
Mitofusin-Mediated Mitochondrial Fusion Inhibits Pseudorabies Virus Infection in Porcine Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Plasmids
2.3. RNA Interference
2.4. Chemical Reagents
2.5. Western Blot
2.6. RNA Extraction and qRT-PCR
2.7. Immunofluorescence Assay (IFA)
2.8. Flow Cytometry
2.9. Viral Titer Assays
2.10. Viral Replication Levels Assays
2.11. Statistical Analysis
3. Results
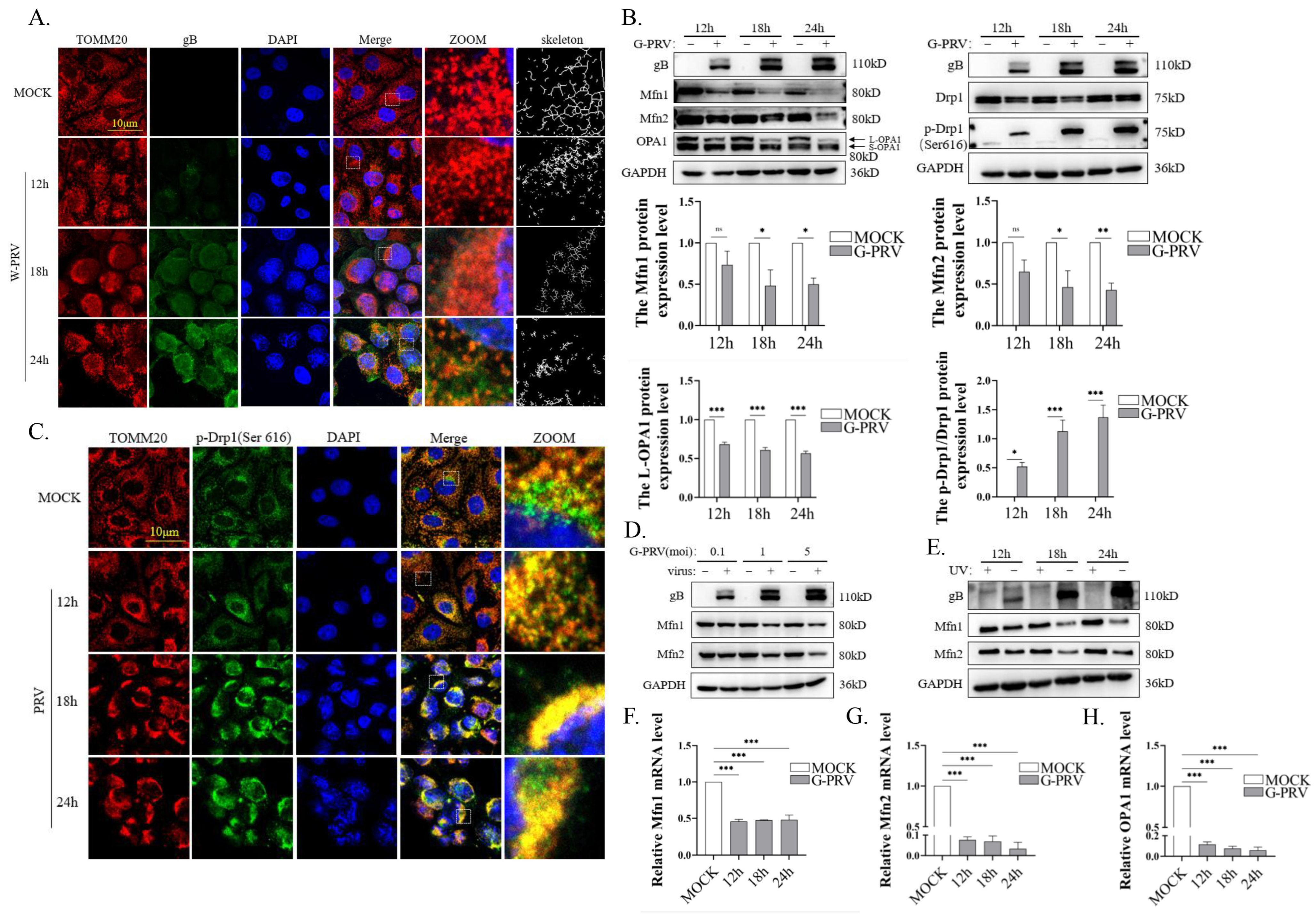
3.1. PRV Infection Impairs Mitochondrial Dynamics
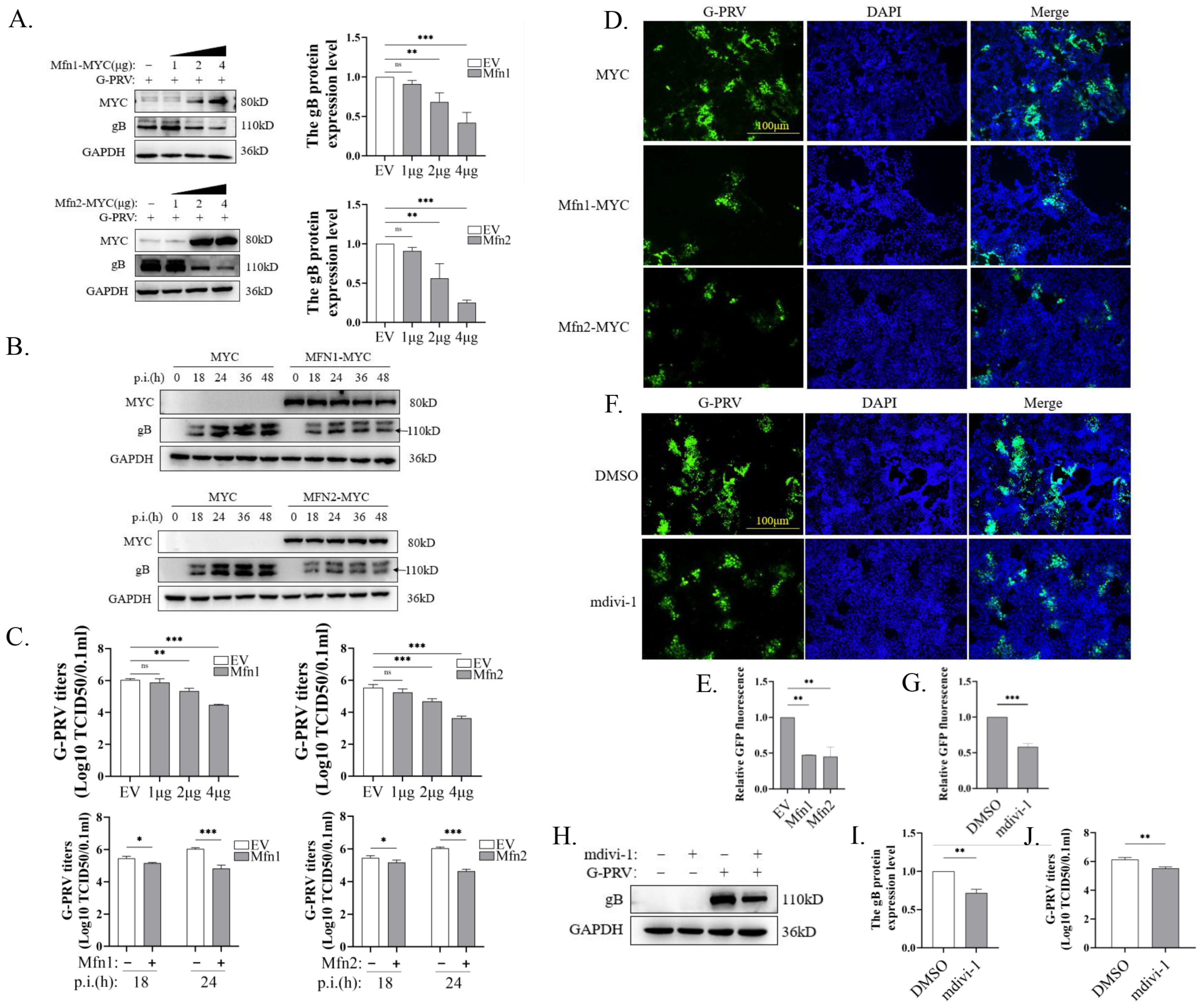
3.2. Promotion of Mitochondrial Fusion Inhibits PRV Replication
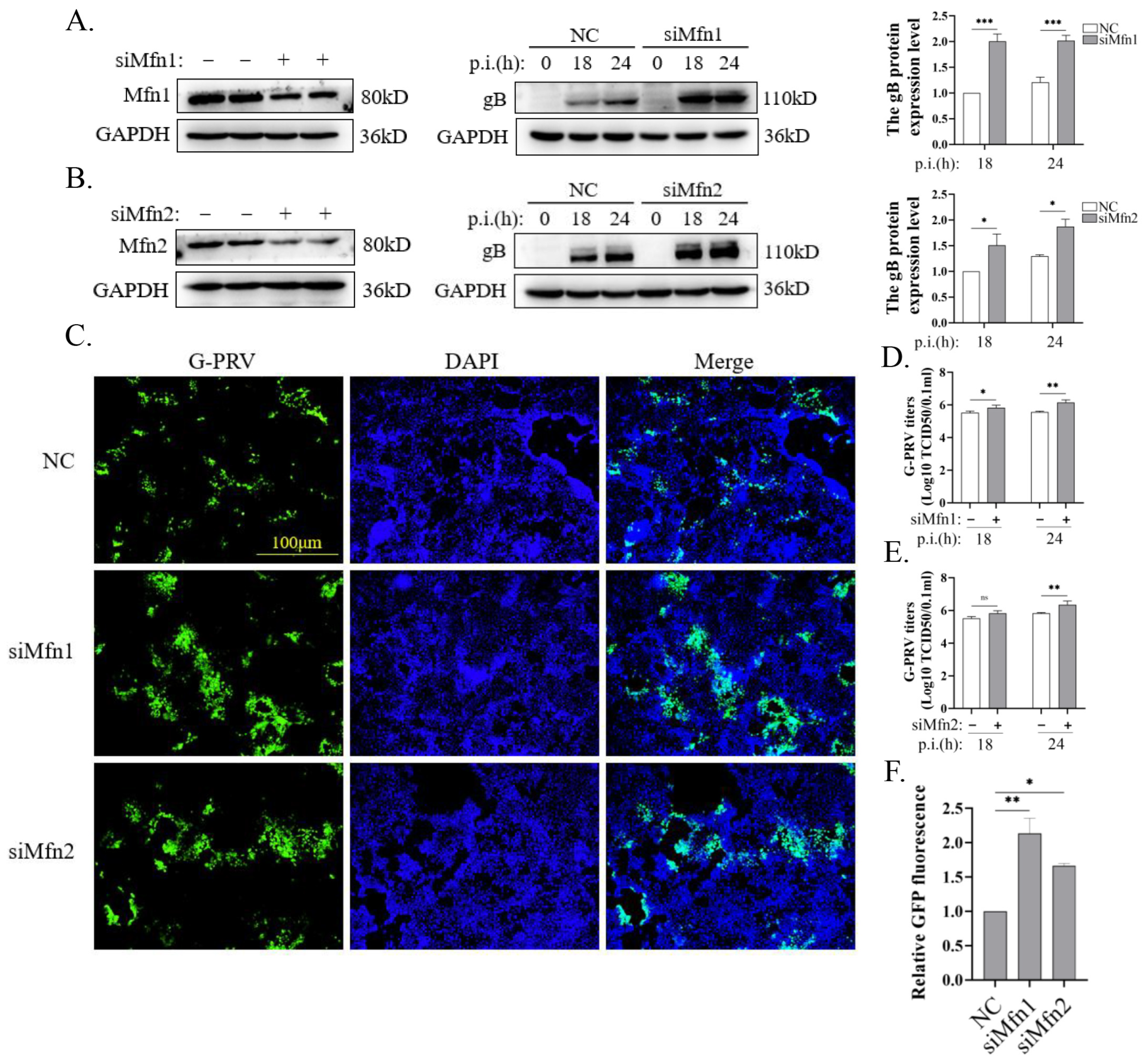
3.3. Blocking Mitochondrial Fusion Promotes PRV Infection
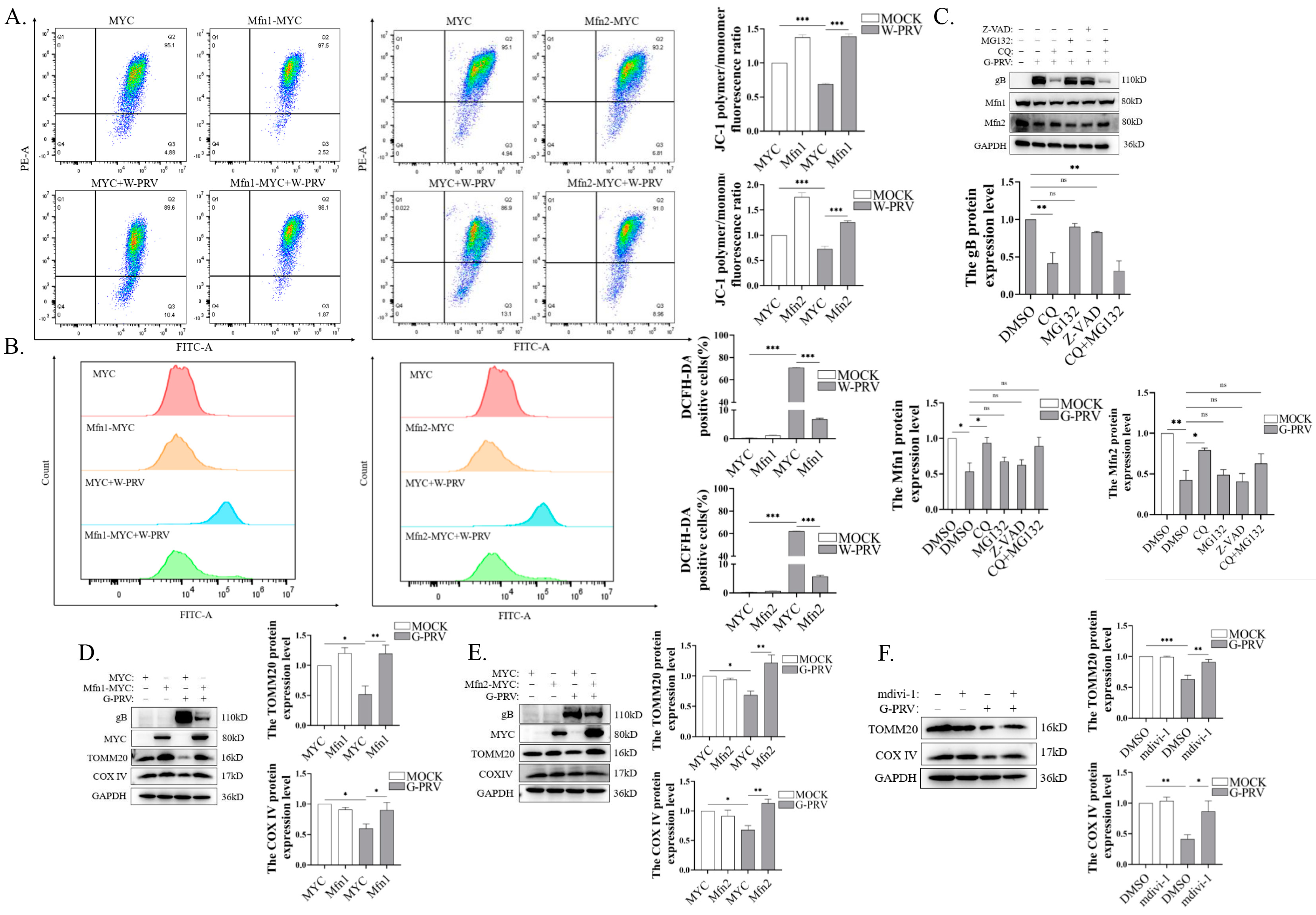
3.4. Promotion of Mitochondrial Fusion Alleviates Mitochondrial Damage and Decreases Mitochondrial Number Caused by PRV Infection
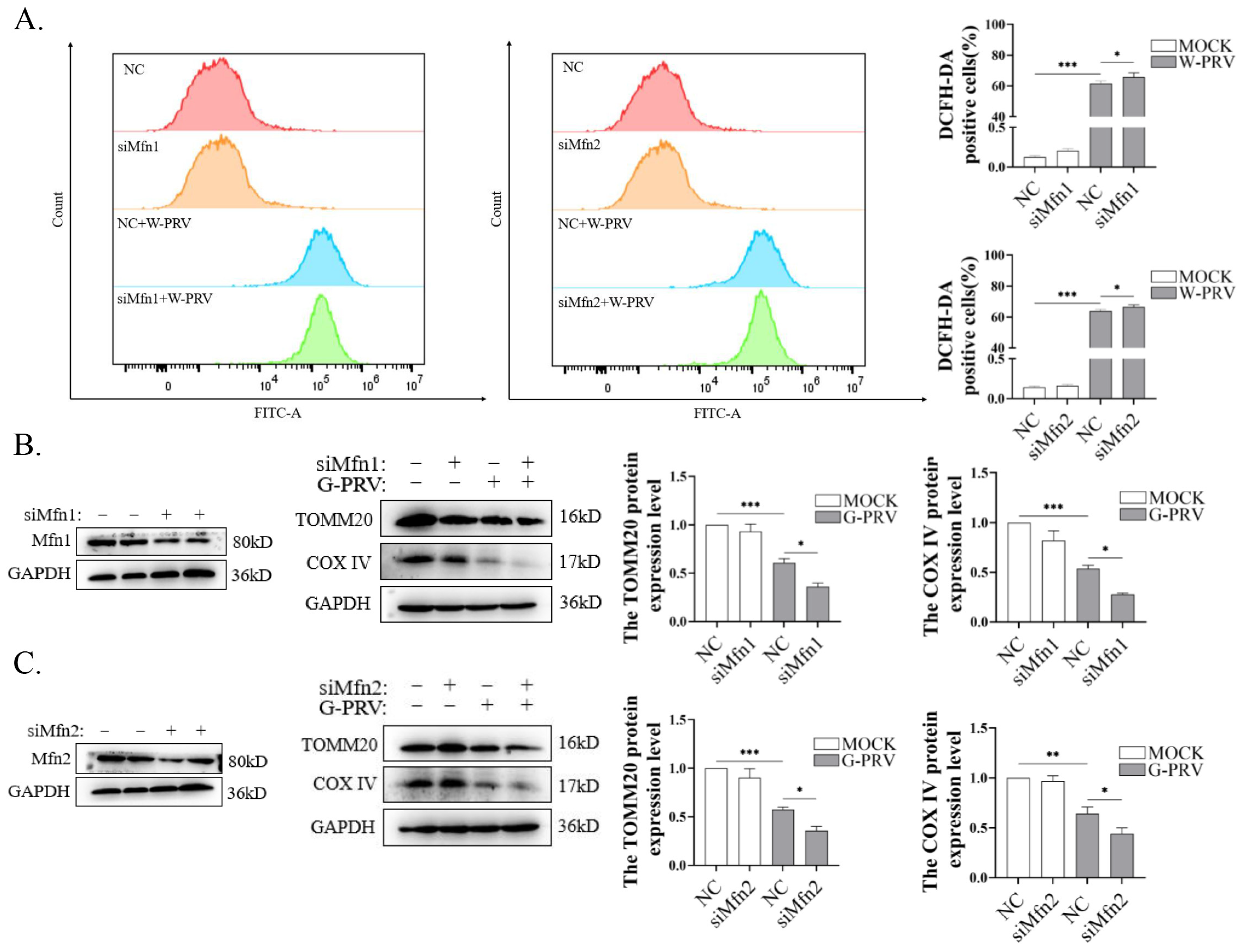
3.5. Blocking Mitochondrial Fusion Enhances PRV-Induced Mitochondrial Damage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laval, K.; Enquist, L.W. The Neuropathic Itch Caused by Pseudorabies Virus. Pathogens 2020, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.F.; Teuffert, J.; Zellmer, R.; Conraths, F.J. Experimental infection of European wild boars and domestic pigs with pseudorabies viruses with differing virulence. Am. J. Vet. Res. 2001, 62, 252–258. [Google Scholar] [CrossRef]
