
Machine Learning and Canine Chronic Enteropathies: A New Approach to Investigate FMT Effects


 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
Study Populations
2.2. Donor Dogs Selection
2.3. FMT Preparation and Administration
2.4. Microbiome Analysis
2.5. Bioinformatic Analysis
2.5.1. Pre-Processing
2.5.2. Diversity Analysis
2.5.3. Differential Abundance Analysis
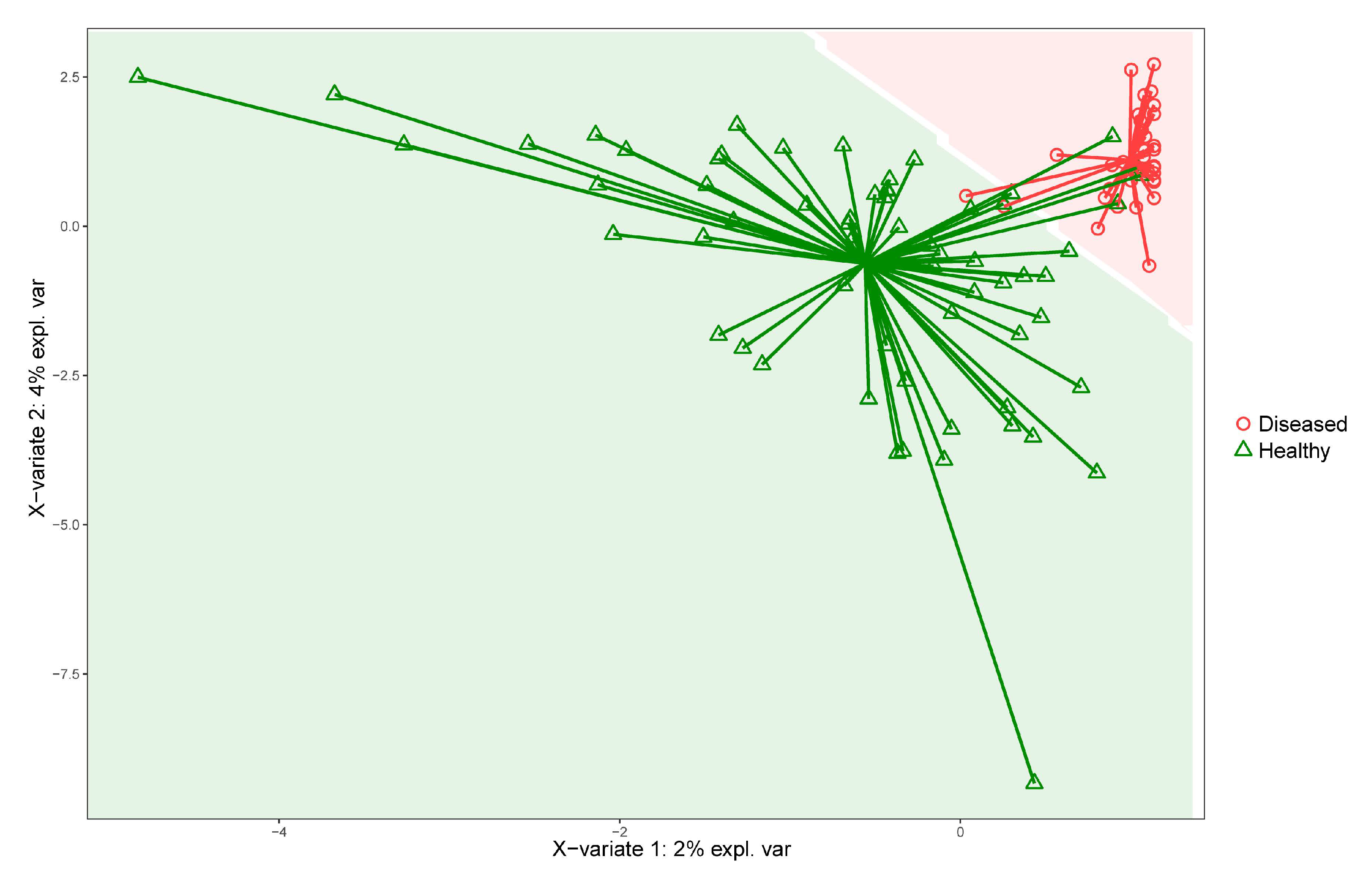
2.5.4. Supervised Machine Learning
2.5.5. Key Features Identification
3. Results
3.1. Microbiome Sequence Data
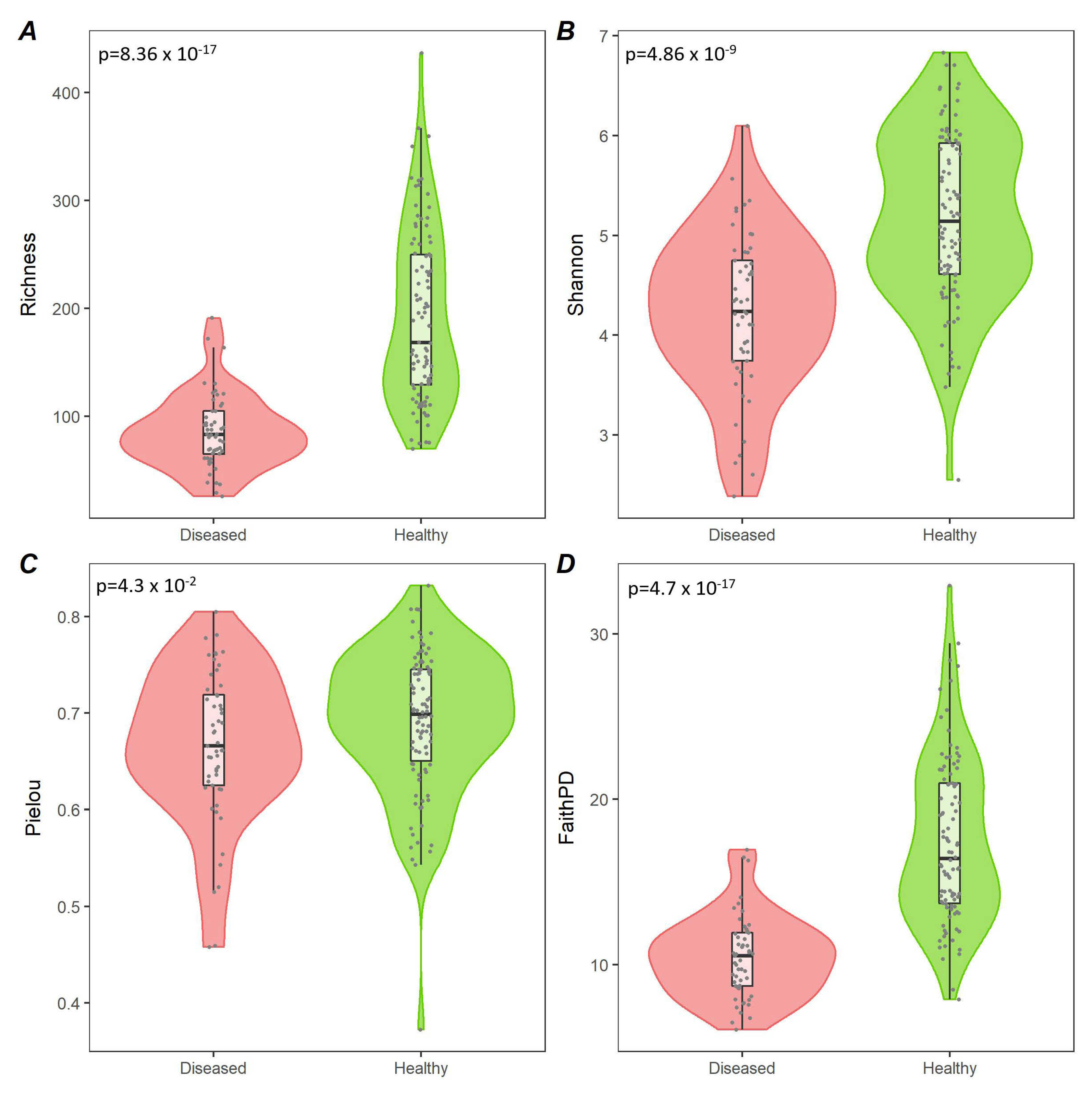
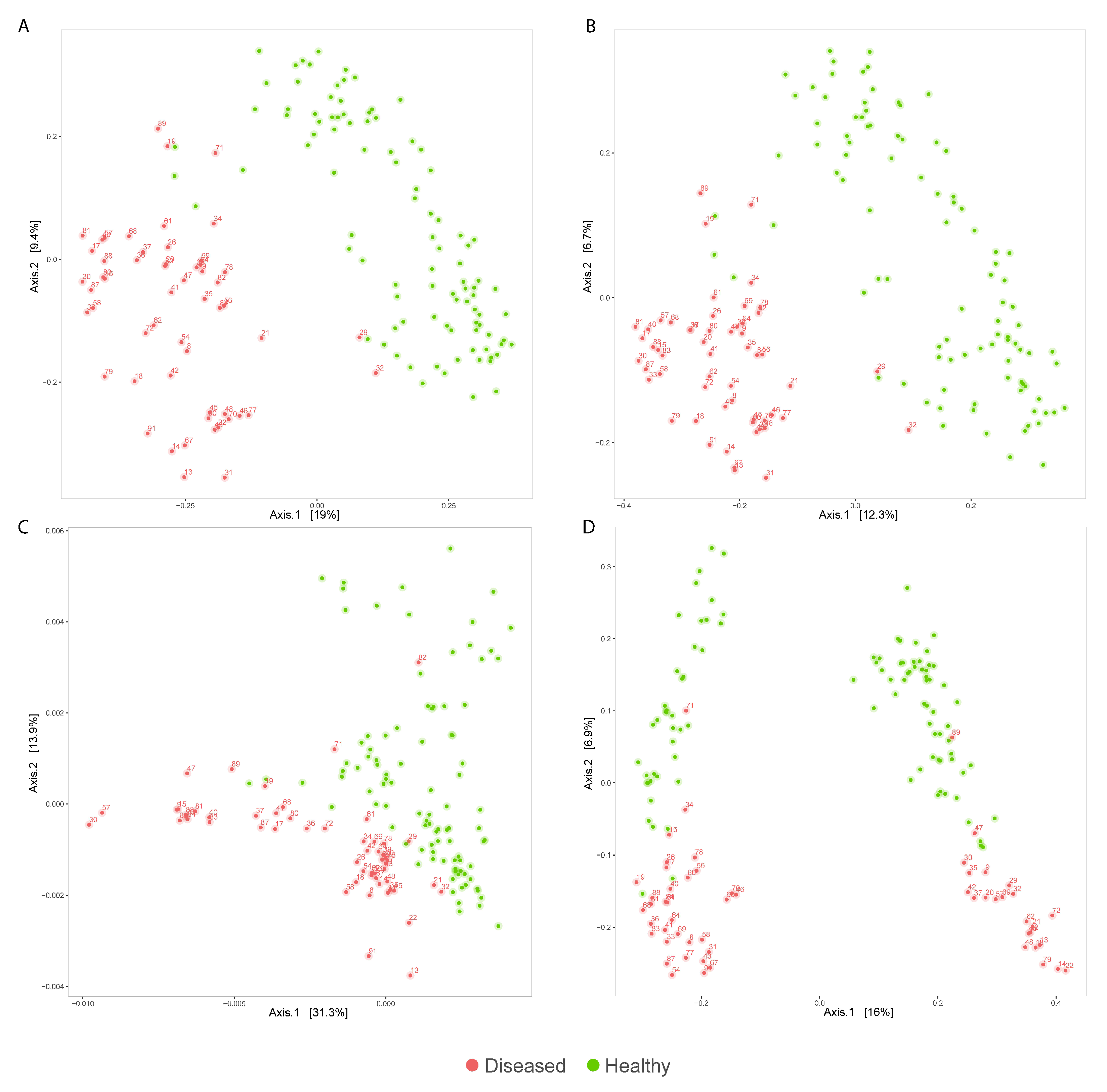
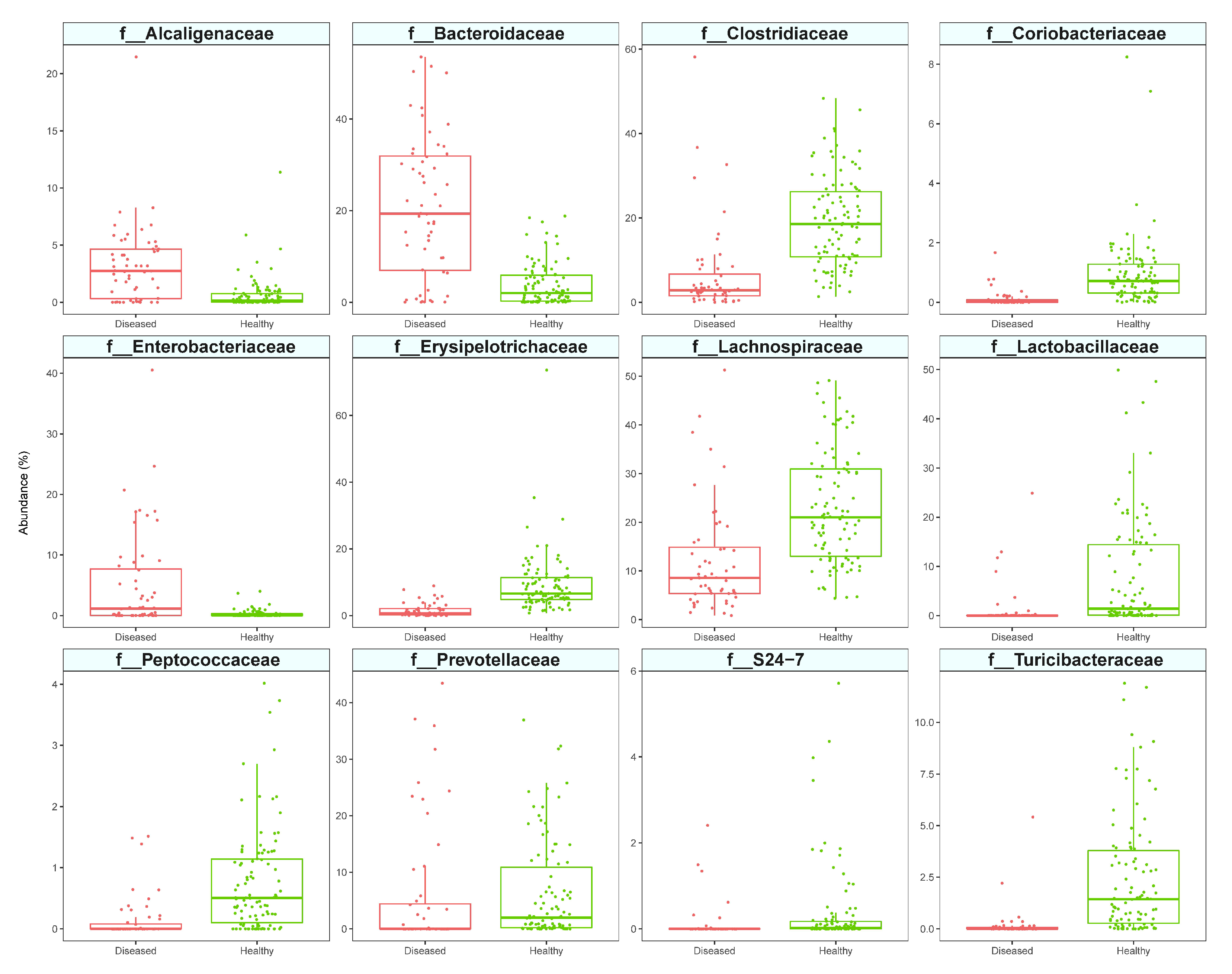
3.2. Analysis of Diseased vs. Healthy Population
3.3. Effect of the FMT on Clinical Response and Microbiota
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pilla, R.; Suchodolski, J.S. The Role of the Canine Gut Microbiome and Metabolome in Health and Gastrointestinal Disease. Front. Vet. Sci. 2019, 6, 498. [Google Scholar] [CrossRef] [PubMed]
- Honneffer, J.B.; Steiner, J.M.; Lidbury, J.A.; Suchodolski, J.S. Variation of the Microbiota and Metabolome along the Canine Gastrointestinal Tract. Metabolomics 2017, 13, 26. [Google Scholar] [CrossRef]
- Hand, D.; Wallis, C.; Colyer, A.; Penn, C.W. Pyrosequencing the Canine Faecal Microbiota: Breadth and Depth of Biodiversity. PLoS ONE 2013, 8, e53115. [Google Scholar]
- Middelbos, I.S.; Vester Boler, B.M.; Qu, A.; White, B.A.; Swanson, K.S.; Fahey, G.C., Jr. Phylogenetic Characterization of Fecal Microbial Communities of Dogs Fed Diets with or without Supplemental Dietary Fiber Using 454 Pyrosequencing. PLoS ONE 2010, 5, e9768. [Google Scholar] [CrossRef]
- Dandrieux, J.R.S. Inflammatory Bowel Disease versus Chronic Enteropathy in Dogs: Are They One and the Same? J. Small Anim. Pract. 2016, 57, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, Y.; Minamoto, T.; Isaiah, A.; Sattasathuchana, P.; Buono, A.; Rangachari, V.R.; McNeely, I.H.; Lidbury, J.; Steiner, J.M.; Suchodolski, J.S. Fecal Short-Chain Fatty Acid Concentrations and Dysbiosis in Dogs with Chronic Enteropathy. J. Vet. Intern. Med. 2019, 33, 1608–1618. [Google Scholar] [CrossRef]
- Tizard, I.R.; Jones, S.W. The Microbiota Regulates Immunity and Immunologic Diseases in Dogs and Cats. Vet. Clin. N. Am. Small Anim. Pract. 2018, 48, 307–322. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilić-Stojanović, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European Consensus Conference on Faecal Microbiota Transplantation in Clinical Practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef]
