1. Introduction
Cell viability is the main parameter to be evaluated when fresh osteochondral grafts are preserved for clinical applications [
1,
2,
3]. Major studies have used fluorescent confocal microscopy to analyze the viability of chondrocytes in their natural three-dimensional arrangement, but only a few have compared different analytical techniques used to quantify this parameter [
4,
5]. Tetrazolium salt has been used to assess chondrocyte viability in cartilage preserved in several types of culture medium [
6,
7]. Other studies have assessed chondrocyte viability by fluorescent confocal microscopy, establishing it as a better technique than the use of colorimetric biochemical methods such as lactate dehydrogenase (LDH), calcein AM-ethidium homodimer-1 or trypan blue [
5,
8]. Depending on the technique used to evaluate cell viability, the percentages of living or dead cells will differ due to the enzymes responsible for the activation of calcein AM or LDH, because biologically, these molecules have different metabolic pathways. Moreover, the mathematical approximations used to analyze the experimental data obtained with the various techniques differ: a hemocytometer is used to quantify the number of viable cells stained with trypan blue, while a custom macro is used to quantify histological or confocal fluorescent images. In our study, we compare flow cytometry and confocal fluorescence microscopy techniques. Flow cytometry is considered to be an invasive technique, since the method used for sample preparation involves extracellular matrix enzymatic digestion to isolate the cells in order to quantify the percentage of live/dead cells. However, other studies have shown that imaging techniques overestimate the number of viable cells [
5]. Furthermore, calcein AM and 7-AAD viability probes are incubated within the native cartilage by passive transport from the dye buffer solution to the chondrocytes embedded in the extracellular matrix, without modifying the native cartilage. Calcein AM produces fluorescence due to the esterase activity of viable cells, while 7-AAD is incorporated into nucleic acids of non-viable cells as a result of plasma membrane compromise.
There is currently no systematic test for assessing cell viability in osteochondral grafts, nor studies that compare the use of flow cytometry with traditional confocal fluorescent microscopy. In the present study, femoral condyles were preserved, cell viability was monitored, and the percentage of viable chondrocytes was quantified using semi-quantitative confocal fluorescence microscopy and quantitative flow cytometry techniques. Establishing a reference method to assess viability would allow standard assessment of the quality of osteochondral allografts, facilitating the safe distribution of tissues for transplantation in young patients with large osteochondral defects who currently do not have effective alternative treatment to recover functionality of the affected joints.
The aim of our study was to compare these two analytical techniques for quantitative determination of cell viability of cartilage chondrocytes present in osteochondral allografts for clinical use. We hypothesize that flow cytometry analysis will allow, using a simpler technology, a higher degree of system characterization for the detection of live, dead and early apoptotic cells compared with confocal fluorescence microscopy, which is unable to distinguish between the double staining that occurs between live and early apoptotic cells.
2. Materials and Methods
2.1. Ethical Considerations
Human samples were obtained, processed, and analyzed according to current guidelines in relation to the collection and preservation of human tissues for clinical use (EEC regulations 2004/23/CE and 2006/17/CE) and in accordance with the protocol and legal requirements for the use of biological samples and biomedical research in Spain (Law 14/2007 and RD 1716/2011). In addition, the acquisition, processing, and preservation of the tissues used in this study were carried out in accordance with Spanish law on the development and applications of organ transplants (RD 9/2014). All the information provided before donation, together with informed consent, guaranteed that the samples obtained were to be used for clinical applications and/or applied research. The use, protection, communication, and transfer of personal data complied with local regulations (Law 15/1999).
2.2. Reagents
The following reagents were used in the study: Ringer’s lactate (Fresenius Kabi, IL, USA, cat # 628339.4), calcein AM (Fisher Scientific SL, MA, USA, cat # 10462052), 7-AAD (BD Biosciences, NJ, USA, cat # 559925 ), Annexin V (Annexin V–APC, BD Biosciences, NJ, USA, cat # 550474), Vybrant®® DyeCycleTM Ruby Stain (Invitrogen, MA, USA, cat # V10273), 0.9% NaCl (Braun, Kronberg, Germany, cat # 3570470), PBS (Gibco, MA, USA, cat # 14190-094), tobramycin (Medicon HealthCare, Maharashtra, India, cat # 672722.5), vancomycin (Laboratorios Reig Jofré, Barcelona, Spain, cat # 606390.3), amphotericin A (XalabarderFarma, Barcelona, Spain, cat # 820152268), cotrimoxazole (Almirall, Barcelona, Spain, cat # 656754.8), pronase (Merck Life Science, Darmstadt, Germany, cat # 10165921001), type II collagenase (Sigma-Aldrich, Darmstadt, Germany, cat # C1764-50MG), DMEM (Gibco, cat # 619965059), FBS (Biowest, Nuaillé, France, cat # S1860-500), HEPES (Sigma-Aldrich, cat # H0887-20ML), Glutamine (Biowest, cat # X0550-100), and a penicillin/streptomycin/amphotericin cocktail (Antibiotic Antimycotic Solution, Merck, Darmstadt, Germany, cat # A5955-20ML).