- Ezura, K.; Usami, Y.; Tajima, K.; Komaniwa, H.; Nagai, S.; Narita, M.; Kawashima, K. Gastrointestinal and skin lesions in piglets naturally infected with pseudorabies virus. J. Vet. Diagn. Investig. 1995, 7, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Freuling, C.M.; Muller, T.F.; Mettenleiter, T.C. Vaccines against pseudorabies virus (PrV). Vet. Microbiol. 2017, 206, 3–9. [Google Scholar] [CrossRef]
- Wu, R.; Bai, C.; Sun, J.; Chang, S.; Zhang, X. Emergence of virulent pseudorabies virus infection in northern China. J. Vet. Sci. 2013, 14, 363–365. [Google Scholar] [CrossRef]
- An, T.Q.; Peng, J.M.; Tian, Z.J.; Zhao, H.Y.; Li, N.; Liu, Y.M.; Chen, J.Z.; Leng, C.L.; Sun, Y.; Chang, D.; et al. Pseudorabies virus variant in Bartha-K61-vaccinated pigs, China, 2012. Emerg. Infect. Dis. 2013, 19, 1749–1755. [Google Scholar] [CrossRef]
- Tong, W.; Liu, F.; Zheng, H.; Liang, C.; Zhou, Y.J.; Jiang, Y.F.; Shan, T.L.; Gao, F.; Li, G.X.; Tong, G.Z. Emergence of a Pseudorabies virus variant with increased virulence to piglets. Vet. Microbiol. 2015, 181, 236–240. [Google Scholar] [CrossRef]
