Simple Summary
China has a high prevalence of bovine tuberculosis. In addition, nontuberculous mycobacteria infections may impact the quarantine of bovine tuberculosis. This study investigated the contamination status of Mycobacterium abscessus in cattle farms and slaughterhouses across 12 provinces in China using high-throughput sequencing technology to examine herds with high positivity rates for caudal-fold skin tests and interferon-gamma release assays. The results indicate that M. abscessus contamination was present in the cattle farms and slaughterhouses, and the detection rate of NTM was higher in CFT-positive herds compared to CFT-negative herds, suggesting a potential zoonotic risk. This emphasizes the necessity of enhancing relevant testing protocols for NTM.
Abstract
Nontuberculous mycobacteria (NTM) are environmental organisms that can cause opportunistic infections in humans and animals. Mycobacterium abscessus (Mab) is a rapidly growing Mycobacterium known for its resistance to multiple antibiotics and ability to cause respiratory, skin, and mucosal infections. Understanding the distribution and prevalence of NTM, particularly Mab, in cattle farms and slaughterhouses is crucial for developing effective prevention and control measures. We collected environmental swabs from various surfaces (e.g., feed troughs, sinks, walls, floors, feces, and padding) in cattle farms and slaughterhouses across multiple provinces. High-throughput sequencing technology was utilized to analyze the 16S rDNA V3–V4 region of bacterial DNA extracted from the samples, and qPCR methods were employed to detect and quantify Mycobacterium abscessus in the collected samples. Bioinformatics analysis was performed to identify and classify the NTM species present in the samples. This study compared the abundance and diversity of NTM in different environments and assessed the potential zoonotic risk. A total of 1648 environmental swabs were collected from cattle farms and slaughterhouses in 12 provinces of China in 2023, of which 12 samples tested positive for Mab qPCR detection, yielding a detection rate of 0.73% (12/1648). Among them, the detection rate of environmental samples from cattle farms and slaughterhouses was 0.42% (3/720) and 0.87% (9/928), respectively. This study provides valuable information on the epidemiology of NTM in cattle farms and slaughterhouses, contributing to developing effective strategies for preventing and controlling NTM infections. It also enhances our understanding of the zoonotic potential of Mycobacterium abscessus and other NTM species.
1. Introduction
Nontuberculous mycobacteria (NTM) are all species of bacteria within the genus Mycobacterium (M.), except M. tuberculosis and M. leprae; to date, more than 180 species have been identified [1]. Widely distributed in the environment, NTM predominantly comprise species harmless to humans, but certain strains pose risks as opportunistic pathogens, particularly among immunocompromised individuals [2]. Additionally, some species of mycobacteria are associated with increased mortality in domestic and wild animals or aquatic organisms, contributing to high economic damage and negative environmental impact [3,4]. M. abscessus (Mab) is recognized as a significant pathogen capable of causing respiratory, skin, and mucosal infections, with treatment-challenging regimens due to its resistance [2,5].
Since the first isolation of Mab in 1952 and its recognition as a new species of NTM [6], the nomenclature and species/subspecies identification of Mab have undergone multiple revisions. In 1972, following an international collaborative study by the International Working Group on Mycobacterial Taxonomy, Mab was granted subspecies status. In 1992, DNA hybridization technology was used to identify Mab as an independent strain [6,7]. In 2006, based on rpoB gene sequence analysis, two new species of Mab subsp. massiliense and Mab subsp. bolletii were identified. These, along with Mab, form the three subspecies of the Mycobacterium abscessus complex (MABC) [8].
MABC poses a great threat due to its high degree of resistance [4,9]. In China, MABC is one of the most common NTM pathogens isolated clinically, accounting for 22.2~23.1% of all NTM strains [10,11,12,13]. Globally, MABC is also the most frequently encountered pathogen in hospitals. Notably, Mab accounts for about 80% of NTM that cause respiratory tract infections. It is more common in immunocompromised populations such as patients with cystic fibrosis (CF), human immunodeficiency virus (HIV)-positive individuals, and those with chronic obstructive pulmonary disease or bronchiectasis. Importantly, Mab has also been detected in the lymph nodes of some wild animals, including a lion that tested positive for Mycobacterium tuberculosis [14]. These findings not only suggest Mab as an emerging veterinary pathogen but also highlight its potential interference with the immunodiagnosis of tuberculosis.
Next-Generation Sequencing (NGS) has demonstrated performances comparable to traditional methods in precisely identifying NTM species in clinical specimens, showing its promising potential and efficiency as an alternative tool for the rapid diagnosis of NTM disease [15].
This study investigates the distribution of Mab in Chinese cattle farms and slaughterhouses, aiming to provide a scientific basis for developing effective prevention and control measures. The results are expected to help reduce Mab transmission risks in cattle populations, safeguard livestock industry health, and support public health.
2. Materials and Methods
2.1. Sample Collection
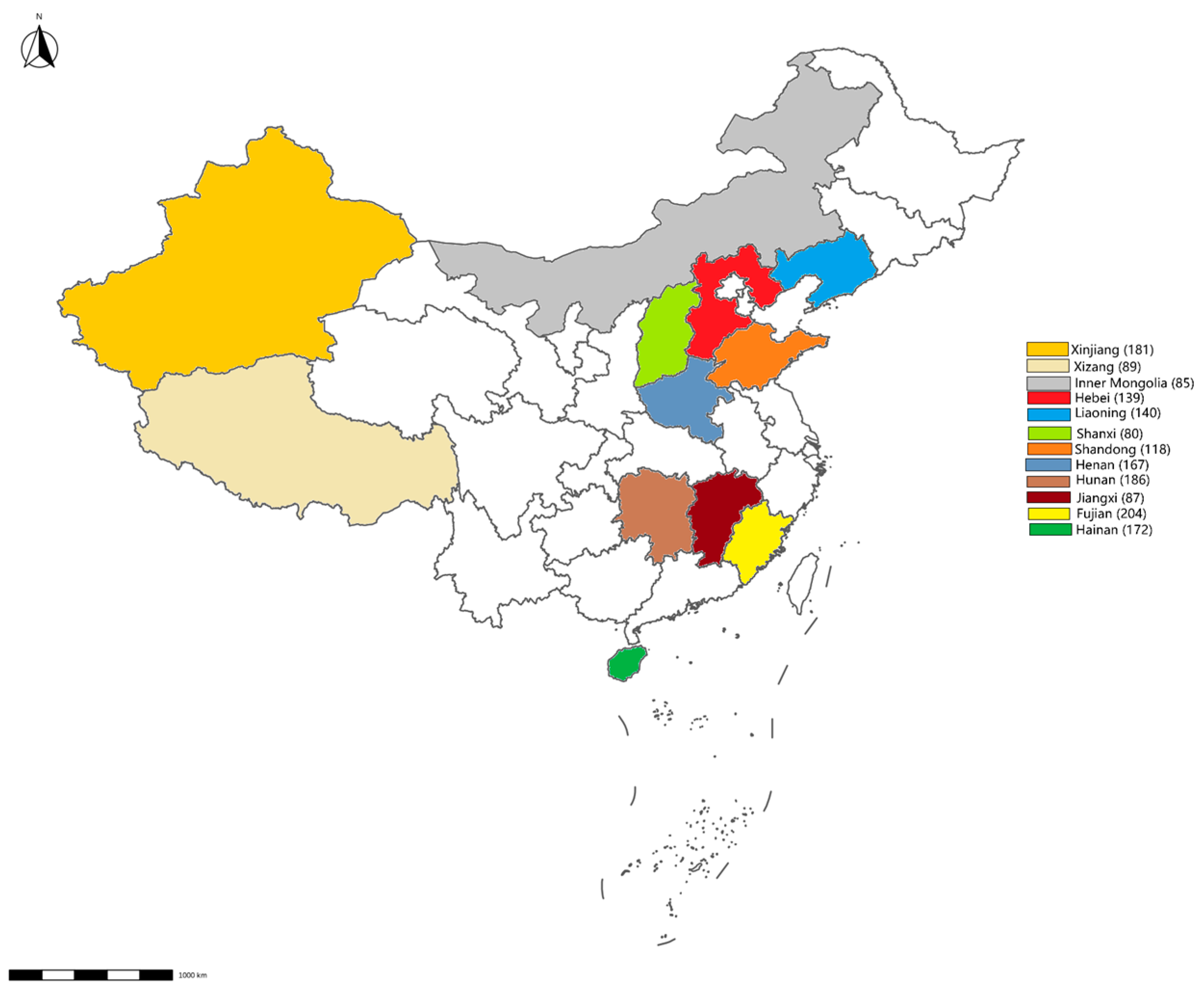
From January to December 2023, 1648 environmental swabs were collected from 54 cattle farms and 24 slaughterhouses across 12 provinces (Figure 1). These cattle farms and slaughterhouses were selected through stratified random sampling to represent China’s major dairy production regions. The sampling targeted high-contact surfaces (feed troughs, floors, walls) and biological materials (feces, nasal/rectal swabs). After collection, the environmental swabs were placed in 0.5 mL of PSB and stored at −20 °C.