- Kang, D.-W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy Alters Gut Ecosystem and Improves Gastrointestinal and Autism Symptoms: An Open-Label Study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Kootte, R.S.; Levin, E.; Salojärvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619.e6. [Google Scholar] [CrossRef]
- Gal, A.; Barko, P.C.; Biggs, P.J.; Gedye, K.R.; Midwinter, A.C.; Williams, D.A.; Burchell, R.K.; Pazzi, P. One Dog’s Waste Is Another Dog’s Wealth: A Pilot Study of Fecal Microbiota Transplantation in Dogs with Acute Hemorrhagic Diarrhea Syndrome. PLoS ONE 2021, 16, e0250344. [Google Scholar] [CrossRef]
- Bottero, E.; Benvenuti, E.; Ruggiero, P. Ruggiero Trapianto Del Microbiota Fecale (FMT) in 16 Cani Affetti Da IBD Idiopatica. Veterinaria 2017, 31, 31–45. [Google Scholar]
- Sugita, K.; Yanuma, N.; Ohno, H.; Takahashi, K.; Kawano, K.; Morita, H.; Ohmori, K. Oral Faecal Microbiota Transplantation for the Treatment of Clostridium Difficile-Associated Diarrhoea in a Dog: A Case Report. BMC Vet. Res. 2019, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Niina, A.; Kibe, R.; Suzuki, R.; Yuchi, Y.; Teshima, T.; Matsumoto, H.; Kataoka, Y.; Koyama, H. Improvement in Clinical Symptoms and Fecal Microbiome After Fecal Microbiota Transplantation in a Dog with Inflammatory Bowel Disease. Vet. Med. 2019, 10, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.Q.; Gomes, L.A.; Santos, I.S.; Alfieri, A.F.; Weese, J.S.; Costa, M.C. Fecal Microbiota Transplantation in Puppies with Canine Parvovirus Infection. J. Vet. Intern. Med. 2018, 32, 707–711. [Google Scholar] [CrossRef]
- Gerbec, Z. Evaluation of Therapeutic Potential of Restoring Gastrointestinal Homeostasis by a Fecal Microbiota Transplant in Dogs. Master’s Thesis, University of Ljubljana, Ljubljana, Slovenia, 2016. [Google Scholar]
- Sugita, K.; Shima, A.; Takahashi, K.; Matsuda, Y.; Miyajima, M.; Hirokawa, M.; Kondo, H.; Kimura, J.; Ishihara, G.; Ohmori, K. Successful Outcome after a Single Endoscopic Fecal Microbiota Transplantation in a Shiba Dog with Non-Responsive Enteropathy during the Treatment with Chlorambucil. J. Vet. Med. Sci. 2021, 83, 984–989. [Google Scholar] [CrossRef]
- Niina, A.; Kibe, R.; Suzuki, R.; Yuchi, Y.; Teshima, T.; Matsumoto, H.; Kataoka, Y.; Koyama, H. Fecal Microbiota Transplantation as a New Treatment for Canine Inflammatory Bowel Disease. Biosci. Microbiota Food Health 2021, 40, 98–104. [Google Scholar] [CrossRef]