- Wang, C.H.; Yuan, J.; Qin, H.Y.; Luo, Y.; Cong, X.; Li, Y.; Chen, J.; Li, S.; Sun, Y.; Qiu, H.J. A novel gE-deleted pseudorabies virus (PRV) provides rapid and complete protection from lethal challenge with the PRV variant emerging in Bartha-K61-vaccinated swine population in China. Vaccine 2014, 32, 3379–3385. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, X.; Wang, X.; Wang, W.; Gao, S.; Liu, X.; Kai, Y.; Chen, C. Efficacy of the Bartha-K61 vaccine and a gE(-)/gI(-)/TK(-) prototype vaccine against variant porcine pseudorabies virus (vPRV) in piglets with sublethal challenge of vPRV. Res. Vet. Sci. 2020, 128, 16–23. [Google Scholar] [CrossRef]
- Wang, D.; Tao, X.; Fei, M.; Chen, J.; Guo, W.; Li, P.; Wang, J. Human encephalitis caused by pseudorabies virus infection: A case report. J. Neurovirol 2020, 26, 442–448. [Google Scholar] [CrossRef]
- Yang, X.; Guan, H.; Li, C.; Li, Y.; Wang, S.; Zhao, X.; Zhao, Y.; Liu, Y. Characteristics of human encephalitis caused by pseudorabies virus: A case series study. Int. J. Infect. Dis. 2019, 87, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Fan, S.; Wu, K.; Zhao, M.; Yuan, J.; Ma, S.; Zhu, E.; Chen, Y.; Ding, H.; Yi, L.; Chen, J. LDHB inhibition induces mitophagy and facilitates the progression of CSFV infection. Autophagy 2021, 17, 2305–2324. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef]
