
Copper(II)-Promoted Reactions of α-Pyridoin Oxime: A Dodecanuclear Cluster and a 2D Coordination Polymer


 ,
,  ,
,  , ,
, ,  ,
, 

Abstract
1. Introduction
2. Results and Discussion
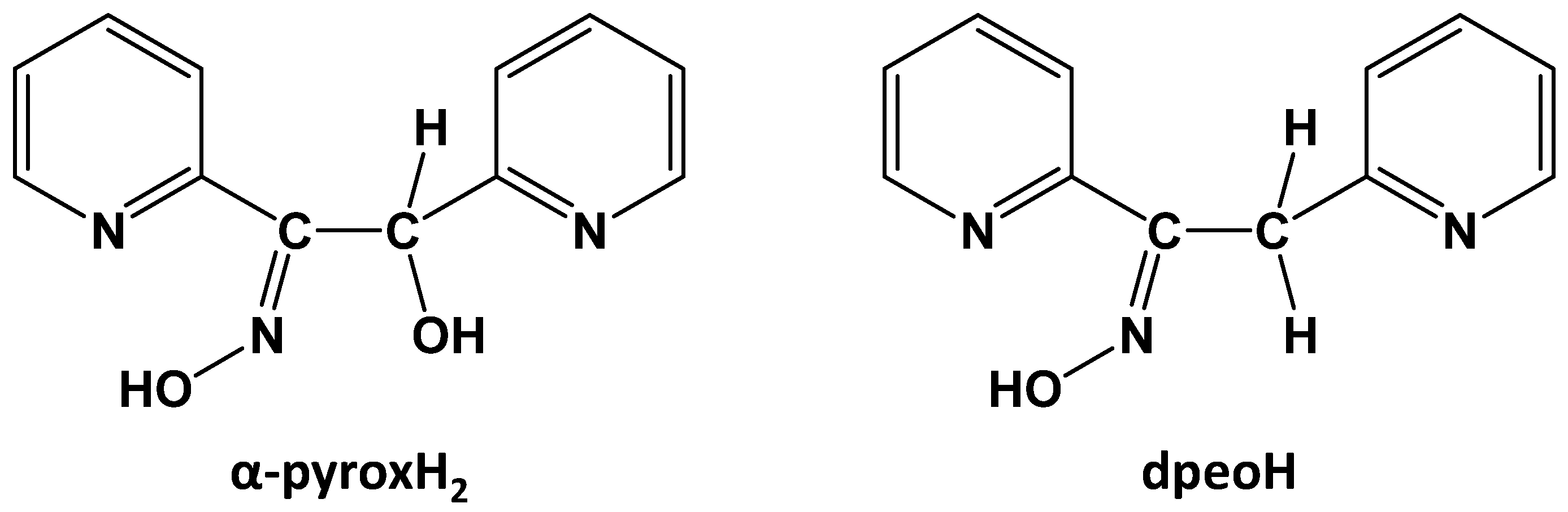
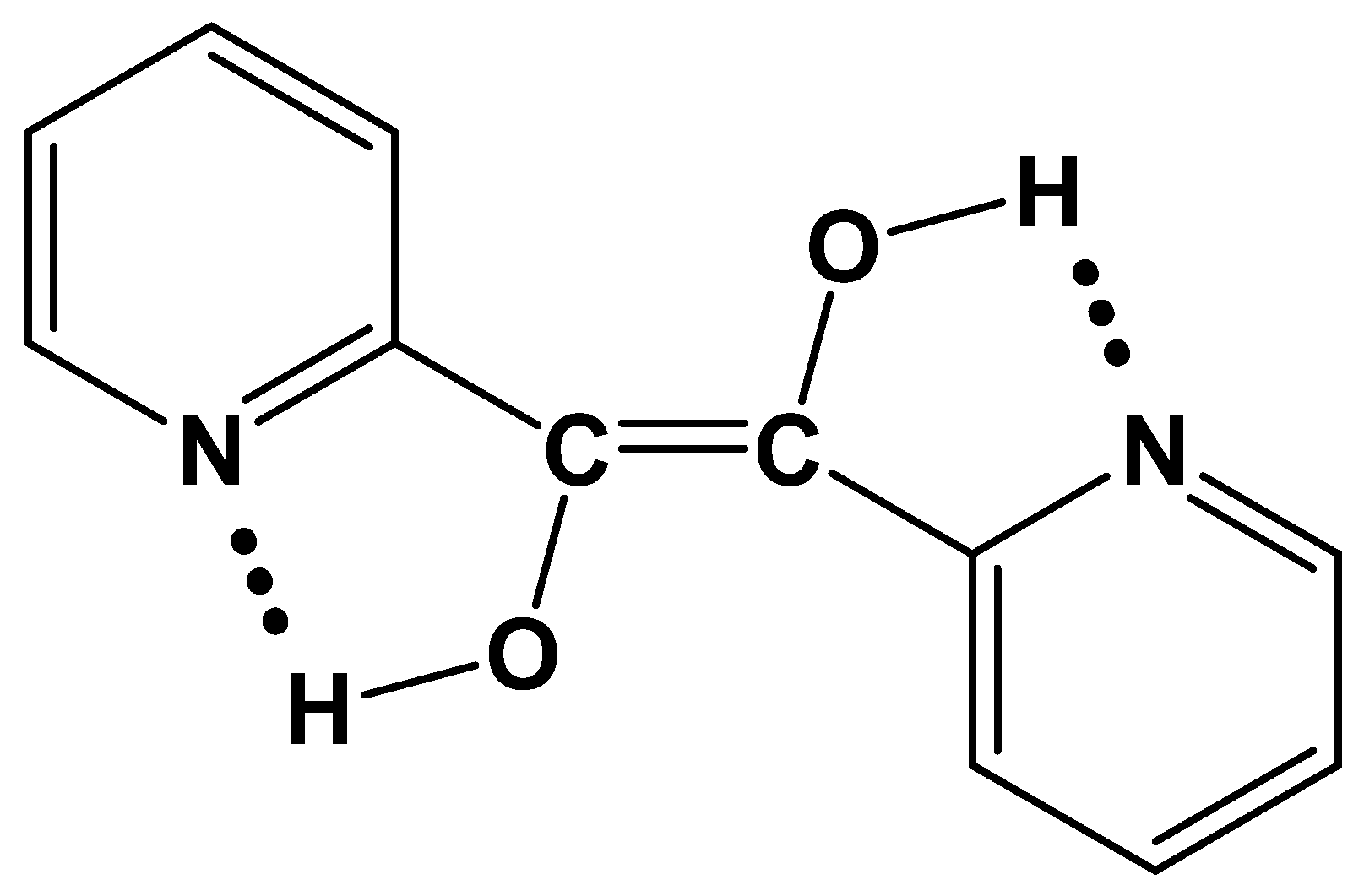