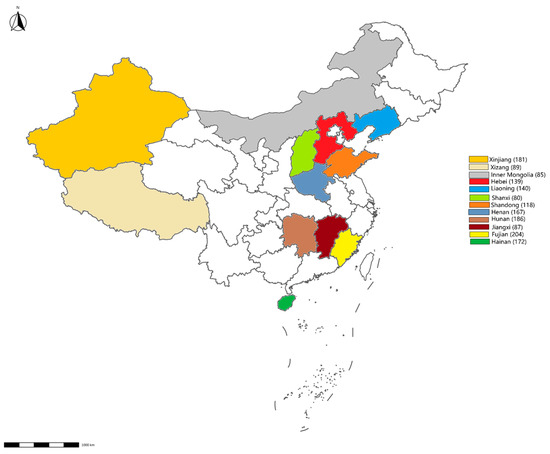
Figure 1.
Map showing the sampling sites. The number of samples collected from each province is displayed in brackets.
A Xinjiang cattle farm with high caudal-fold skin test (CFT)/interferon-gamma release assay (IGRA) positivity rates was selected for comparative analysis. The CFT and IGRA testing methodology was based on the protocol listed in the Manual of Diagnostic Tests and Vaccines for Terrestrial Animals by WOAH [16]. The cattle were sampled into 4 groups (Table 1). From each group, 20 cattle were randomly chosen, and nasal and rectal swabs were gathered. Specimens were collected using sterile cotton swabs. The dry swab was first moistened with PBS, and the moistened swab was then used to collect cow nasal secretions and rectal feces. The obtained samples were transferred to 5 mL centrifuge tubes containing sterile physiological saline, stored at 4 °C, and transported to the laboratory.

Table 1.
Group information on high-throughput sequencing.
2.2. Environmental Samples Detection
The environmental samples were inactivated at 95 °C for 20 min. Then, they were centrifuged, and the qPCR method established by Salvatore et al. [17] was used to detect Mab, with double-distilled water and standard plasmids serving as controls. PCR primers and probes were synthesized by the Beijing Genomics Institute (Beijing, China), and their sequences are detailed in Table 2. All environmental samples were amplified in triplicate on a QuantStudio™ Real-Time PCR System (Thermo Fisher Scientific, Shanghai, China). Real-time PCR amplification was carried out in a 20 μL reaction mixture consisting of 10 μL Premix Ex Taq (Takara, Beijing, China), 15 pM of each primer, and 1 μL of inactivated environmental sample. The cycling conditions were as follows: initial denaturation for 3 min at 95 °C, followed by 15 s at 95 °C, 30 s at 56 °C, and 30 s at 72 °C (40 cycles).
Table 2.
qPCR primer sequences for Mab.
2.3. High-Throughput Sequencing of 16S rDNA V3–V4 Region
Total bacterial DNA extraction from nasal swabs and rectal swabs was carried out following the instructions of the QIAamp DNA Stool Mini Kit (Lumiprobe, Shenzhen, China). The concentration of total DNA was determined using NanoDrop2000 (Thermo Fisher Scientific, Shanghai, China) and recorded. The extracted DNA was then sent to Beijing Novogene Company (Beijing, China) for the high-throughput sequencing of the 16S rDNA V3–V4 region. The sequencing primers used were 341F (5′ CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNNGGGTATCTAAT-3′). Small fragment libraries were constructed based on the characteristics