- Lee, J.K.; Shin, O.S. Zika virus modulates mitochondrial dynamics, mitophagy, and mitochondria-derived vesicles to facilitate viral replication in trophoblast cells. Front. Immunol. 2023, 14, 1203645. [Google Scholar] [CrossRef]
- Gong, Y.; Tang, N.; Liu, P.; Sun, Y.; Lu, S.; Liu, W.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; et al. Newcastle disease virus degrades SIRT3 via PINK1-PRKN-dependent mitophagy to reprogram energy metabolism in infected cells. Autophagy 2021, 18, 1503–1521. [Google Scholar] [CrossRef]
- Chodkowski, M.; Serafińska, I.; Brzezicka, J.; Golke, A.; Słońska, A.; Krzyżowska, M.; Orłowski, P.; Bąska, P.; Bańbura, M.W.; Cymerys, J. Human herpesvirus type 1 and type 2 disrupt mitochondrial dynamics in human keratinocytes. Arch. Virol. 2018, 163, 2663–2673. [Google Scholar] [CrossRef]
- Song, X.; Wang, Y.; Zou, W.; Wang, Z.; Cao, W.; Liang, M.; Li, F.; Zeng, Q.; Ren, Z.; Wang, Y.; et al. Inhibition of mitophagy via the EIF2S1-ATF4-PRKN pathway contributes to viral encephalitis. J. Adv. Res. 2024, S2090-1232(24)00326-6. [Google Scholar] [CrossRef]
- Kramer, T.; Enquist, L.W. Alphaherpesvirus infection disrupts mitochondrial transport in neurons. Cell Host Microbe 2012, 11, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ding, C.; Zhu, Z.; Wang, W.; Wen, W.; Favoreel, H.W.; Li, X. Pseudorabies virus infection triggers mitophagy to dampen the interferon response and promote viral replication. J. Virol. 2024, 98, e0104824. [Google Scholar] [CrossRef]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef]