2.1. Synthesis of the Free Ligand
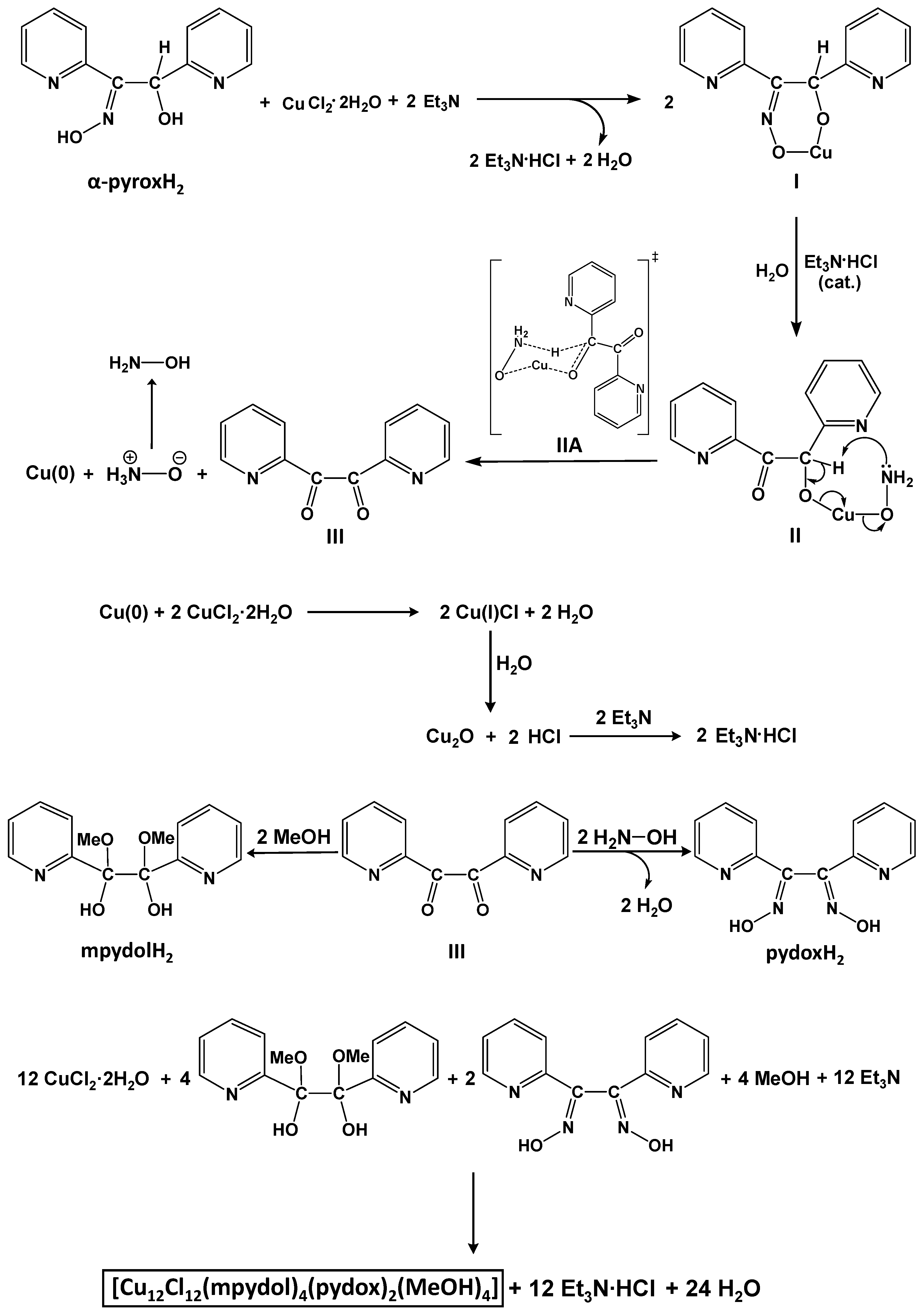