- Chaitman, J.; Ziese, A.-L.; Pilla, R.; Minamoto, Y.; Blake, A.B.; Guard, B.C.; Isaiah, A.; Lidbury, J.A.; Steiner, J.M.; Unterer, S.; et al. Fecal Microbial and Metabolic Profiles in Dogs With Acute Diarrhea Receiving Either Fecal Microbiota Transplantation or Oral Metronidazole. Front. Vet. Sci. 2020, 7, 192. [Google Scholar] [CrossRef]
- Burton, E.N.; O’Connor, E.; Ericsson, A.C.; Franklin, C.L. Evaluation of Fecal Microbiota Transfer as Treatment for Postweaning Diarrhea in Research-Colony Puppies. J. Am. Assoc. Lab. Anim. Sci. 2016, 55, 582–587. [Google Scholar]
- Berlanda, M.; Innocente, G.; Simionati, B.; Di Camillo, B.; Facchin, S.; Giron, M.C.; Savarino, E.; Sebastiani, F.; Fiorio, F.; Patuzzi, I. Faecal Microbiome Transplantation as a Solution to Chronic Enteropathies in Dogs: A Case Study of Beneficial Microbial Evolution. Animals 2021, 11, 1433. [Google Scholar] [CrossRef]
- Allenspach, K.; Wieland, B.; Gröne, A.; Gaschen, F. Chronic Enteropathies in Dogs: Evaluation of Risk Factors for Negative Outcome. J. Vet. Intern. Med. 2007, 21, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Pengo, G.; Caldin, M.; Piccionello, A.P.; Steiner, J.M.; Cohen, N.D.; Jergens, A.E.; Suchodolski, J.S. Comparison of Microbiological, Histological, and Immunomodulatory Parameters in Response to Treatment with Either Combination Therapy with Prednisone and Metronidazole or Probiotic VSL#3 Strains in Dogs with Idiopathic Inflammatory Bowel Disease. PLoS ONE 2014, 9, e94699. [Google Scholar]
- Staley, C.; Hamilton, M.J.; Vaughn, B.P.; Graiziger, C.T.; Newman, K.M.; Kabage, A.J.; Sadowsky, M.J.; Khoruts, A. Successful Resolution of Recurrent Clostridium Difficile Infection Using Freeze-Dried, Encapsulated Fecal Microbiota; Pragmatic Cohort Study. Am. J. Gastroenterol. 2017, 112, 940–947. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Scarsella, E.; Stefanon, B.; Cintio, M.; Licastro, D.; Sgorlon, S.; Dal Monego, S.; Sandri, M. Learning Machine Approach Reveals Microbial Signatures of Diet and Sex in Dog. PLoS ONE 2020, 15, e0237874. [Google Scholar] [CrossRef]