2.3. Osteochondral Tissue Processing
Osteochondral grafts were harvested from deceased donors within 24 h after death and stored at 4 °C. The study used 12 femoral condyles from three human cadaveric donors, obtained after consent to be used for scientific purposes. Femoral condyles were processed in class B cleanrooms under a class A laminar flow cabinet in our classified facilities (Barcelona Tissue Bank (BTB)—Banc de Sang i Teixits (BST);
www.bancsang.net (accessed on 27 August 2022). Soft tissues adjacent to the joint area were removed and the bone area of the graft was washed with 0.9% NaCl to remove any remaining blood and fat residues. Condyles were stored for three weeks in physiological medium (Ringer’s lactate) supplemented with an antibiotic cocktail (tobramycin, vancomycin, cotrimoxazole) and anti-fungal agent (amphotericin A).
2.4. Cell Viability by Flow Cytometry
For the flow cytometry analysis, cartilage samples were obtained by sagittal cutting (400 mg) and chopped into small fragments. Pre-digestion was carried out with 1 mg/mL pronase dissolved in culture medium (DMEM, 1% HEPES, 1% Glutamine, 1% penicillin/streptomycin/amphotericin supplement). Cartilage fragments were incubated for 1h at 37 °C with gentle agitation. A second digestion was subsequently performed by adding 3 mg/mL type II collagenase dissolved in complete culture medium (DMEM, 10% FBS, 1% HEPES, 1% glutamine, 1% penicillin/streptomycin/amphotericin supplement), and incubated for 3 h at 37 °C with gentle agitation. After digestion, the cell suspension was filtered through a 70-µm pore membrane filter to remove undigested cartilage fragments. The cell suspension was then centrifuged at 2400 rpm for 5 min and the pellet was resuspended in PBS and divided equally into 6 aliquots. These were incubated at room temperature (RT) in darkness with the following probes: (i) calcein AM (4 µM for 30 min), (ii) 7-AAD (0.5 µg/mL for 10 min), (iii) Annexin V (0.5 µg/mL for 10 min), and (iv) Vybrant®® DyeCycleTM Ruby Stain (5 µM for 15 min). The cell fluorescence signal (5 × 105 cells/mL and 5000 events per measurement) was acquired using a BD LSR Fortessa cytometer (Becton Dickinson, San José, CA, USA) using the following lasers and detectors: (i) Blu-ray/violet, 405 nm and 50 mW; (ii) Pacific blue, 488 nm and 50 mW; (iii) FITC, 640 nm and 40 mW; and (iv) Alexa Fluor, 647 nm and 50 mW. The data obtained were analyzed using FACS Diva software (Becton Dickinson).