of the amplified regions, and paired-end sequencing was performed on the libraries using the Illumina NovaSeq (Illumina, San Diego, CA, USA) sequencing platform. The raw sequencing data were demultiplexed based on unique barcodes to separate reads corresponding to each sample, during which the barcodes and primer sequences were trimmed from the reads. Subsequently, the paired-end reads (R1 and R2) were concatenated or merged into longer contiguous sequences using FLASH (1.2.11) software. FastQC (v0.11.9) software was used to perform quality control on the merged tags, obtain clean tags, and filter out chimeric sequences to obtain effective tags for subsequent analysis, and DADA2 was used for denoising and determining each amplicon sequence variant (ASV). Data processing included quality control (FastQC), chimera removal (DADA2), and taxonomic classification (QIIME2, Silva v132).
2.4. Specific ASV Screening, Sequence Comparison, and Strain Identification
Based on the amplicon sequence variant (ASV) annotation results, an abundance table was compiled encompassing taxonomic levels from kingdom to species. Subsequently, this study focused on analyzing and screening processes related to the Mycobacterium genus from the genus-level abundance table. The representative amplified sequences of these specific ASVs were identified in the corresponding original data, and NCBI-BLAST was utilized for the alignment analysis of these extracted sequences for strain-level identification.
3. Results
3.1. Detection Results of Environmental Samples
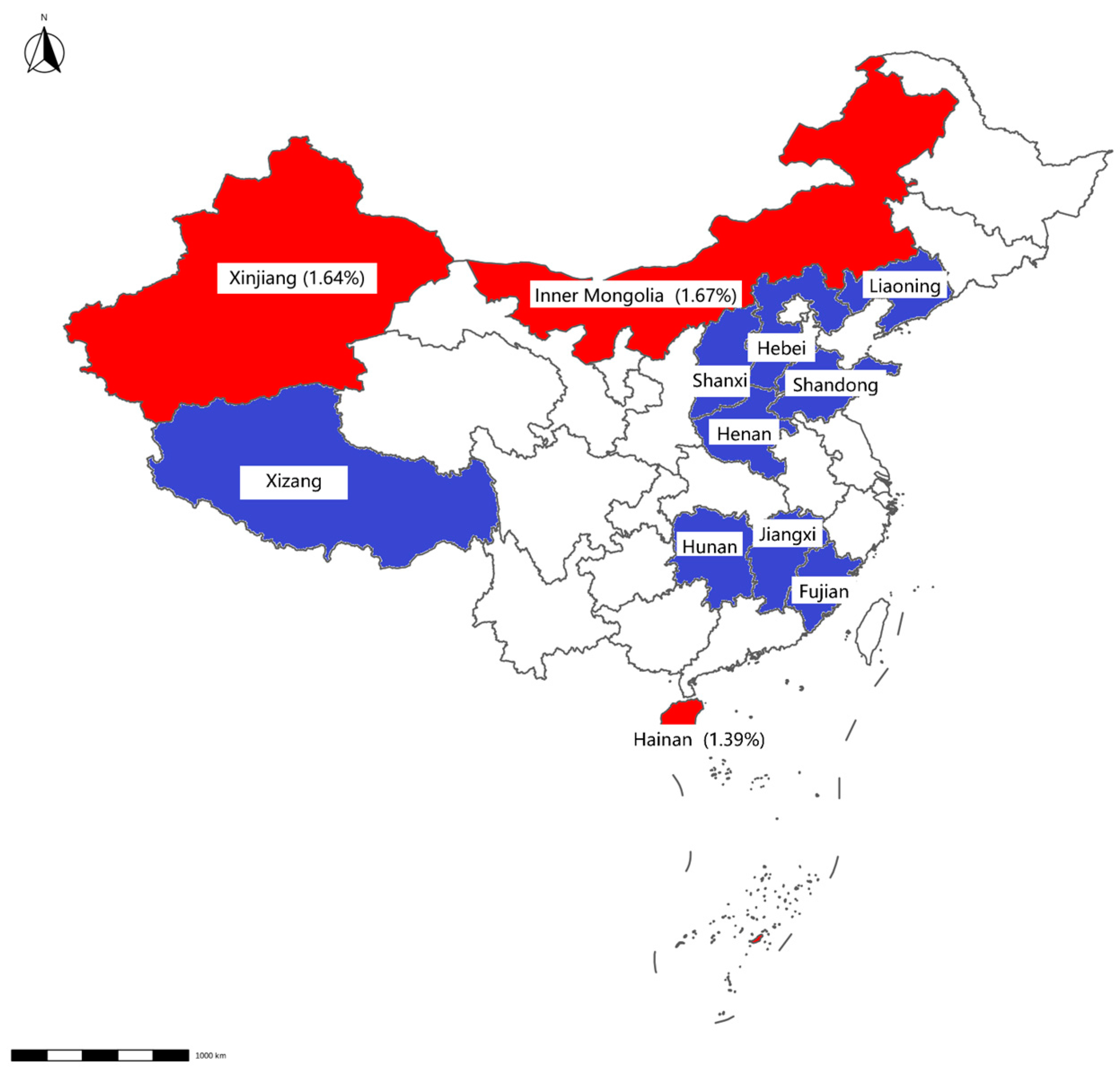
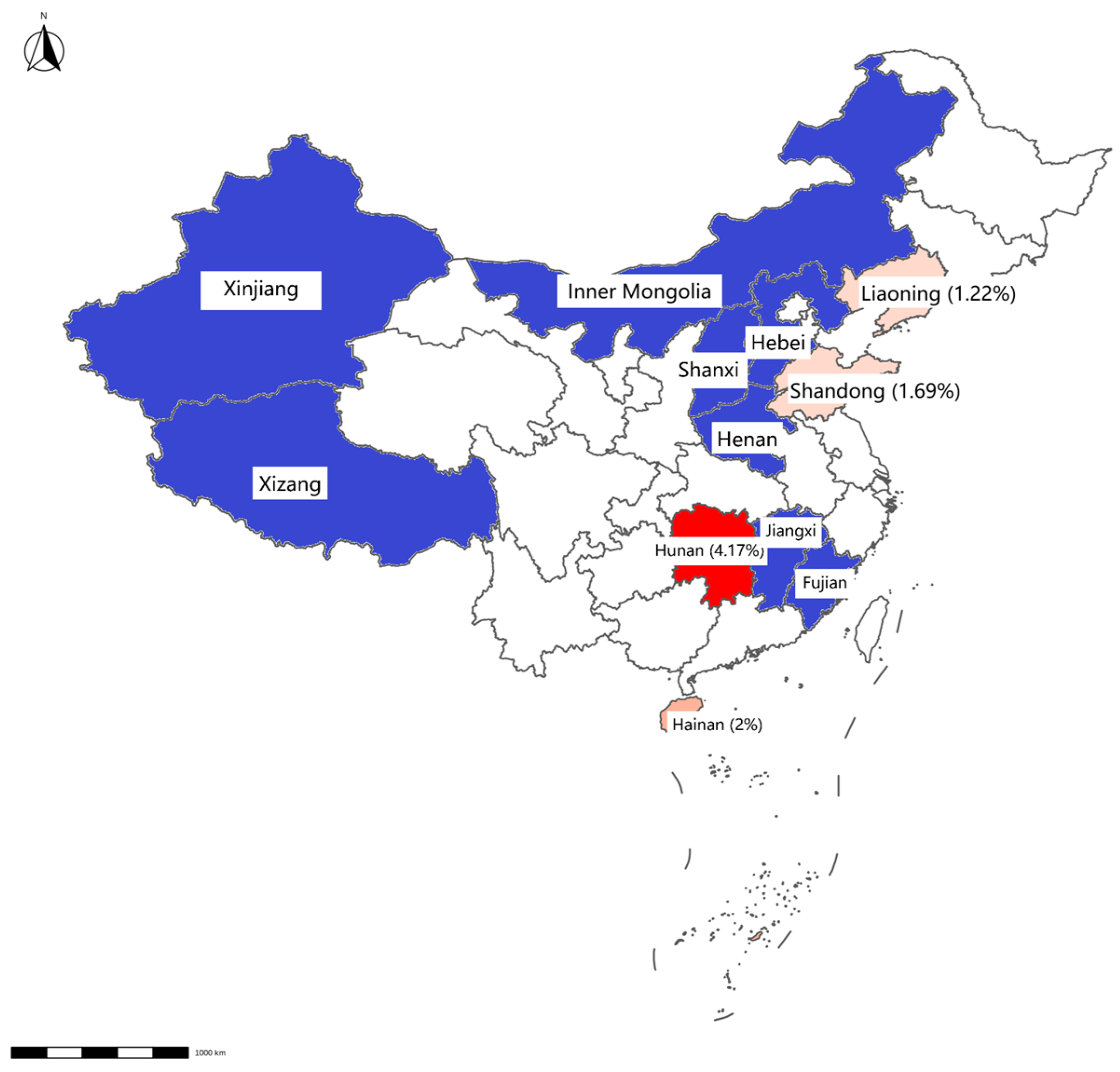
Mab was detected in 0.73% of samples (12/1648), with higher prevalence in slaughterhouses (0.87%, 9/928) than in farms (0.42%, 3/720). Geographically, Mab-positive samples were clustered in Hainan, Inner Mongolia, Hunan, Shandong, and Liaoning (Table 3, Figure 2 and Figure 3).
Table 3.
qPCR detection of Mab in environmental samples from 12 provinces in China.
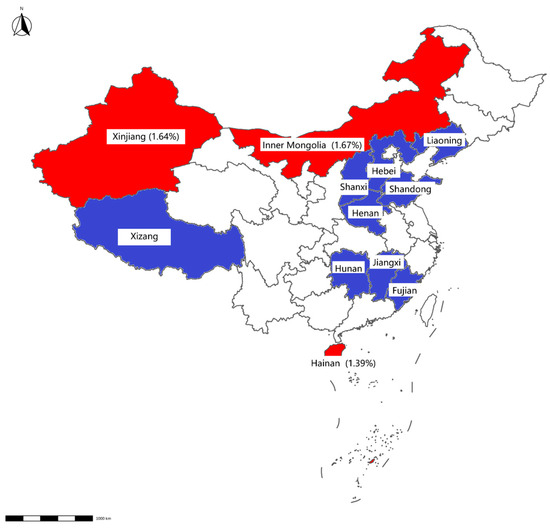
Figure 2.
Map displaying detection results of swab samples from cattle farms. The positive rate for each province is displayed in brackets. Provinces with negative Mab results detected by qPCR are highlighted in blue, whereas those with positive results are indicated in red.
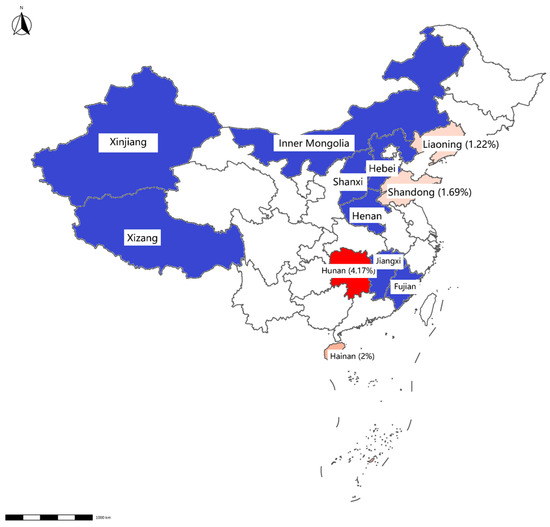
Figure 3.
Map displaying detection results of swab samples from slaughterhouses. The positive rate for each province is displayed in brackets. Provinces with negative Mab results detected by qPCR are highlighted in blue, whereas those with positive results are indicated in red.