- Oanh, N.T.K.; Park, Y.Y.; Cho, H. Mitochondria elongation is mediated through SIRT1-mediated MFN1 stabilization. Cell Signal 2017, 38, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, J.; Chen, J.; Zhang, Z.; Xu, P.; Qi, H.; Chen, Z.; Liu, J.; Lu, J.; Shi, M.; et al. Ginsenoside compound K protects against cerebral ischemia/reperfusion injury via Mul1/Mfn2-mediated mitochondrial dynamics and bioenergy. J. Ginseng Res. 2023, 47, 408–419. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; To, R.K.; Spector, S.A. TREM-1 Protects HIV-1-Infected Macrophages from Apoptosis through Maintenance of Mitochondrial Function. mBio 2019, 10, e02638-02619. [Google Scholar] [CrossRef]
- Yu, C.Y.; Liang, J.J.; Li, J.K.; Lee, Y.L.; Chang, B.L.; Su, C.I.; Huang, W.J.; Lai, M.M.; Lin, Y.L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef]
- Fu, P.F.; Cheng, X.; Su, B.Q.; Duan, L.F.; Wang, C.R.; Niu, X.R.; Wang, J.; Yang, G.Y.; Chu, B.B. CRISPR/Cas9-based generation of a recombinant double-reporter pseudorabies virus and its characterization in vitro and in vivo. Vet. Res. 2021, 52, 95. [Google Scholar] [CrossRef]