2.5. Cell Viability by Confocal Fluorescence Microscopy
For the confocal fluorescence microscopy assay, fragments of cartilage were obtained by sagittal cutting of the cartilage into 2 × 2 mm pieces. Samples were first incubated with 1 mL of 4 µM calcein AM dissolved in PBS for 30 min at RT in darkness with soft stirring. They were then washed twice with PBS and incubated with 1 mL of 0.5 µg/mL 7-AAD for 10 min at RT in darkness with soft stirring. They were then washed twice with PBS. The cell fluorescence signal was acquired by a Leica TCS SP5 confocal inverted microscope (Leica Microsystems, Mannheim, Germany) on a DMI6000 stand, with the Leica Application Suite Advanced Fluorescence software (Leica LAS AF, version 2.6.3.8173), using a 10×/0.4 NA HC PL APO objective. Calcein images were taken with the 488 nm laser line and emission detection between 495 and 550 nm, taking two averages. Images of the 7-AAD were taken with the 561 nm laser line and emission detection between 574 and 666 nm, taking two averages. The transmitted light channel was used to set the limits of the tissue block in the tile scan area, but not for image acquisition. The channels of 3D stacks were taken sequentially in stack-by-stack acquisition mode. Scanner speed was set at 600 Hz in bidirectional mode. The cartilage fragment was scanned from the surface to the deepest area adjacent to the bone, and cartilage images of the superficial, middle, and deep cartilage area were acquired. The images were bidirectional, captured with a 10× magnification objective, at 1024 × 1024-pixel resolution (speed of 600 Hz) using digital capture software. Images were analyzed using ImageJ Fiji software (1.52p version;
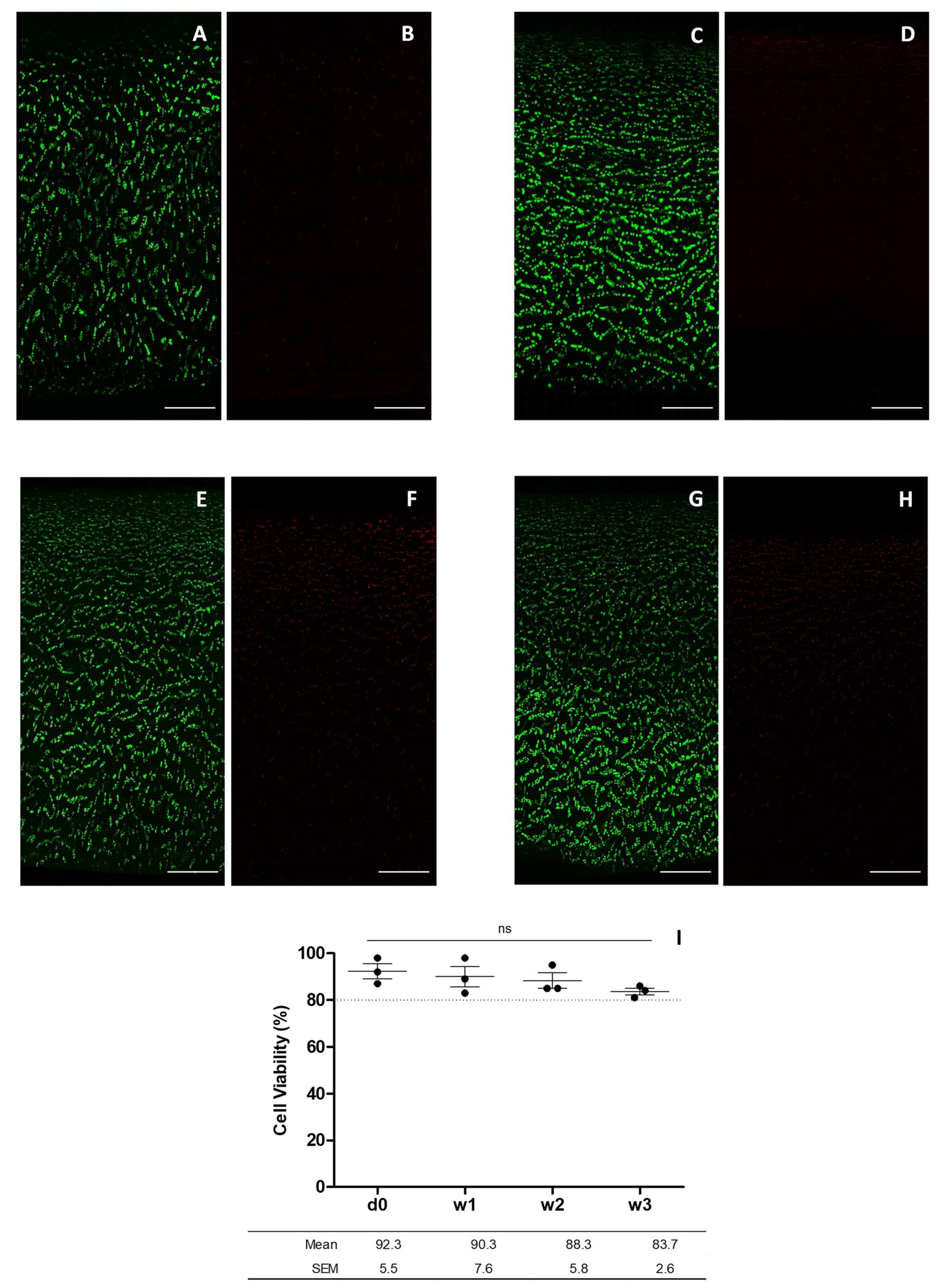
https://imagej.net (accessed on 27 August 2022) through hand-made custom macros that quantified live and dead cells individually. Projection of an image of all z-stack planes together was split into 2 channels, to analyze the calcein and 7-AAD signals, respectively. A threshold value was selected in the histogram to define the value from which the signal would be counted as a particle. The segmentation tool was then applied to separate all sets of cells and differentiate them as a single unit. Finally, the particle analysis tool was applied to count the particles collected in the green and red channels, corresponding to the living and dead cells, respectively.