3.2. Dilution Curve
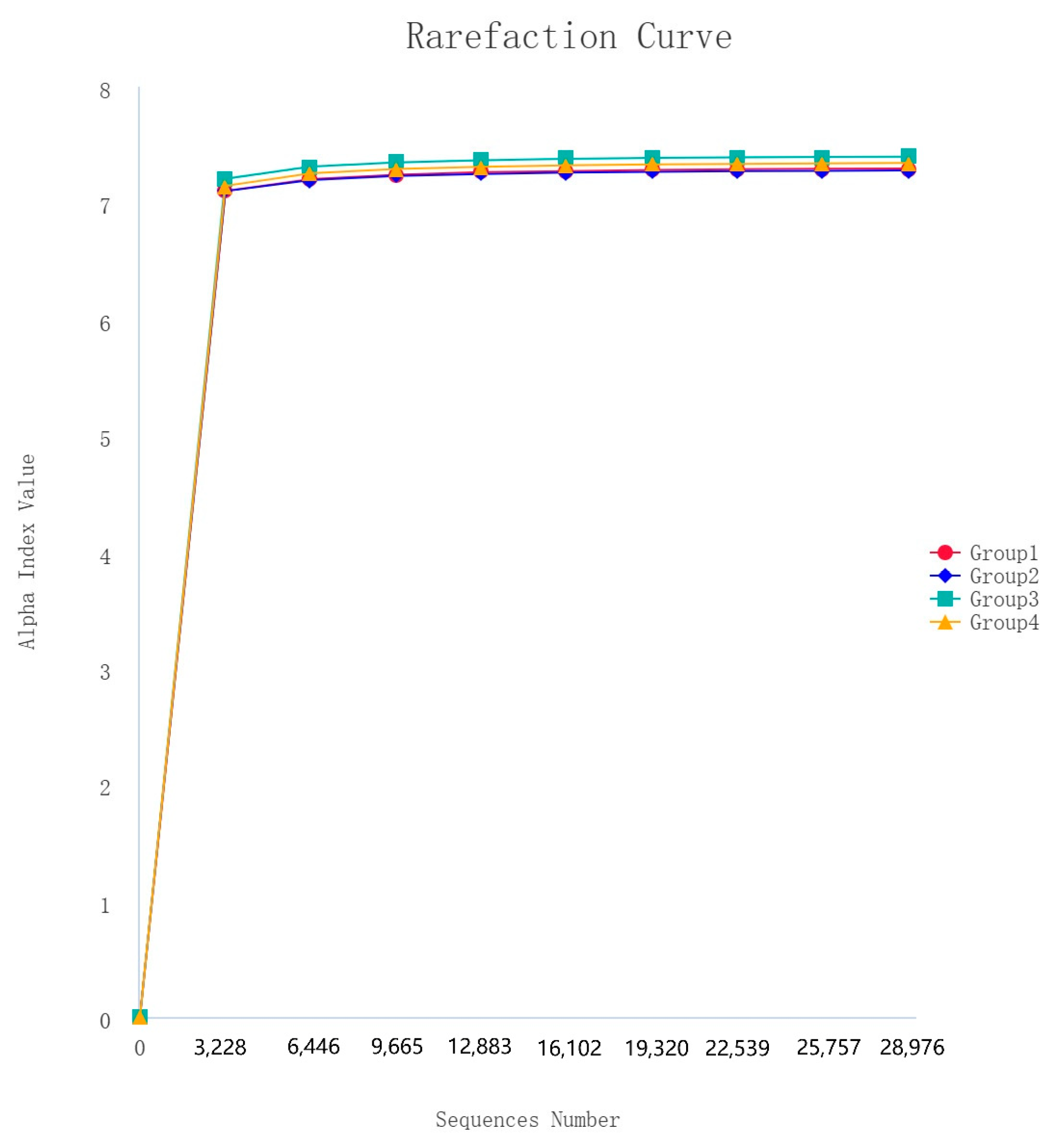
After 16S rDNA sequencing, 160 samples underwent quality control, splicing, and noise reduction in the double-ended sequences, resulting in a total of 20,472 ASVs. The observed rarefaction curves for the ASVs and the Shannon alpha diversity index approached those of the saturation phase, indicating good sample coverage, and the asymptotic distribution of the curves suggests that they are comparable (Figure 4).
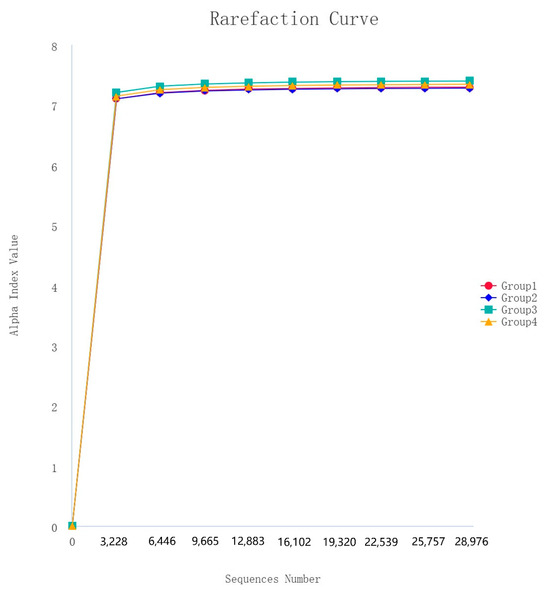
Figure 4.
Rarefaction curves of alpha diversity index (Shannon).
3.3. Analysis of Intergroup Differences in Nasal Swab Samples and Rectal Swab Samples
Beta diversity analysis was carried out on high-throughput sequencing data using QIIME 2.
The linear discriminant analysis effect size (LEfSe) was used to assess the differences between different groups of samples both at the genus or higher taxonomic level.
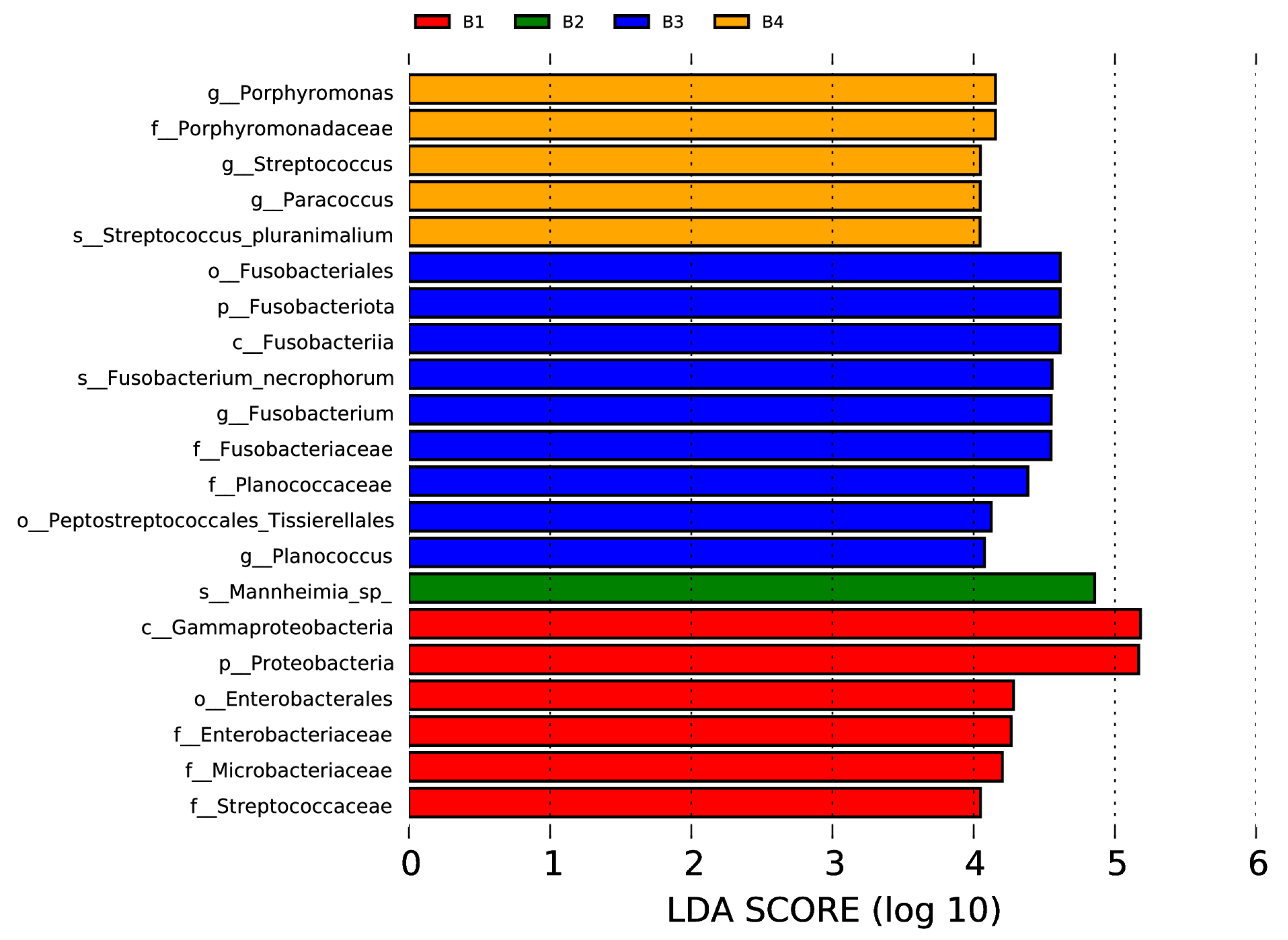
In the nasal swab samples, there were two phyla (Fusobacteriota and Proteobacteria), two classes (Fusobacteriia and Gammaproteobacteria), three orders (including Fusobacteriales, Peptostreptococcales, Tissierellales, and Enterobacterales), six families (Porphyromonadaceae, Fusobacteriaceae, Planococcaceae, Enterobacteriaceae, Microbacteriaceae, and Streptococcaceae), five genera (Porphyromonas, Streptococcus, Paracoccus, Fusobacterium, and Planococcus), and three species (Streptococcus pluranimalium, Fusobacterium necrophorum, and Mannheimia sp.) with significant intergroup differences, as shown in Figure 5 (p < 0.05, LDA > 4).