- Chen, L.; Reeve, J.; Zhang, L.; Huang, S.; Wang, X.; Chen, J. GMPR: A Robust Normalization Method for Zero-Inflated Count Data with Application to Microbiome Sequencing Data. PeerJ 2018, 6, e4600. [Google Scholar] [CrossRef]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the Analysis of High-Throughput Sequencing Datasets: Characterizing RNA-Seq, 16S rRNA Gene Sequencing and Selective Growth Experiments by Compositional Data Analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Analysis of Compositions of Microbiomes with Bias Correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Rohart, F.; Gautier, B.; Singh, A.; Cao, K.-A.L. mixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- AlShawaqfeh, M.K.; Wajid, B.; Minamoto, Y.; Markel, M.; Lidbury, J.A.; Steiner, J.M.; Serpedin, E.; Suchodolski, J.S. A Dysbiosis Index to Assess Microbial Changes in Fecal Samples of Dogs with Chronic Inflammatory Enteropathy. FEMS Microbiol. Ecol. 2017, 93, fix136. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.G.; Wu, J.R.; Gloor, G.B. Expanding the UniFrac Toolbox. PLoS ONE 2016, 11, e0161196. [Google Scholar]
- Roggenkamp, A. Phylogenetic Analysis of Enteric Species of the Family Enterobacteriaceae Using the oriC-Locus. Syst. Appl. Microbiol. 2007, 30, 180–188. [Google Scholar] [CrossRef]
- Lluch, J.; Servant, F.; Païssé, S.; Valle, C.; Valière, S.; Kuchly, C.; Vilchez, G.; Donnadieu, C.; Courtney, M.; Burcelin, R.; et al. The Characterization of Novel Tissue Microbiota Using an Optimized 16S Metagenomic Sequencing Pipeline. PLoS ONE 2015, 10, e0142334. [Google Scholar] [CrossRef]
- Minamoto, Y.; Otoni, C.C.; Steelman, S.M.; Büyükleblebici, O.; Steiner, J.M.; Jergens, A.E.; Suchodolski, J.S. Alteration of the Fecal Microbiota and Serum Metabolite Profiles in Dogs with Idiopathic Inflammatory Bowel Disease. Gut Microbes 2015, 6, 33–47. [Google Scholar] [CrossRef]
- Bresciani, F.; Minamoto, Y.; Suchodolski, J.S.; Galiazzo, G.; Vecchiato, C.G.; Pinna, C.; Biagi, G.; Pietra, M. Effect of an Extruded Animal Protein-Free Diet on Fecal Microbiota of Dogs with Food-Responsive Enteropathy. J. Vet. Intern. Med. 2018, 32, 1903–1910. [Google Scholar] [CrossRef]
- Suchodolski, J.S. Fecal Microbiome in Dogs with Acute Diarrhea and Idiopathic Inflammatory Bowel Disease. In Encyclopedia of Metagenomics; Springer: Boston, MA, USA, 2015; pp. 183–186. [Google Scholar]
- Suchodolski, J.S.; Markel, M.E.; Garcia-Mazcorro, J.F.; Unterer, S.; Heilmann, R.M.; Dowd, S.E.; Kachroo, P.; Ivanov, I.; Minamoto, Y.; Dillman, E.M.; et al. The Fecal Microbiome in Dogs with Acute Diarrhea and Idiopathic Inflammatory Bowel Disease. PLoS ONE 2012, 7, e51907. [Google Scholar] [CrossRef] [PubMed]