- Lian, Z.; Liu, P.; Zhu, Z.; Sun, Z.; Yu, X.; Deng, J.; Li, R.; Li, X.; Tian, K. Isolation and Characterization of a Novel Recombinant Classical Pseudorabies Virus in the Context of the Variant Strains Pandemic in China. Viruses 2023, 15, 1966. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef]
- Li, L.; Qi, R.; Zhang, L.; Yu, Y.; Hou, J.; Gu, Y.; Song, D.; Wang, X. Potential biomarkers and targets of mitochondrial dynamics. Clin. Transl. Med. 2021, 11, e529. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Song, L.; Yu, J.; Huang, F.; Li, Y.; Ma, C. Mdivi-1: A promising drug and its underlying mechanisms in the treatment of neurodegenerative diseases. Histol. Histopathol. 2022, 37, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Liu, Z.; Peng, L.; Tang, X.; Meng, F.; Ao, Y.; Zhou, M.; Wang, M.; Cao, X.; Qin, B.; et al. Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. Elife 2018, 7, e34836. [Google Scholar] [CrossRef]
- Chaikeeratisak, V.; Nguyen, K.; Khanna, K.; Brilot, A.F.; Erb, M.L.; Coker, J.K.; Vavilina, A.; Newton, G.L.; Buschauer, R.; Pogliano, K.; et al. Assembly of a nucleus-like structure during viral replication in bacteria. Science 2017, 355, 194–197. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, T.; Zhong, L.; Zhang, W.; Zhang, Y.; Yu, X.; Yuan, S.; Ni, T. Molecular architecture of coronavirus double-membrane vesicle pore complex. Nature 2024, 633, 224–231. [Google Scholar] [CrossRef]