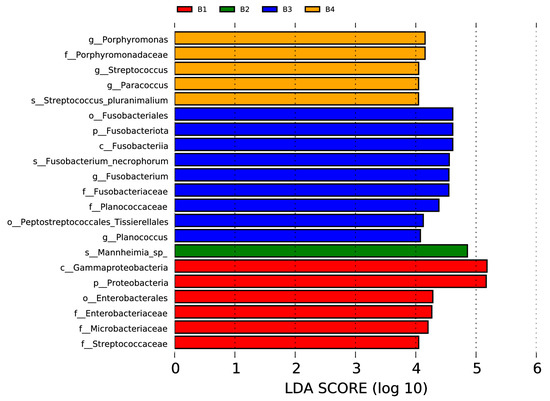
Figure 5.
Visualization of most significant taxa (genus or higher level) that differentiate between different groups in bovine nasal swab sample microbiomes.
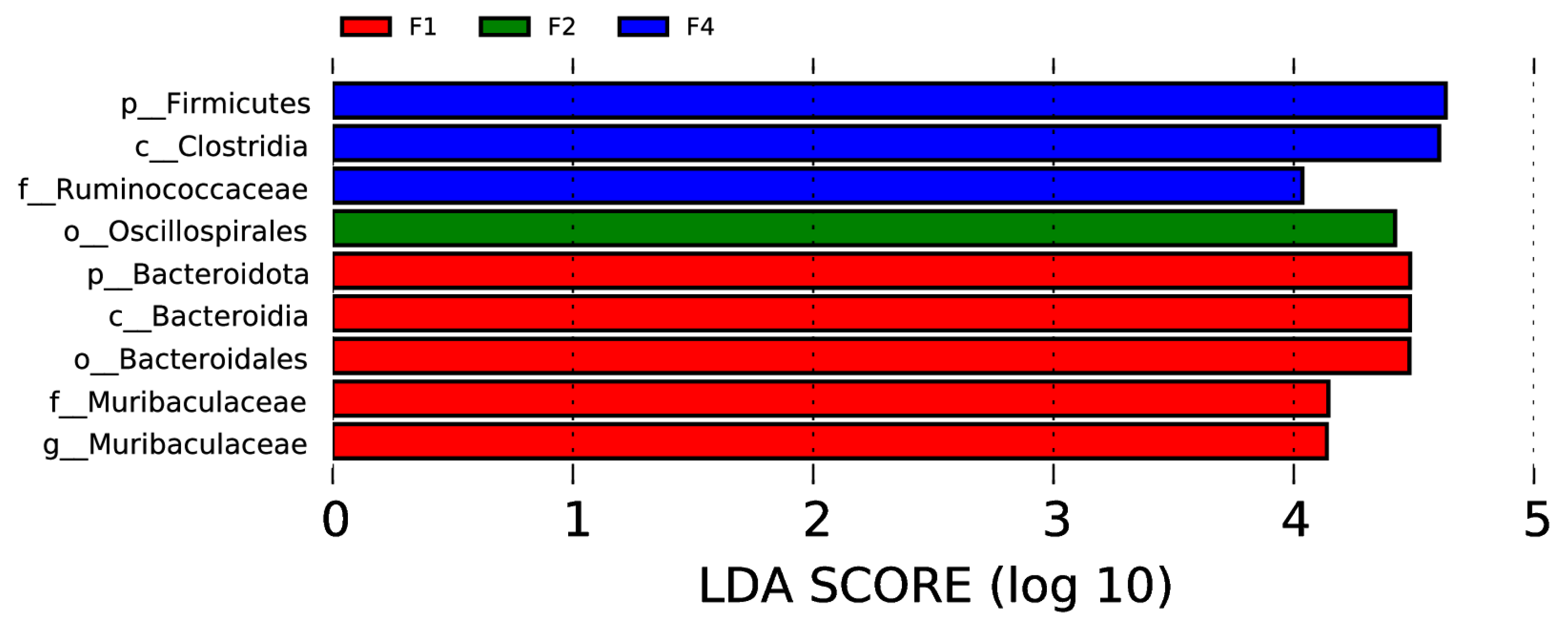
In the rectal samples, there were two phyla (Firmicutes and Bacteroidota), two classes (Clostridia and Bacteroidia), two orders (Oscillospirales and Bacteroidales), two families (Ruminococcaceae and Muribaculaceae), and one genus (Muribaculaceae) with significant intergroup differences, as shown in Figure 6 (p < 0.05, LDA > 4).
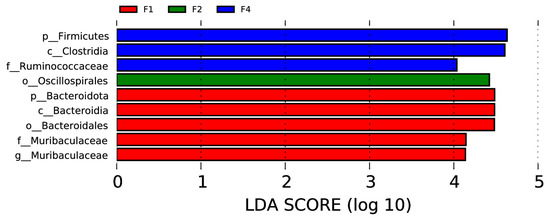
Figure 6.
Visualization of most significant taxa (genus or higher level) that differentiate between different groups in bovine rectal swab sample microbiomes.