2.6. Statistical Analysis
Experiments were performed with cartilage from femoral condyles of at least three different donors, and each assay was performed with three technical replicates. The percentage of cell viability is presented as the mean ± standard error of the mean (MD ± SEM) for: number of living cells, dead cells, and apoptotic cells, referring in all cases to the total cell numbers. ANOVA analysis followed by Dunnett’s post hoc test was used for comparisons;
p values less than 0.05 were considered statistically significant. The statistics were determined using commercial software (GraphPad Prism version 5.00 Windows, GraphPad Software, San Diego, CA, USA,
www.graphpad.com (accessed on 27 August 2022).
4. Discussion
Our study compared two methods for assessing cell viability in osteochondral grafts. As a result, the cell viability of chondrocytes measured by confocal fluorescence microscopy and flow cytometry was 83.7 ± 2.6% and 55.8 ± 7.8% for week three, respectively. Contrary to our hypothesis, confocal fluorescence microscopy approach is more advantageous and accurate, as it correlates better with actual cell viability values for monitoring osteochondral graft preservation, detecting only the cells that died a natural death associated with the preservation method.
Although several scientific studies have explored the best method for preserving fresh osteochondral grafts, no consensus has yet been reached regarding the ideal preservation media [
4,
9,
10,
11,
12,
13,
14,
15,
16,
17]. Considering these previous studies as a whole, several preservation media appear to be suitable for maintaining the viability of osteochondral allografts, but it is unclear which medium facilitates better chondrocyte survival, owing to the heterogeneity of results among studies. We hypothesize that these different cell viability results are due to the complexity of analyzing fluorescent confocal microscopy images, since there is no systematic standardized method to determine the cell count using microscopy images. Several approaches have been used to determine chondrocyte viability, such as LDH histochemical staining, calcein AM-ethidium homodimer-1 (CaAM-EthH) staining measured with confocal microscopy and cartilage digestion followed by trypan blue assay [
5]. In relation to the quantification of living and dead cells, the study showed that CaAM-EthH was the only technique to overestimate the cell numbers; however, it was the most appropriate for providing valuable information related with the native cartilage. Furthermore, when LDH histochemical staining was compared with calcein AM-ethidium homodimer-1 staining, measured with confocal microscopy, it resulted in a greater percentage of viable cells analyzed with confocal microscopy than with LDH histochemical staining [
8]. In flow cytometry, the cell solution resulting from digested cartilage is mixed with calcein AM and 7-AAD, which selectively enter living and dead cells, respectively, thus enabling their quantification. The final percentage of cell viability corresponds to the sum of the cells that died due to the stress suffered during the digestion process plus the number of cells that died due to the graft preservation process itself. In contrast, the confocal fluorescent microscopy technique overcomes these limitations methods used maintain its original histological architecture. The cell viability results obtained differed depending on the analysis technique used. Fluorescent confocal microscopy presented 83.7 ± 2.6% viability compared with 55.8 ± 7.8% viability obtained using flow cytometry, after three weeks’ tissue preservation. Flow cytometry results were lower, as previous enzymatic digestion of the cartilage matrix causes cell death inherent to the sample preparation, but not values determined compared with the real cell viability, due to natural causes associated with the preservation method used. In contrast, analysis using confocal fluorescence microscopy allows the preserved osteochondral allografts to maintain their cell viability as described previously [
18,
19,
20]. Evaluation of cell viability by confocal fluorescence imaging enables analysis of cartilage samples in their real state of preservation because cartilage does not require significant processing steps prior to analysis. Conversely, the post-analysis methodology used to count living and dead cells is less systematic, producing variations between studies [
4,
9,
10,
11,
12,
13,
14,
15,
16,
17]. Mathematical treatment of fluorescence images to obtain a live-dead ratio involves a hand-made design of macro templates that include particle cluster fragmentation, and it establishes a fluorescence signal threshold from which the signal is considered a particle (and counted as a cell) or is discarded. Other scientific studies support this evidence, concluding that overestimation of the total cell count by confocal fluorescence microscopy can be explained due to random factors derived from inaccurate calculation of the effective volume of the scanned cartilage fragment, the refractive index of the cartilaginous tissue, and the water content and the angle of the scans by which the fluorescence images are acquired [
5]. Thus, quantification of cell viability using the laser scanning confocal microscopy technique can give poorly reproducible results in cell detection as a result of the water content, extracellular matrix, and the optical properties of the cartilage sample.