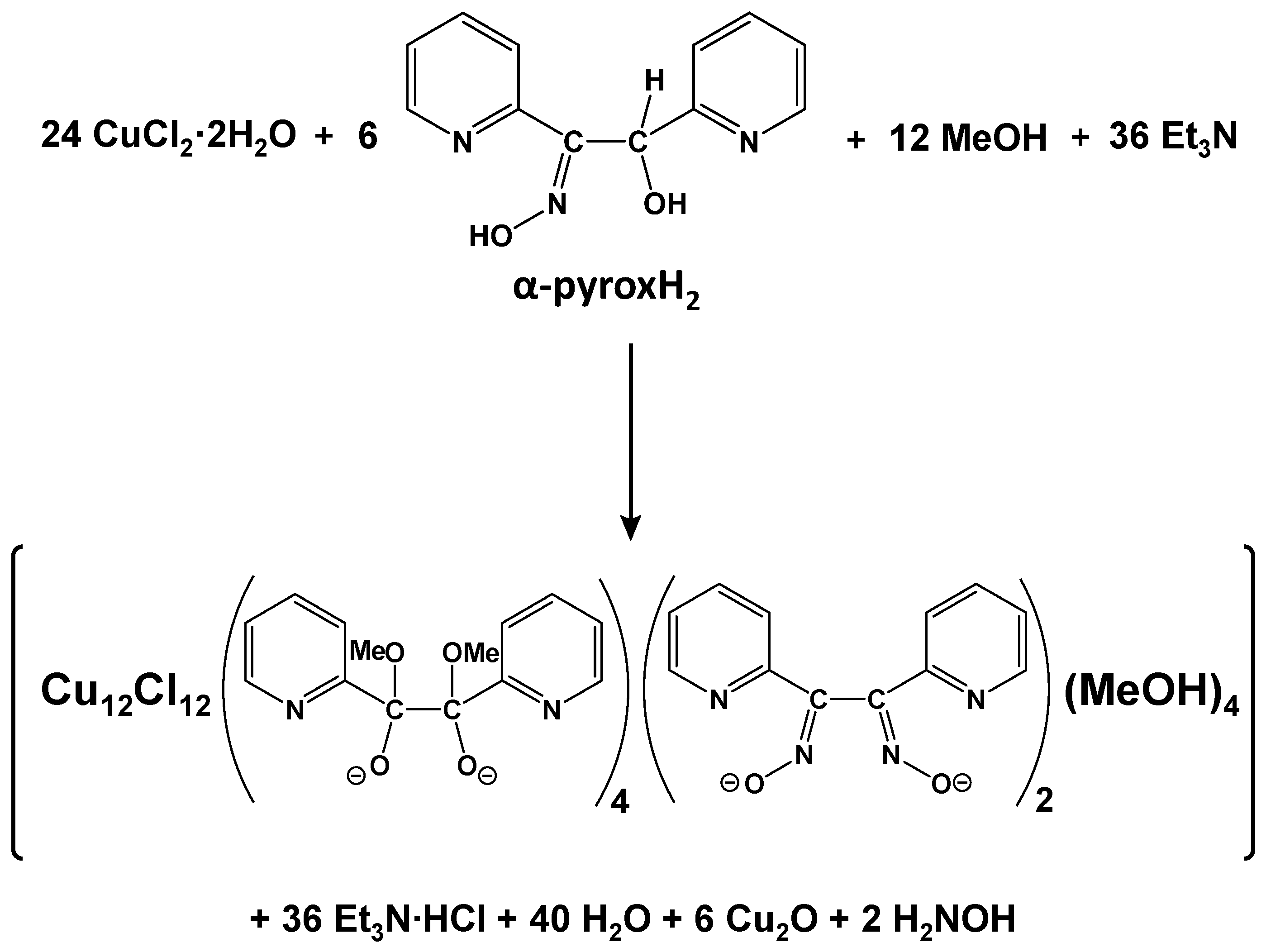
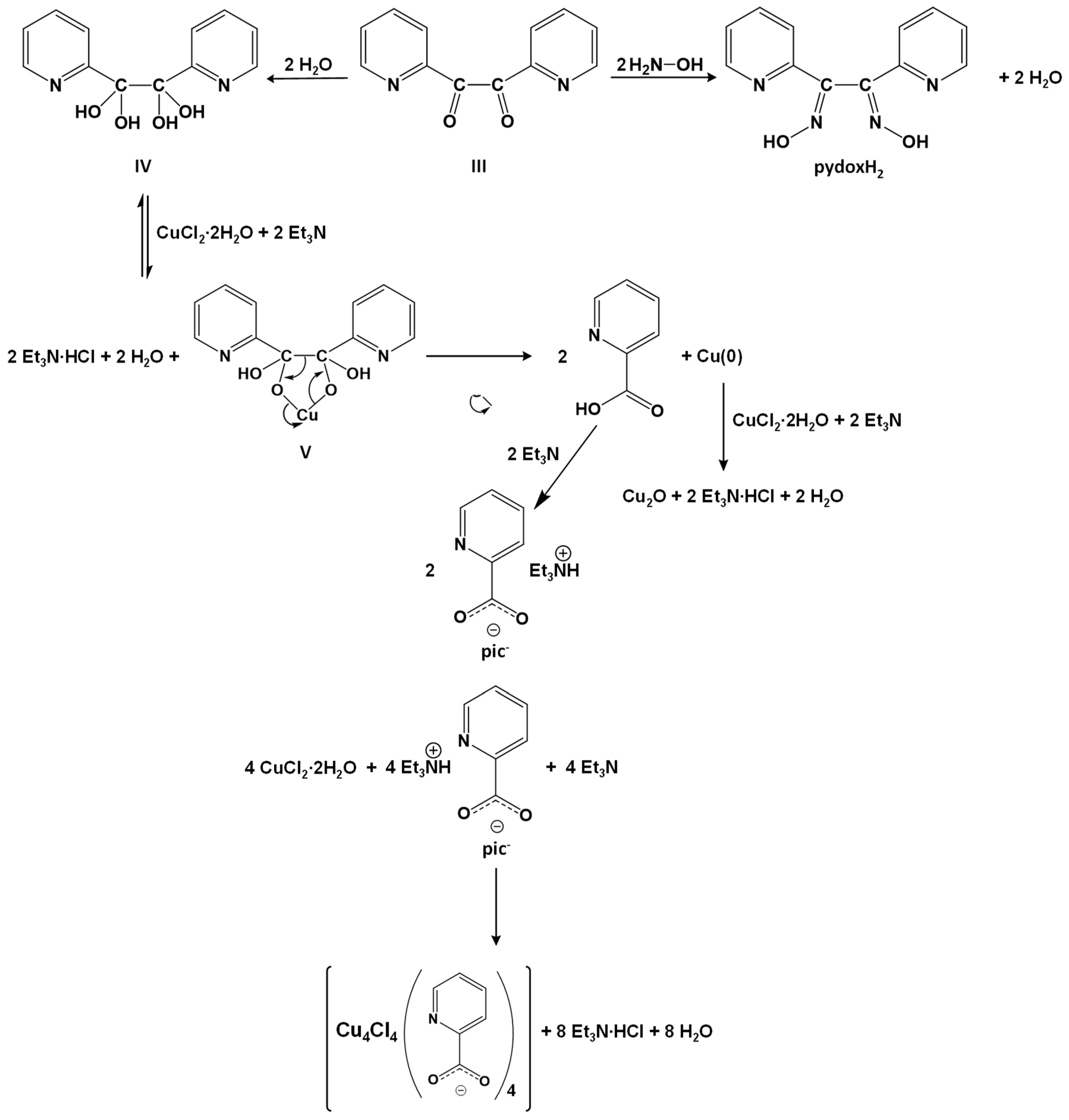
