rosetta, AlphaFold, and mass spectrometry covalent labeling. Nat. Commun. 2022, 13, 7846. [Google Scholar] [CrossRef]
- Holcomb, M.; Santos-Martins, D.; Tillack, A.F.; Forli, S. Performance evaluation of flexible macrocycle docking in AutoDock. QRB Discov. 2022, 3, e18. [Google Scholar] [CrossRef] [PubMed]
- Meeko: Preparation of Small Molecules for AutoDock. Docking Covalent Ligands as Flexible Sidechains. Available online: https://github.com/forlilab/Meeko?tab=readme-ov-file#docking-covalent-ligands-as-flexible-sidechains (accessed on 28 December 2024).
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock4 with GPUs and Gradient-Based Local Search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef]
- Yousef, M.; Allmer, J. Deep learning in bioinformatics. Turk. J. Biol. 2023, 47, 366–382. [Google Scholar] [CrossRef]
- Lee, K.; Famiglietti, M.L.; McMahon, A.; Wei, C.-H.; MacArthur, J.A.L.; Poux, S.; Breuza, L.; Bridge, A.; Cunningham, F.; Xenarios, I.; et al. Scaling up data curation using deep learning: An application to literature triage in genomic variation resources. PLoS Comput. Biol. 2018, 14, e1006390. [Google Scholar] [CrossRef]
- Krishna, R.; Wang, J.; Ahern, W.; Sturmfels, P.; Venkatesh, P.; Kalvet, I.; Lee, G.R.; Morey-Burrows, F.S.; Anishchenko, I.; Humphreys, I.R.; et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 2024, 384, eadl2528. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Ruidong Wu; Fan Ding; Rui Wang; Rui Shen; Xiwen Zhang; Shitong Luo; Chenpeng Su; Zuofan Wu; Qi Xie; Bonnie Berger; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022. bioRxiv:2022.07.21.500999.
- chai-1. Available online: https://chaiassets.com/chai-1/paper/technical_report_v1.pdf (accessed on 28 December 2024).
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Ertelt, M.; Mulligan, V.K.; Maguire, J.B.; Lyskov, S.; Moretti, R.; Schiffner, T.; Meiler, J.; Schoeder, C.T. Combining machine learning with structure-based protein design to predict and engineer post-translational modifications of proteins. PLoS Comput. Biol. 2024, 20, e1011939. [Google Scholar] [CrossRef] [PubMed]
- Leman, J.K.; Weitzner, B.D.; Renfrew, P.D.; Lewis, S.M.; Moretti, R.; Watkins, A.M.; Mulligan, V.K.; Lyskov, S.; Adolf-Bryfogle, J.; Labonte, J.W.; et al. Better together: Elements of successful scientific software development in a distributed collaborative community. PLoS Comput. Biol. 2020, 16, e1007507. [Google Scholar] [CrossRef]
- Glukhov, E.; Averkava, V.; Kotelnikov, S.; Stepanenko, D.; Nguyen, T.; Mitchell, J.C.; Simmerling, C.; Vajda, S.; Emili, A.; Padhorny, D.; et al. Phospho-Tune: Enhanced Structural Modeling of Phosphorylated Protein Interactions. bioRxiv 2024. bioRxiv:2024.02.29.582580. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, J.-Y.; Fu, M.; Wang, D.; Pelletier, A.R.; Sigdel, D.; Ng, D.C.M.; Wang, W.; Ping, P. MIND-S is a deep-learning prediction model for elucidating protein post-translational modifications in human diseases. Cell Rep. Methods 2023, 3, 100430. [Google Scholar] [CrossRef]
- Cao, C.; Magalhães, P.; Krapp, L.F.; Bada Juarez, J.F.; Mayer, S.F.; Rukes, V.; Chiki, A.; Lashuel, H.A.; Dal Peraro, M. Deep Learning-Assisted Single-Molecule Detection of Protein Post-translational Modifications with a Biological Nanopore. ACS Nano 2023, 18, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- AI Applications in PTM Research. Created in BioRender. Kim, D. 2024. Available online: https://app.biorender.com/citation/670d390a8a4a644fef1a948a (accessed on 28 December 2024).
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef]
- Pakhrin, S.C.; Pokharel, S.; Pratyush, P.; Chaudhari, M.; Ismail, H.D.; KC, D.B. LMPhosSite: A Deep Learning-Based Approach for General Protein Phosphorylation Site Prediction Using Embeddings from the Local Window Sequence and Pretrained Protein Language Model. J. Proteome Res. 2023, 22, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Schussheim, B.; Chatterjee, P. PTM-Mamba: A PTM-Aware Protein Language Model with Bidirectional Gated Mamba Blocks. bioRxiv 2024. bioRxiv:2024.02.28.581983. [Google Scholar] [CrossRef]
- Pokharel, S.; Pratyush, P.; Heinzinger, M.; Newman, R.H.; Kc, D.B. Improving protein succinylation sites prediction using embeddings from protein language model. Sci. Rep. 2022, 12, 16933. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Kandel, J.; Tayara, H.; Chong, K.T. Post-translational modification prediction via prompt-based fine-tuning of a GPT-2 model. Nat. Commun. 2024, 15, 6699. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Chan, W.-S.; Huang, L.; Liu, L.; Chen, X.; Zhang, W.; Wang, F.; Cheng, K.; Sun, H.; Wong, K.-C. Mini-review: Recent advances in post-translational modification site prediction based on deep learning. Comput. Struct. Biotechnol. J. 2022, 20, 3522–3532. [Google Scholar] [CrossRef]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020, 48, W140–W146. [Google Scholar] [CrossRef]
- Henikoff, J.G.; Henikoff, S. Using substitution probabilities to improve position-specific scoring matrices. Comput. Appl. Biosci. CABIOS 1996, 12, 135–143. [Google Scholar] [CrossRef]
- Wu, M.; Yang, Y.; Wang, H.; Xu, Y. A deep learning method to more accurately recall known lysine acetylation sites. BMC Bioinform. 2019, 20, 49. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Zhang, Q.; Liu, Z.; Du, Y.; Gao, X.; Zhao, Q.; Cheng, H.; Li, X.; Liu, Z.-X. Deep learning based prediction of reversible HAT/HDAC-specific lysine acetylation. Brief. Bioinform. 2020, 21, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Histone-Net: A Multi-Paradigm Computational Framework for Histone Occupancy and Modification Prediction|Complex & Intelligent Systems. Available online: https://link.springer.com/article/10.1007/s40747-022-00802-w (accessed on 11 June 2024).