Aside from the method used to perform post analysis, another point that has been taken into account is the ability of the technique to distinguish between living and apoptotic cells. Calcein AM is a probe that has the ability to penetrate the plasma membrane of cells. In contrast, 7-AAD is a large molecular size probe that is able to intercalate to the nucleic acids of non-viable cells with a compromised extracellular membrane. When intercalated, the 7-AAD/DNA complex allows the quantification of non-viable cells. Annexin V is a specific marker of apoptotic cells that binds to the phosphatidylserine protein when it translocates from the inside to the outside of the plasma membrane. This occurs when the cell enters in apoptosis, since phosphatidylserine has the function of signaling to the macrophages that the apoptotic cell must be phagocytized [
21]. When intercalated, the Annexin V/phosphatidylserine complex permits the quantification of apoptotic cells. In addition, in the flow cytometry technique, chondrocytes can be labeled with a cell cycle probe as a control for the correct allocation of subpopulations of living or apoptotic cells. This analysis evaluates the phase of the cell cycle in which the cells are found by staining them with the Ruby probe because of its ability to penetrate the extracellular membrane of viable cells and intercalate with their DNA. When viable cells are in one of the different phases of the cell cycle (G0/G1, S or G2/M), a DNA fluorescence spectrum is obtained after irradiation. The signal results in a double peak emission pattern of different intensity depending on the amount of DNA contained in the cells, according to the phase of the cell cycle in which the cells are found. In contrast, apoptotic cells have their DNA so fragmented that Ruby staining is negative. Furthermore, necrotic cells appear in the initial broadband, corresponding to low DNA concentrations. Calcein AM produces double staining as this probe is incorporated by both living and early apoptotic cells, but the post-analysis method does not have the ability to differentiate between this double staining. Thus, it counts a fraction of the cell population that becomes apoptotic really early as a living cell, since the former also produce stain positive for calcein. In contrast, flow cytometry detects early apoptotic cells in the Ruby histogram area, delimiting the wide peak of degraded DNA and the double peak corresponding to the cell cycle. In addition, it is also possible to differentiate early apoptotic cells, as these stain positive for Annexin V. However, although flow cytometry makes it possible to distinguish between live and early apoptotic cells, the cell viability result is lower compared with the real value, since this technique counts dead and early apoptotic cells produced by the enzymatic digestion of the sample and not by the viability resulting from the graft preservation method.
Each analytical technique has its advantages and limitations, and it must be assumed that, depending on the analytical technique used and the parameters fixed by the analyst, the cell viability threshold value obtained after preserving the allograft will be different. Confocal fluorescence microscopy is more advantageous or convenient to flow cytometry in terms of simplicity and required laboratory time, since the sample does not require pre-treatment. This means that the cell viability results obtained correlate better with the state of viability of the cells at the time of preservation. In addition, it provides information on cell distribution and morphology. However, it has two limitations: (1) the inability to discern apoptotic cells, including them as living cells and therefore overestimating counting them as living; and (2) the subjectivity of the post-analysis method used is a function of the hand-made macros used, which produces a deviation between the results found in different studies. The flow cytometry technique is superior in terms of accuracy, since it can distinguish between living, dead and apoptotic cells. However, it has more limitations than confocal fluorescence microscopy. The results obtained using this technique do not accurately reflect the real cell viability values, as it underestimates the number of living cells by counting cells affected by enzymatic digestion of the sample, which are not associated with the natural death due to the cartilage preservation method, as dead or apoptotic. In addition, it provides no information about cell distribution or morphology. It is less advantageous in terms of time and simplicity, since it requires a long protocol for digestion of the cartilage fragments, as well as incubation of the different fluorescence probes. Both stages require a total time of approximately 6 h until cytometry measurements can be performed. In contrast, the confocal fluorescence microscopy technique only requires about 45 min of sample preparation until the images can be acquired.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}