3.4. Analysis of Mycobacterium ASVs in Different Groups
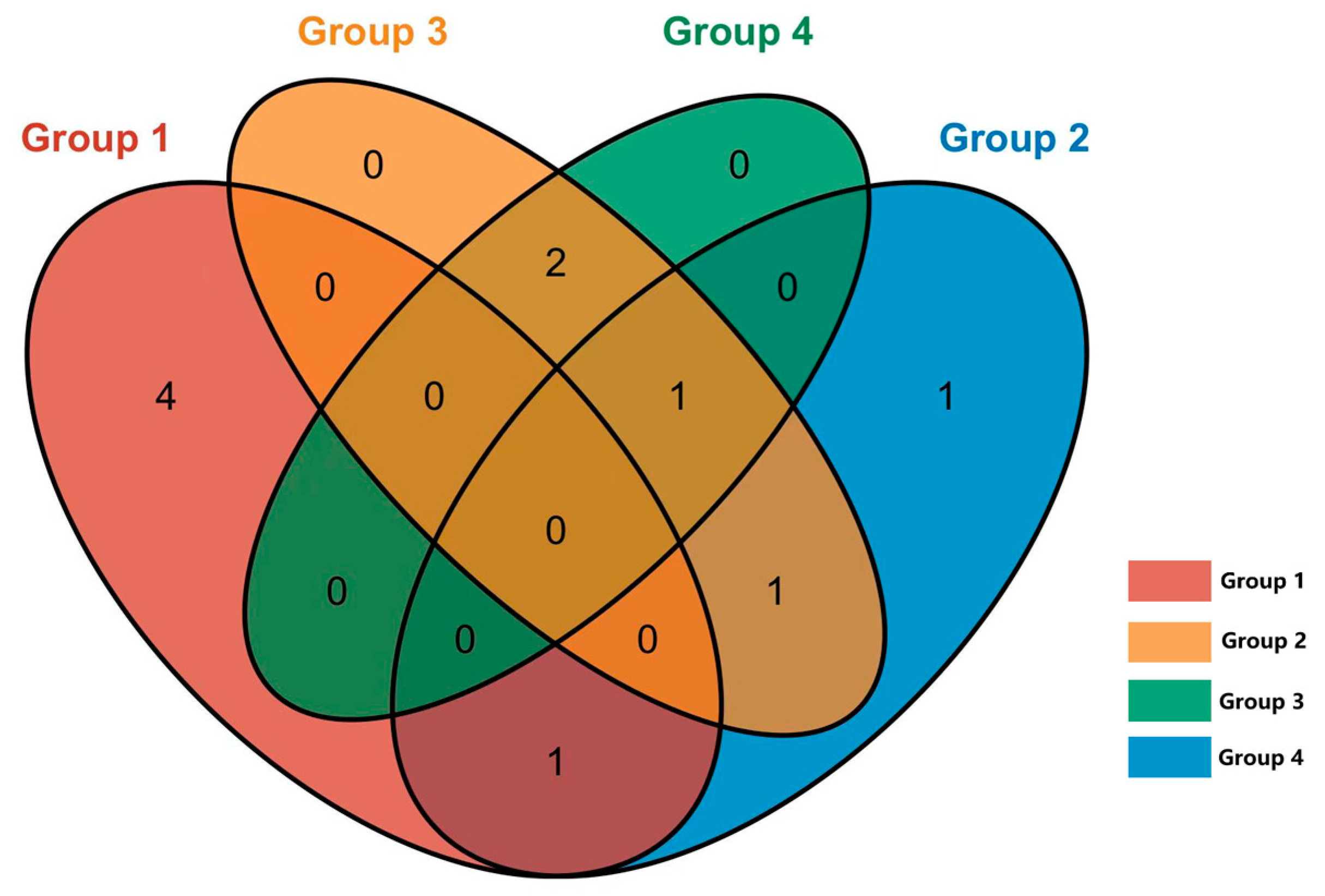
In the nasal and rectal swabs, the genus- and species-level assessments indicated a low abundance of Mycobacterium species (less than 0.1% of reads in the sample). These Mycobacterium genus-specific reads were isolated manually for further BLAST-based analysis. A total of 40 ASV-representative bacterial genera were identified as Mycobacterium. Through NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi, 14 December 2024) comparison, a total of 11 NTM species were identified, with no detection of the Mycobacterium tuberculosis complex. The distribution of NTM among different groups is shown in Figure 7.
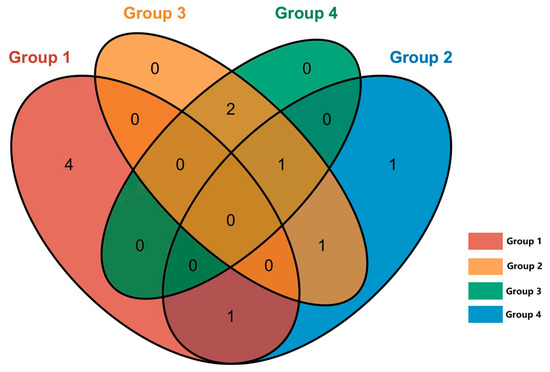
Figure 7.
Venn diagram of NTM shared by different groups. Group 1: M. bovis positive (IGRA), positive (CFT); Group 2: M. avium positive (IGRA), positive (CFT); Group 3: negative (IGRA), positive (CFT); Group 4: negative (IGRA), negative (CFT).
NTM were detected in 22 out of 60 cows (36.7%) in groups 1–3 (CFT-positive), compared to only 4 out of 20 cows (20%) in group 4 (CFT-negative). Notably, the detection rate of NTM in groups 1, 2, and 3 was significantly higher compared to in group 4.
In group 1, where cows tested positive for CFT and M. bovis in IGRA, NTM was detected in 8 cows out of 20, yielding a detection rate of 40%. In group 2, cows that tested positive for CFT and M. avium in IGRA, NTM was detected in 7 of the total 20 cows (35%). On the other hand, in groups 3 and 4, where cows tested negative in IGRA, NTM was detected in 12 of the total 40 cows (30%). Notably, there were no statistically significant differences observed in the NTM detection rates among these groups.
In total, 26 nasal swab samples and 3 rectal swab samples tested positive for NTM, revealing that NTM are more likely to be found in the respiratory tract than in the digestive tract. Further analysis through BLAST comparisons identified 11 NTM species: M. phlei, M. aichiense, M. abscessus, M. saskatchewanense, M. smegmatis, M. kansasii, M. confluentis, M. vaccae, M. jacquezzii, M. celeriflavum, and M. stelleraae.
4. Discussion
This study confirms Mab’s presence in Chinese livestock environments, particularly in regions with warm climates and an active animal trade. Xinjiang and Inner Mongolia are two of the main milk supply provinces in China; in these regions the detection rate is lower in slaughterhouses than in livestock farms. However, Hunan and the other provinces mostly supply beef cattle, and in these regions the detection rate in slaughterhouses is significantly higher than that in breeding farms. Our findings reveal significant differences in the NTM contamination on these farms compared to previous reports, which may be attributed to climatic and regional variations [18].
This study sampled 12 major cattle breeding provinces in China. In previous years, multiple studies have shown that the detection rate of M. bovis in Xinjiang has always been higher than in other provinces. Therefore, we chose Xinjiang as the main research province and conducted sampling and mNGS analysis on a highly CFT- and IGRA-positive cattle farm in Xinjiang. LEfSe analysis showed distinct microbial communities in the respiratory and digestive tracts across groups. However, these differences mainly resulted from variations in bacterial counts on swabs and were not linked to pathogenic effects. In this study, it appears that cattle infected or latently infected with tuberculosis or NTM do not exhibit a significant effect on the microbiota of their respiratory tract infection. Higher NTM detection rates in CFT-positive herds suggest diagnostic interference, necessitating species-specific testing, but M. smegmatis and M. kansasii are known to confound tuberculosis tests [19,20]. Although M. saskatchewanense and Mab have not been reported as pathogenic to cows [21,22,23], the zoonotic potential of M. saskatchewanense and Mab warrants further investigation.
Mab is an important pathogen that causes respiratory, skin, and mucous membrane infections, and its resistance to multiple antibiotics makes it notoriously challenging to treat [2,5]. Due to the lack of widely accessible antibody detection products, this study employed molecular biological techniques to identify the existence of Mab in cattle farms and slaughterhouses. Although at a low detection rate, the presence of Mab was confirmed across numerous provinces in China, and there is a risk of transmission from animals or animal products to humans [24,25]. This report marks the first survey of its kind in the country.
In veterinary medicine, livestock diagnosed with tuberculosis infections are predominantly managed through culling protocols. However, this approach may lead to diagnostic misclassification due to cross-reactivity with infection caused by NTM, imposing substantial economic burdens on global livestock industries. Current evidence suggests that implementing multi-modal diagnostic assays—including interferon-gamma release assays, mycobacterial culture speciation, and molecular differentiation techniques—could effectively distinguish NTM-infected herds from true TB-positive populations [26,27,28].
In veterinary practice, livestock populations misdiagnosed with tuberculosis due to nontuberculous mycobacterial (NTM) infections may be managed through antimicrobial protocols analogous to those in human medicine, typically involving combination therapies with macrolides (e.g., clarithromycin), rifamycins (rifampicin), fluoroquinolones (ciprofloxacin), and aminoglycosides (amikacin) [29,30,31,32,33,34]. Staged culling protocols should be implemented only after confirmed therapeutic failure through mycobacterial culture sensitivity testing and clinical outcome monitoring. This evidence-based approach synergistically preserves productive herds while minimizing zoonotic transmission risks through sustained bacteriological clearance. The transition from blanket depopulation to precision veterinary medicine frameworks enables the maintenance of elite genetic reservoirs and enhancement of herd productivity metrics, concurrently reducing annual agricultural economic losses in affected regions through preserved breeding stock value and optimized antimicrobial stewardship outcomes.