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.-M.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Chen, X.; Cheng, K.; Chen, N.; Zheng, Z.; Wang, F.; Sun, H.; Wong, K.-C. TransPTM: A Transformer-Based Model for Non-Histone Acetylation Site Prediction. Brief. Bioinform. 2024, 25, bbae219. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef] [PubMed]
- Day, N.J.; Zhang, T.; Gaffrey, M.J.; Zhao, R.; Fillmore, T.L.; Moore, R.J.; Rodney, G.G.; Qian, W.-J. A deep redox proteome profiling workflow and its application to skeletal muscle of a Duchenne Muscular Dystrophy model. Free Radic. Biol. Med. 2022, 193, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Skowronek, P.; Thielert, M.; Voytik, E.; Tanzer, M.C.; Hansen, F.M.; Willems, S.; Karayel, O.; Brunner, A.-D.; Meier, F.; Mann, M. Rapid and In-Depth Coverage of the (Phospho-)Proteome With Deep Libraries and Optimal Window Design for dia-PASEF. Mol. Cell. Proteom. 2022, 21, 100279. [Google Scholar] [CrossRef] [PubMed]
- Joyce, A.W.; Searle, B.C. Computational approaches to identify sites of phosphorylation. Proteomics 2024, 24, 2300088. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Teo, G.C.; Kong, A.T.; Haynes, S.E.; Avtonomov, D.M.; Geiszler, D.J.; Nesvizhskii, A.I. Identification of modified peptides using localization-aware open search. Nat. Commun. 2020, 11, 4065. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Wang, Y.; Yang, Y.; Zhao, D.; Wang, X.; Shen, C.; Qiao, L. DeepFLR facilitates false localization rate control in phosphoproteomics. Nat. Commun. 2023, 14, 2269. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Wang, Y.; Zheng, Y.; Liu, Z.; Zhang, Q.; Wang, S.; Zhao, Q.; Zhang, X.; Li, X.; Xu, R.-H.; et al. qPTM: An updated database for PTM dynamics in human, mouse, rat and yeast. Nucleic Acids Res. 2023, 51, D479–D487. [Google Scholar] [CrossRef]
- Boatner, L.M.; Palafox, M.F.; Schweppe, D.K.; Backus, K.M. CysDB: A human cysteine database based on experimental quantitative chemoproteomics. Cell Chem. Biol. 2023, 30, 683–698.e3. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef]
- Craveur, P.; Rebehmed, J.; de Brevern, A.G. PTM-SD: A database of structurally resolved and annotated posttranslational modifications in proteins. Database 2014, 2014, bau041. [Google Scholar] [CrossRef] [PubMed]
- Borowiec, M.L.; Dikow, R.B.; Frandsen, P.B.; McKeeken, A.; Valentini, G.; White, A.E. Deep learning as a tool for ecology and evolution. Methods Ecol. Evol. 2022, 13, 1640–1660. [Google Scholar] [CrossRef]
- Bradley, D. The evolution of post-translational modifications. Curr. Opin. Genet. Dev. 2022, 76, 101956. [Google Scholar] [CrossRef]
- Yin, Q.; Wu, M.; Liu, Q.; Lv, H.; Jiang, R. DeepHistone: A deep learning approach to predicting histone modifications. BMC Genom. 2019, 20, 193. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J.; Xin, F.; Radivojac, P. Allosteric post-translational modification codes. Trends Biochem. Sci. 2012, 37, 447–455. [Google Scholar] [CrossRef]
- Ochoa, D.; Jarnuczak, A.F.; Viéitez, C.; Gehre, M.; Soucheray, M.; Mateus, A.; Kleefeldt, A.A.; Hill, A.; Garcia-Alonso, L.; Stein, F.; et al. The functional landscape of the human phosphoproteome. Nat. Biotechnol. 2020, 38, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Jung, Y.; Hwang, G.-S.; Han, H.; Cho, M. Phosphorylation alters backbone conformational preferences of serine and threonine peptides. Proteins Struct. Funct. Bioinform. 2011, 79, 3155–3165. [Google Scholar] [CrossRef]
- Tholey, A.; Lindemann, A.; Kinzel, V.; Reed, J. Direct Effects of Phosphorylation on the Preferred Backbone Conformation of Peptides: A Nuclear Magnetic Resonance Study. Biophys. J. 1999, 76, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Needham, E.J.; Parker, B.L.; Burykin, T.; James, D.E.; Humphrey, S.J. Illuminating the dark phosphoproteome. Sci. Signal. 2019, 12, eaau8645. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).