- Kim, S.J.; Ahn, D.G.; Syed, G.H.; Siddiqui, A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion 2018, 41, 21–27. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [PubMed]
- Gou, H.; Zhao, M.; Xu, H.; Yuan, J.; He, W.; Zhu, M.; Ding, H.; Yi, L.; Chen, J. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget 2017, 8, 39382–39400. [Google Scholar] [CrossRef] [PubMed]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Vo, M.T.; Smith, B.J.; Nicholas, J.; Choi, Y.B. Activation of NIX-mediated mitophagy by an interferon regulatory factor homologue of human herpesvirus. Nat. Commun. 2019, 10, 3203. [Google Scholar] [CrossRef]
- Oteiza, A.; Mechti, N. FoxO4 negatively controls Tat-mediated HIV-1 transcription through the post-transcriptional suppression of Tat encoding mRNA. J. Gen. Virol. 2017, 98, 1864–1878. [Google Scholar] [CrossRef]
- Ono, E.; Tomioka, Y.; Taharaguchi, S. Possible roles of transcription factors of pseudorabies virus in neuropathogenicity. Fukuoka Igaku Zasshi 2007, 98, 364–372. [Google Scholar]
- Luo, Z.; Liu, L.F.; Jiang, Y.N.; Tang, L.P.; Li, W.; Ouyang, S.H.; Tu, L.F.; Wu, Y.P.; Gong, H.B.; Yan, C.Y.; et al. Novel insights into stress-induced susceptibility to influenza: Corticosterone impacts interferon-beta responses by Mfn2-mediated ubiquitin degradation of MAVS. Signal Transduct. Target. Ther. 2020, 5, 202. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Aujeszky’s disease (pseudorabies) virus: The virus and molecular pathogenesis--state of the art, June 1999. Vet. Res. 2000, 31, 99–115. [Google Scholar] [CrossRef]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Zhao, Y.; Zhu, Z.; Wen, W.; Li, X. Mitofusin-Mediated Mitochondrial Fusion Inhibits Pseudorabies Virus Infection in Porcine Cells. Vet. Sci. 2025, 12, 368. https://doi.org/10.3390/vetsci12040368
Xu X, Zhao Y, Zhu Z, Wen W, Li X. Mitofusin-Mediated Mitochondrial Fusion Inhibits Pseudorabies Virus Infection in Porcine Cells. Veterinary Sciences. 2025; 12(4):368. https://doi.org/10.3390/vetsci12040368
Chicago/Turabian StyleXu, Xiuhan, Yuan Zhao, Zhenbang Zhu, Wei Wen, and Xiangdong Li. 2025. "Mitofusin-Mediated Mitochondrial Fusion Inhibits Pseudorabies Virus Infection in Porcine Cells" Veterinary Sciences 12, no. 4: 368. https://doi.org/10.3390/vetsci12040368
APA StyleXu, X., Zhao, Y., Zhu, Z., Wen, W., & Li, X. (2025). Mitofusin-Mediated Mitochondrial Fusion Inhibits Pseudorabies Virus Infection in Porcine Cells. Veterinary Sciences, 12(4), 368. https://doi.org/10.3390/vetsci12040368