- Bottero, E.; Ferriani, R.; Benvenuti, E.; Ruggiero, P.; Astorina, S.; Giraldi, M.; Bertoldi, L.; Benvenuto, G.; Sattin, E.; Gianella, P.; et al. Clinical Evaluation and Microbiota Analysis in 9 Dogs with Antibiotic-Responsive Enteropathy: A Prospective Comparison Study. J. Vet. Intern. Med. 2022, 36, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Pilla, R.; Guard, B.C.; Blake, A.B.; Ackermann, M.; Webb, C.; Hill, S.; Lidbury, J.A.; Steiner, J.M.; Jergens, A.E.; Suchodolski, J.S. Long-Term Recovery of the Fecal Microbiome and Metabolome of Dogs with Steroid-Responsive Enteropathy. Animals 2021, 11, 2498. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Maeda, S.; Igarashi, H.; Ohno, K.; Sakai, K.; Yonezawa, T.; Horigome, A.; Odamaki, T.; Matsuki, N. Fecal Microbiome in Dogs with Inflammatory Bowel Disease and Intestinal Lymphoma. J. Vet. Med. Sci. 2017, 79, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Martins, R.; Sullivan, M.C.; Friedman, E.S.; Misic, A.M.; El-Fahmawi, A.; De Martinis, E.C.P.; O’Brien, K.; Chen, Y.; Bradley, C.; et al. Diet-Induced Remission in Chronic Enteropathy Is Associated with Altered Microbial Community Structure and Synthesis of Secondary Bile Acids. Microbiome 2019, 7, 126. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Hayes, M.G.; MacDonald, J.; Desai, D.K.; Allward, N.; Jones, C.M.A.; Wright, R.J.; Dhanani, A.S.; Comeau, A.M.; et al. Microbiome Differential Abundance Methods Produce Different Results across 38 Datasets. Nat. Commun. 2022, 13, 342. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Hyde, E.R.; Suchodolski, J.S.; Knight, R. Dog and Human Inflammatory Bowel Disease Rely on Overlapping yet Distinct Dysbiosis Networks. Nat. Microbiol. 2016, 1, 16177. [Google Scholar] [CrossRef]
- Félix, A.P.; Souza, C.M.M.; de Oliveira, S.G. Biomarkers of Gastrointestinal Functionality in Dogs: A Systematic Review and Meta-Analysis. Anim. Feed. Sci. Technol. 2022, 283, 115183. [Google Scholar] [CrossRef]
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The Gastrointestinal Microbiome: A Review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef]
- Zaneveld, J.R.; McMinds, R.; Thurber, R.V. Stress and Stability: Applying the Anna Karenina Principle to Animal Microbiomes. Nat. Microbiol. 2017, 2, 17121. [Google Scholar] [CrossRef] [PubMed]
- Toresson, L.; Steiner, J.M.; Spillmann, T.; Lidbury, J.A.; Ludvigsson, U.; Suchodolski, J.S. Clinical Effects of Fecal Microbiota Transplantation in Dogs with Chronic Enteropathies. In Proceedings of the ECVIM-CA Congress, Goteborg, Sweden, 1–3 September 2022. [Google Scholar]