2.2. Comments for the Preparation of the Complexes—Mechanism Proposals
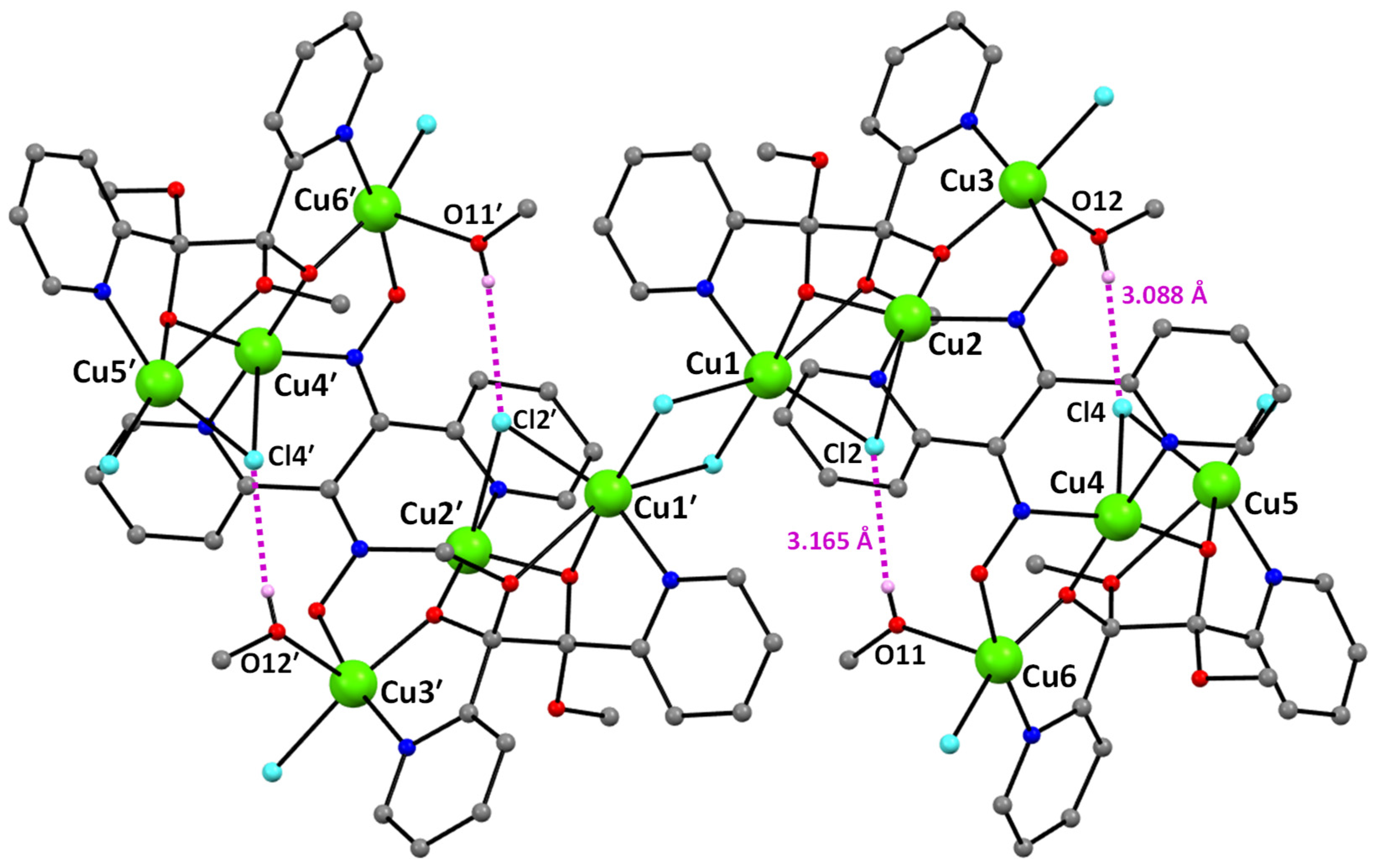
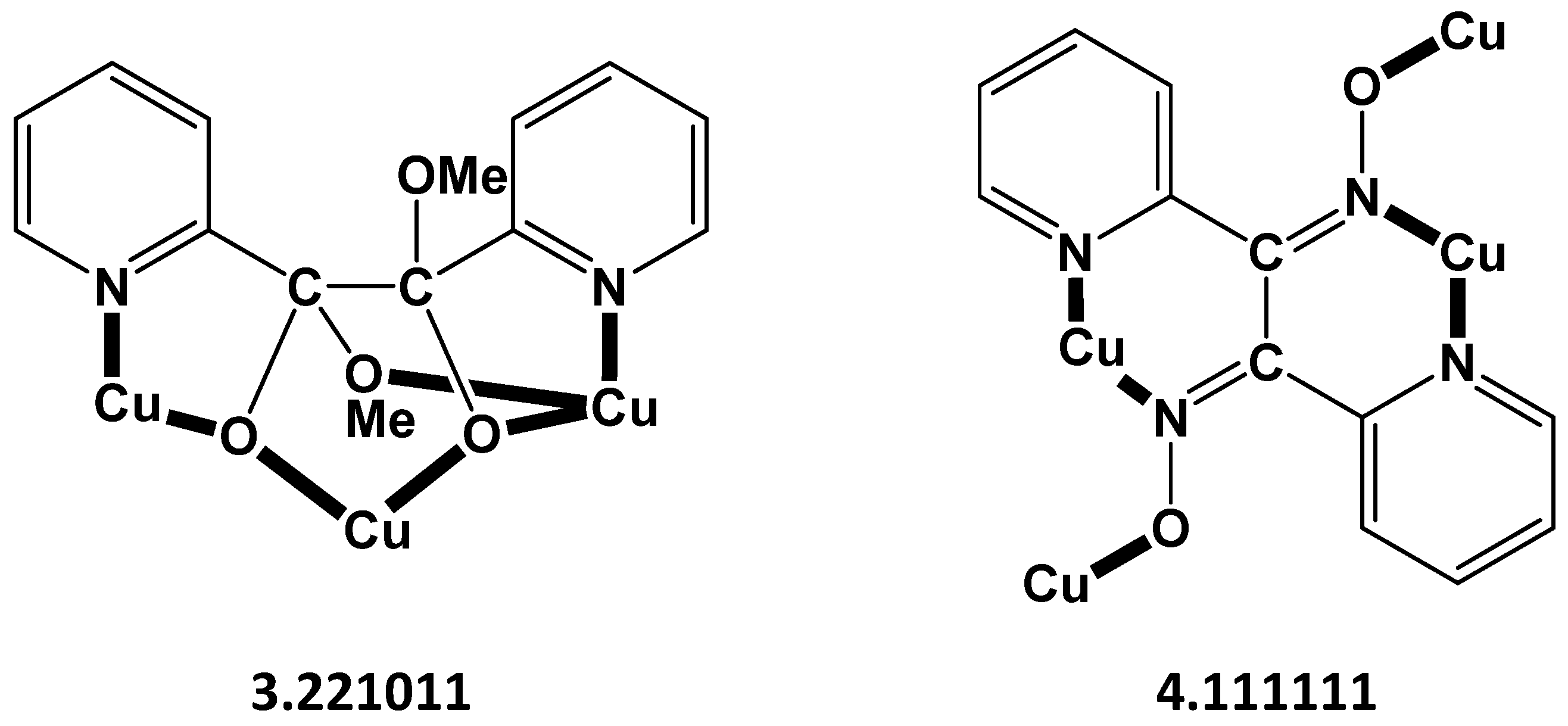