At present, there are many studies indicating that M. tuberculosis or M. bovis can be transmitted from humans to animals [25,35,36,37,38,39]. In Spain, the United States, and India, the human-to-animal transmission of M. tuberculosis or M. bovis is well-documented [35,37,40], and most reports on cattle with M. tuberculosis are from countries where human M. tuberculosis prevalence is very high and likely to have been the source of introduction into the cattle populations [41,42,43]. However, although NTM only poses a potential risk of transmission to humans from animals or animal-derived products [24], there is a lack of research on the transmission of NTM from humans to animals. Unfortunately, the current understanding of NTM’s infectious characteristics, transmission routes, and control strategies in wildlife and livestock remains limited. Given the global rise in zoonotic diseases, Mab represents a significant public health threat. Globalization, climate change, wildlife migration, livestock trade, and human activities may accelerate the spread of NTM. Hence, research on NTM carries not just substantial scientific value but also urgent practical importance.
5. Conclusions
Mab and other NTM species pose significant challenges to livestock health and public health. Enhanced surveillance, climate-adapted hygiene protocols, and molecular diagnostics are critical to mitigating transmission risks. This study provides the first national-scale epidemiological data on NTM in Chinese cattle, informing future research and policy development.
Author Contributions
S.C.: data curation, investigation, validation, and writing—original draft. M.L.: investigation, supervision, validation, resources, and writing—review and editing. Y.L. (Yan Li): methodology, validation, resources, and writing—review and editing. J.Z.: investigation and writing—review and editing. Y.L. (Yanfang Li): formal analysis, validation, funding acquisition, and writing—review and editing. Y.L. (Yan Liang): investigation, validation, and writing—review and editing. X.F.: supervision, funding acquisition, resources, and writing—review and editing. Y.Q.: conceptualization, supervision, formal analysis, project administration, validation, funding acquisition, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Bintuan Science and Technology Program Corps (2020AB015), the Research Initiation Program for High-level Talents at Shihezi University (RCZK201902), the National Key Research and Development Program of China (Grant No. 2022YFD1800702), and the earmarked fund for CARS (Grant No. CARS36).
Institutional Review Board Statement
Ethical review and approval were waived for this study because the research was solely involved routine animal sampling, did not induce pain or suffering in the animals, and did not involve euthanasia.
Informed Consent Statement
Informed Consent was obtained from all animals owners involved in this study.
Data Availability Statement
The data that support the findings of this study are available from NCBI, reference number PRJNA1120236.
Acknowledgments
The authors thank the College of Animal Science and Technology for facilitating the study and the aforementioned funding bodies for their financial support.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Fedrizzi, T.; Meehan, C.J.; Grottola, A.; Giacobazzi, E.; Serpini, G.F.; Tagliazucchi, S.; Fabio, A.; Bettua, C.; Bertorelli, R.; Sanctis, V.D.; et al. Genomic characterization of nontuberculous Mycobacteria. Sci. Rep. 2017, 7, 45258. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Herrmann, J.-L.; Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 2020, 18, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Cai, Q.; Miao, Q.; Song, Z.; Fang, Y.; Hu, B. High-Throughput Metagenomics for Identification of Pathogens in the Clinical Settings. Small Methods 2020, 5, 2000792. [Google Scholar] [CrossRef] [PubMed]
- Prevots, D.R.; Marras, T.K. Epidemiology of Human Pulmonary Infection with Nontuberculous Mycobacteria. Clin. Chest Med. 2015, 36, 13–34. [Google Scholar] [CrossRef]
- Degiacomi, G.; Sammartino, J.C.; Chiarelli, L.R.; Riabova, O.; Makarov, V.; Pasca, M.R. Mycobacterium abscessus, an emerging and worrisome pathogen among cystic fibrosis patients. Int. J. Mol. Sci. 2019, 20, 5868. [Google Scholar] [CrossRef]
- Lopeman, R.C.; Harrison, J.; Desai, M.; Cox, J.A.G. Mycobacterium abscessus: Environmental bacterium turned clinical nightmare. Microorganisms 2019, 7, 90. [Google Scholar] [CrossRef]
- Lee, M.-R.; Sheng, W.-H.; Hung, C.-C.; Yu, C.-J.; Lee, L.-N.; Hsueh, P.-R. Mycobacterium abscessus complex infections in humans. Emerg. Infect. Dis. 2015, 21, 1638–1646. [Google Scholar] [CrossRef]
- Tortoli, E.; Kohl, T.A.; Brown-Elliott, B.A.; Trovato, A.; Leão, S.C.; Garcia, M.J.; Vasireddy, S.; Turenne, C.Y.; Griffith, D.E.; Philley, J.V.; et al. Emended description of Mycobacterium abscessus, Mycobacterium abscessus subsp. abscessus and Mycobacterium abscessus subsp. bolletii and designation of Mycobacterium abscessus subsp. massiliense comb. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4471–4479. [Google Scholar] [CrossRef]
- Degiacomi, G.; Chiarelli, L.R.; Recchia, D.; Petricci, E.; Gianibbi, B.; Fiscarelli, E.V.; Fattorini, L.; Manetti, F.; Pasca, M.R. The Antimalarial Mefloquine Shows Activity against Mycobacterium abscessus, Inhibiting Mycolic Acid Metabolism. Int. J. Mol. Sci. 2021, 22, 8533. [Google Scholar] [CrossRef]
- Andrew, E.C.; Connell, T.; Robinson, P.; Curtis, N.; Massie, J.; Robertson, C.; Harrison, J.; Shanthikumar, S.; Bryant, P.A.; Starr, M.; et al. Pulmonary Mycobacterium abscessus complex in children with cystic fibrosis: A practical management guideline. J. Paediatr. Child Health 2019, 55, 502–511. [Google Scholar] [CrossRef]
- Mougari, F.; Guglielmetti, L.; Raskine, L.; Sermet-Gaudelus, I.; Veziris, N.; Cambau, E. Infections caused by Mycobacterium abscessus: Epidemiology, diagnostic tools and treatment. Expert. Rev. Anti-Infect. Ther. 2016, 14, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Dorn, A.v. Multidrug-resistant Mycobacterium abscessus threatens patients with cystic fibrosis. Lancet Respir. Med. 2017, 5, 15. [Google Scholar] [CrossRef]
- Honda, J.R.; Virdi, R.; Chan, E.D. Global environmental nontuberculous Mycobacteria and their contemporaneous man-made and natural niches. Front. Microbiol. 2018, 9, 2029. [Google Scholar] [CrossRef]
- Gcebe, N.; Hlokwe, T.M. Non-tuberculous mycobacteria in South African wildlife: Neglected pathogens and potential impediments for bovine tuberculosis diagnosis. Front. Cell. Infect. Microbiol. 2017, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Wang, Q.; Pan, J.; Bao, R.; Jin, W.; Yao, Y.; Fang, T.; Li, N.; Luan, S.; et al. Comparison of molecular testing methods for diagnosing non-tuberculous mycobacterial infections. Eur. J. Clin. Microbiol. Infect. Dis. 2024, 44, 109–116. [Google Scholar] [CrossRef] [PubMed]
- International Office of Epizootics Biological Standards Commission. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 12th ed.; Office International des Épizooties: Paris, France, 2023. [Google Scholar]
- Marras, S.A.E.; Chen, L.; Shashkina, E.; Davidson, R.M.; Strong, M.; Daley, C.L.; Kreiswirth, B.N. A molecular-beacon-based multiplex real-Time PCR assay to distinguish Mycobacterium abscessus subspecies and determine macrolide susceptibility. J. Clin. Microbiol. 2021, 59, e0045521. [Google Scholar] [CrossRef]