- Haifer, C.; Paramsothy, S.; Kaakoush, N.O.; Saikal, A.; Ghaly, S.; Yang, T.; Luu, L.D.W.; Borody, T.J.; Leong, R.W. Lyophilised Oral Faecal Microbiota Transplantation for Ulcerative Colitis (LOTUS): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Gastroenterol. Hepatol. 2022, 7, 141–151. [Google Scholar] [CrossRef]
- Kalenyak, K.; Isaiah, A.; Heilmann, R.M.; Suchodolski, J.S.; Burgener, I.A. Comparison of the Intestinal Mucosal Microbiota in Dogs Diagnosed with Idiopathic Inflammatory Bowel Disease and Dogs with Food-Responsive Diarrhea before and after Treatment. FEMS Microbiol. Ecol. 2018, 94, fix173. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | |||
|---|---|---|---|
| CE | Healthy | ||
| Prediction | CE | 20 | 1 |
| Healthy | 0 | 33 | |
| Species | Methods |
|---|---|
| k__Bacteria;p__Actinobacteria;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Adlercreutzia;s__ | 3 |
| k__Bacteria;p__Actinobacteria;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Collinsella;s__ | 3 |
| k__Bacteria;p__Actinobacteria;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Slackia;s__ | 3 |
| k__Bacteria;p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Bacteroides;s__uniformis | 3 |
| k__Bacteria;p__Firmicutes;c__Bacilli;o__Turicibacterales;f__Turicibacteraceae;g__Turicibacter;s__ | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__[Mogibacteriaceae];g__;s__ | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Clostridiaceae;g__Clostridium;s__hiranonis | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Blautia;s__ | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Blautia;s__producta | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Dorea;s__ | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__;s__ | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Allobaculum;s__ | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Clostridium;s__spiroforme | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Coprobacillus;s__ | 3 |
| k__Bacteria;p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__[Paraprevotellaceae];g__;s__ | 2 |
| k__Bacteria;p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__[Paraprevotellaceae];g__[Prevotella];s__ | 2 |
| k__Bacteria;p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Bacteroides;s__ | 2 |
| k__Bacteria;p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__Prevotellaceae;g__Prevotella;s__copri | 2 |
| k__Bacteria;p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Lactobacillaceae;g__Lactobacillus;s__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__[Ruminococcus];__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__[Ruminococcus];s__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Roseburia;s__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Peptococcaceae;g__Peptococcus;s__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Peptostreptococcaceae;g__;s__ | 2 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__[Eubacterium];s__ | 2 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__[Eubacterium];s__biforme | 2 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Catenibacterium;s__ | 2 |
| k__Bacteria;p__Fusobacteria;c__Fusobacteriia;o__Fusobacteriales;f__Fusobacteriaceae;__;__ | 2 |
| k__Bacteria;p__Proteobacteria;c__Betaproteobacteria;o__Burkholderiales;f__Alcaligenaceae;g__Sutterella;s__ | 2 |
| k__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Aeromonadales;f__Succinivibrionaceae;g__Anaerobiospirillum;s__ | 2 |
| k__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Enterobacteriales;f__Enterobacteriaceae;__;__ | 2 |
| Genera | Methods |
|---|---|
| k__Bacteria;p__Actinobacteria;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Adlercreutzia | 3 |
| k__Bacteria;p__Actinobacteria;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Slackia | 3 |
| k__Bacteria;p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Bacteroides | 3 |
| k__Bacteria;p__Firmicutes;c__Bacilli;o__Lactobacillales;f__Lactobacillaceae;g__Lactobacillus | 3 |
| k__Bacteria;p__Firmicutes;c__Bacilli;o__Turicibacterales;f__Turicibacteraceae;g__Turicibacter | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Clostridiaceae;g__Clostridium | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Blautia | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Dorea | 3 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Peptococcaceae;g__Peptococcus | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Allobaculum | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Catenibacterium | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Clostridium | 3 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__Coprobacillus | 3 |
| k__Bacteria;p__Proteobacteria;c__Betaproteobacteria;o__Burkholderiales;f__Alcaligenaceae;g__Sutterella | 3 |
| k__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Enterobacteriales;f__Enterobacteriaceae;__ | 3 |