- Zeng, W.; Zhang, Y.; Zhao, X.; Huang, G.; Jiang, Y.; Dong, H.; Li, X.; Wan, K.; He, C. Occurrence of non-tuberculous mycobacteria species in livestock from northern China and first isolation of Mycobacterium caprae. Epidemiol. Infect. 2013, 141, 1545–1551. [Google Scholar] [CrossRef]
- Jenkins, A.O.; Gormley, E.; Gcebe, N.; Fosgate, G.T.; Conan, A.; Aagaard, C.; Michel, A.L.; Rutten, V.P.M.G. Cross reactive immune responses in cattle arising from exposure to Mycobacterium bovis and non-tuberculous mycobacteria. Prev. Vet. Med. 2018, 152, 16–22. [Google Scholar] [CrossRef]
- Vordermeier, H.M.; Brown, J.; Cockle, P.J.; Franken, W.P.J.; Drijfhout, J.W.; Arend, S.M.; Ottenhoff, T.H.M.; Jahans, K.; Hewinson, R.G. Assessment of cross-reactivity between Mycobacterium bovis and M. kansasii ESAT-6 and CFP-10 at the T-Cell epitope level. Clin. Vaccine Immunol. 2007, 14, 1203–1209. [Google Scholar] [CrossRef]
- Bisognin, F.; Ferraro, V.; Sorella, F.; Lombardi, G.; Lazzarotto, T.; Monte, P.D. First isolation of Mycobacterium saskatchewanense from medical devices. Sci. Rep. 2023, 13, 21628. [Google Scholar] [CrossRef]
- Kim, M.; Yang, M.-S.; Son, J.; Rhim, H.; Jeong, T.-W.; Kim, B.; Ik-Han, J. Systemic infection of Mycobacterium abscessus in a free-ranging wild Eurasian Eagle Owl (Bubo bubo). J. Wildl. Dis. 2022, 58, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cerdeira, C.; Hernández-Castro, R.; Sánchez-Cárdenas, C.D.; Arenas, R.; Meza-Robles, A.; Toussaint-Caire, S.; Atoche-Diéguez, C.; Martínez-Herrera, E. Atypical Mycobacteriosis due to Mycobacterium abscessus subsp. massiliense: Our experince. Pathogens 2022, 11, 1399. [Google Scholar] [CrossRef]
- Ali, Z.I.; Hanafy, M.; Hansen, C.; Saudi, A.M.; Talaat, A.M. Genotypic analysis of nontuberculous mycobacteria isolated from raw milk and human cases in Wisconsin. J. Dairy Sci. 2021, 104, 211–220. [Google Scholar] [CrossRef]
- Soliman, N.S.; Soliman, M.S.; Khairat, S.M.; Gad, M.A.; Shawky, S.; Elkholy, A.A. Genetic diversities and drug resistance in Mycobacterium bovis isolates from zoonotic tuberculosis using whole genome sequencing. BMC Genom. 2024, 25, 1024. [Google Scholar] [CrossRef]
- Lin, Q.; Yao, Y.; Li, X.; Zhang, S.; Guo, H.; Ma, X.; Chen, W.; Ru, C.; Wang, L.; Wang, B.; et al. The application of nanopore targeted sequencing for pathogen diagnosis in bronchoalveolar lavage fluid of patients with pneumonia: A prospective multicenter study. Infect. Dis. 2024, 56, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, Y.; Wang, Z.; Lin, Y.; Li, Y.; Chen, Y.; Chen, X.; Zhang, C.; Li, W.; Zhang, W.; et al. Multiplex PCR-based next generation sequencing as a novel, targeted and accurate molecular approach for periprosthetic joint infection diagnosis. Front. Microbiol. 2023, 14, 1181348. [Google Scholar] [CrossRef]
- Miao, Q.; Ma, Y.; Wang, Q.; Pan, J.; Zhang, Y.; Jin, W.; Yao, Y.; Su, Y.; Huang, Y.; Wang, M.; et al. Microbiological Diagnostic Performance of Metagenomic Next-generation Sequencing When Applied to Clinical Practice. Clin. Infect. Dis. 2018, 67, S231–S240. [Google Scholar] [CrossRef]
- Choi, H.; Kim, S.-Y.; Lee, H.; Jhun, B.W.; Park, H.Y.; Jeon, K.; Kim, D.H.; Huh, H.J.; Ki, C.-S.; Lee, N.Y.; et al. Clinical Characteristics and Treatment Outcomes of Patients with Macrolide-Resistant Mycobacterium massiliense Lung Disease. Antimicrob. Agents Chemother. 2017, 61, e02189-16. [Google Scholar] [CrossRef]
- Daley, C.L.; Iaccarino, J.M.; Lange, C.; Cambau, E.; Wallace, R.J.; Andrejak, C.; Böttger, E.C.; Brozek, J.; Griffith, D.E.; Guglielmetti, L.; et al. Treatment of nontuberculous mycobacterial pulmonary disease: An official ATS/ERS/ESCMID/IDSA clinical practice guideline. Eur. Respir. J. 2020, 56, 2000535. [Google Scholar] [CrossRef]
- Asakura, T.; Nakagawa, T.; Suzuki, S.; Namkoong, H.; Morimoto, K.; Ishii, M.; Kurashima, A.; Betsuyaku, T.; Ogawa, K.; Hasegawa, N. Efficacy and safety of intermittent maintenance therapy after successful treatment of Mycobacterium avium complex lung disease. J. Infect. Chemother. 2019, 25, 218–221. [Google Scholar] [CrossRef]
- Moon, S.M.; Yoo, I.Y.; Huh, H.J.; Lee, N.Y.; Jhun, B.W. Intermittent Treatment with Azithromycin and Ethambutol for Noncavitary Mycobacterium avium Complex Pulmonary Disease. Antimicrob. Agents Chemother. 2019, 64, e01787-19. [Google Scholar] [CrossRef]
- Yamada, K.; Seki, Y.; Nakagawa, T.; Hayashi, Y.; Yagi, M.; Ogawa, K. Outcomes and risk factors after adjuvant surgical treatments for Mycobacterium avium complex lung disease. Gen. Thorac. Cardiovasc. Surg. 2019, 67, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.D. Surgical Treatment of Pulmonary Nontuberculous Mycobacterial Infections. Thorac. Surg. Clin. 2019, 29, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J.E.; Patton, E.A.; Gibbons-Burgener, S.N.; Klos, R.F.; Tans-Kersten, J.L.; Carlson, B.W.; Keller, S.J.; Pritschet, D.J.; Rollo, S.; Dutcher, T.V.; et al. Human-to-Cattle Mycobacterium tuberculosis Complex Transmission in the United States. Front. Vet. Sci. 2021, 8, 691192. [Google Scholar] [CrossRef]
- Rodwell, T.C.; Kapasi, A.J.; Moore, M.; Milian-Suazo, F.; Harris, B.; Guerrero, L.P.; Moser, K.; Strathdee, S.A.; Garfein, R.S. Tracing the origins of Mycobacterium bovis tuberculosis in humans in the USA to cattle in Mexico using spoligotyping. Int. J. Infect. Dis. 2010, 14 (Suppl. S3), e129–e135. [Google Scholar] [CrossRef]
- Romero, B.; Rodríguez, S.; Bezos, J.; Díaz, R.; Copano, M.F.; Merediz, I.; Mínguez, O.; Marqués, S.; Palacios, J.J.; García de Viedma, D.; et al. Humans as source of Mycobacterium tuberculosis infection in cattle, Spain. Emerg. Infect. Dis. 2011, 17, 2393–2395. [Google Scholar] [CrossRef]
- Gumi, B.; Schelling, E.; Berg, S.; Firdessa, R.; Erenso, G.; Mekonnen, W.; Hailu, E.; Melese, E.; Hussein, J.; Aseffa, A.; et al. Zoonotic transmission of tuberculosis between pastoralists and their livestock in South-East Ethiopia. EcoHealth 2012, 9, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Malama, S.; Munyeme, M.; Mwanza, S.; Muma, J.B. Isolation and characterization of non tuberculous mycobacteria from humans and animals in Namwala District of Zambia. BMC Res. Notes 2014, 7, 622. [Google Scholar] [CrossRef]
- Prasad, H.K.; Singhal, A.; Mishra, A.; Shah, N.P.; Katoch, V.M.; Thakral, S.S.; Singh, D.V.; Chumber, S.; Bal, S.; Aggarwal, S.; et al. Bovine tuberculosis in India: Potential basis for zoonosis. Tuberculosis 2005, 85, 421–428. [Google Scholar] [CrossRef]
- Adesokan, H.K.; Akinseye, V.O.; Streicher, E.M.; Van Helden, P.; Warren, R.M.; Cadmus, S.I. Reverse zoonotic tuberculosis transmission from an emerging Uganda I strain between pastoralists and cattle in South-Eastern Nigeria. BMC Vet. Res. 2019, 15, 437. [Google Scholar] [CrossRef]
- Parsons, S.D.; Warren, R.M.; Ottenhoff, T.H.; Gey van Pittius, N.C.; van Helden, P.D. Detection of Mycobacterium tuberculosis infection in dogs in a high-risk setting. Res. Vet. Sci. 2012, 92, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Payeur, J.B.; Jarnagin, J.L.; Marquardt, J.G.; Whipple, D.L. Mycobacterial isolations in captive elephants in the United States. Ann. N. Y. Acad. Sci. 2002, 969, 256–258. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).