| k__Bacteria;p__Actinobacteria;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Collinsella | 2 |
| k__Bacteria;p__Bacteroidetes;c__Bacteroidia;o__Bacteroidales;f__Prevotellaceae;g__Prevotella | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;__;__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__[Mogibacteriaceae];g__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Roseburia | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Peptostreptococcaceae;g__ | 2 |
| k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Ruminococcaceae;g__Faecalibacterium | 2 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__ | 2 |
| k__Bacteria;p__Firmicutes;c__Erysipelotrichi;o__Erysipelotrichales;f__Erysipelotrichaceae;g__p-75-a5 | 2 |
| k__Bacteria;p__Fusobacteria;c__Fusobacteriia;o__Fusobacteriales;f__Fusobacteriaceae;__ | 2 |
| k__Bacteria;p__Fusobacteria;c__Fusobacteriia;o__Fusobacteriales;f__Fusobacteriaceae;g__Fusobacterium | 2 |
| k__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Aeromonadales;f__Succinivibrionaceae;g__Anaerobiospirillum | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Innocente, G.; Patuzzi, I.; Furlanello, T.; Di Camillo, B.; Bargelloni, L.; Giron, M.C.; Facchin, S.; Savarino, E.; Azzolin, M.; Simionati, B. Machine Learning and Canine Chronic Enteropathies: A New Approach to Investigate FMT Effects. Vet. Sci. 2022, 9, 502. https://doi.org/10.3390/vetsci9090502
Innocente G, Patuzzi I, Furlanello T, Di Camillo B, Bargelloni L, Giron MC, Facchin S, Savarino E, Azzolin M, Simionati B. Machine Learning and Canine Chronic Enteropathies: A New Approach to Investigate FMT Effects. Veterinary Sciences. 2022; 9(9):502. https://doi.org/10.3390/vetsci9090502
Chicago/Turabian StyleInnocente, Giada, Ilaria Patuzzi, Tommaso Furlanello, Barbara Di Camillo, Luca Bargelloni, Maria Cecilia Giron, Sonia Facchin, Edoardo Savarino, Mirko Azzolin, and Barbara Simionati. 2022. "Machine Learning and Canine Chronic Enteropathies: A New Approach to Investigate FMT Effects" Veterinary Sciences 9, no. 9: 502. https://doi.org/10.3390/vetsci9090502
APA StyleInnocente, G., Patuzzi, I., Furlanello, T., Di Camillo, B., Bargelloni, L., Giron, M. C., Facchin, S., Savarino, E., Azzolin, M., & Simionati, B. (2022). Machine Learning and Canine Chronic Enteropathies: A New Approach to Investigate FMT Effects. Veterinary Sciences, 9(9), 502. https://doi.org/10.3390/vetsci9090502