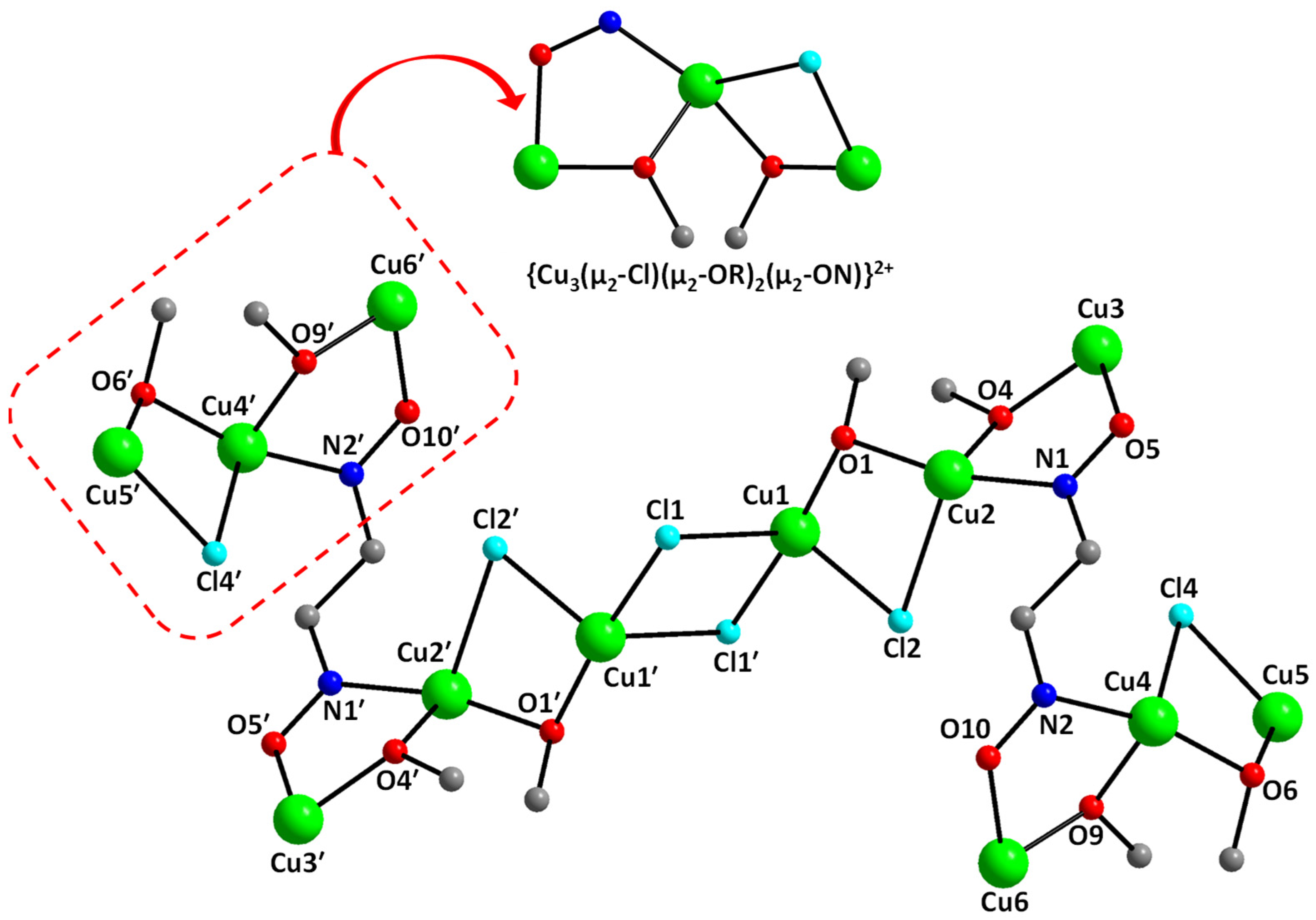
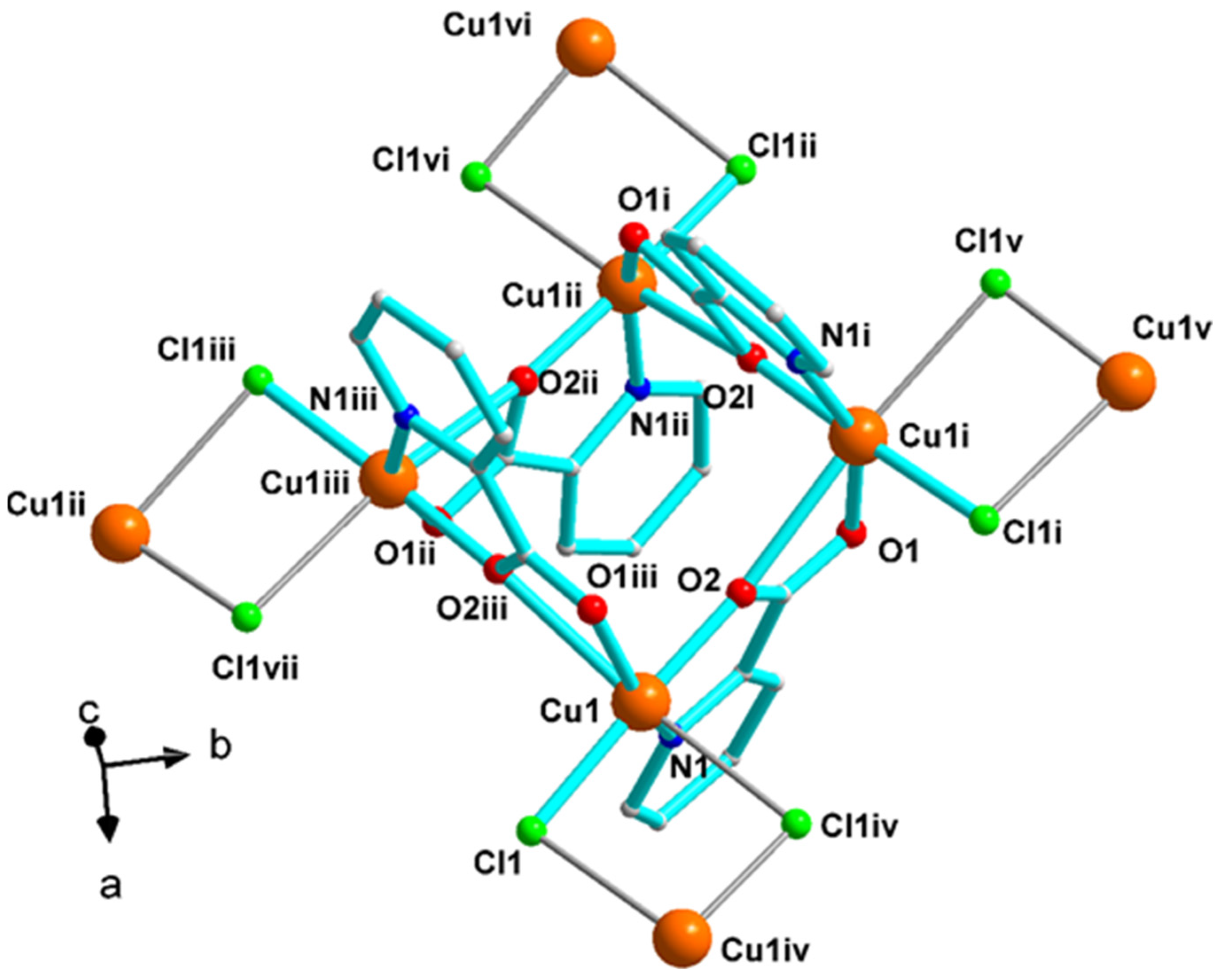
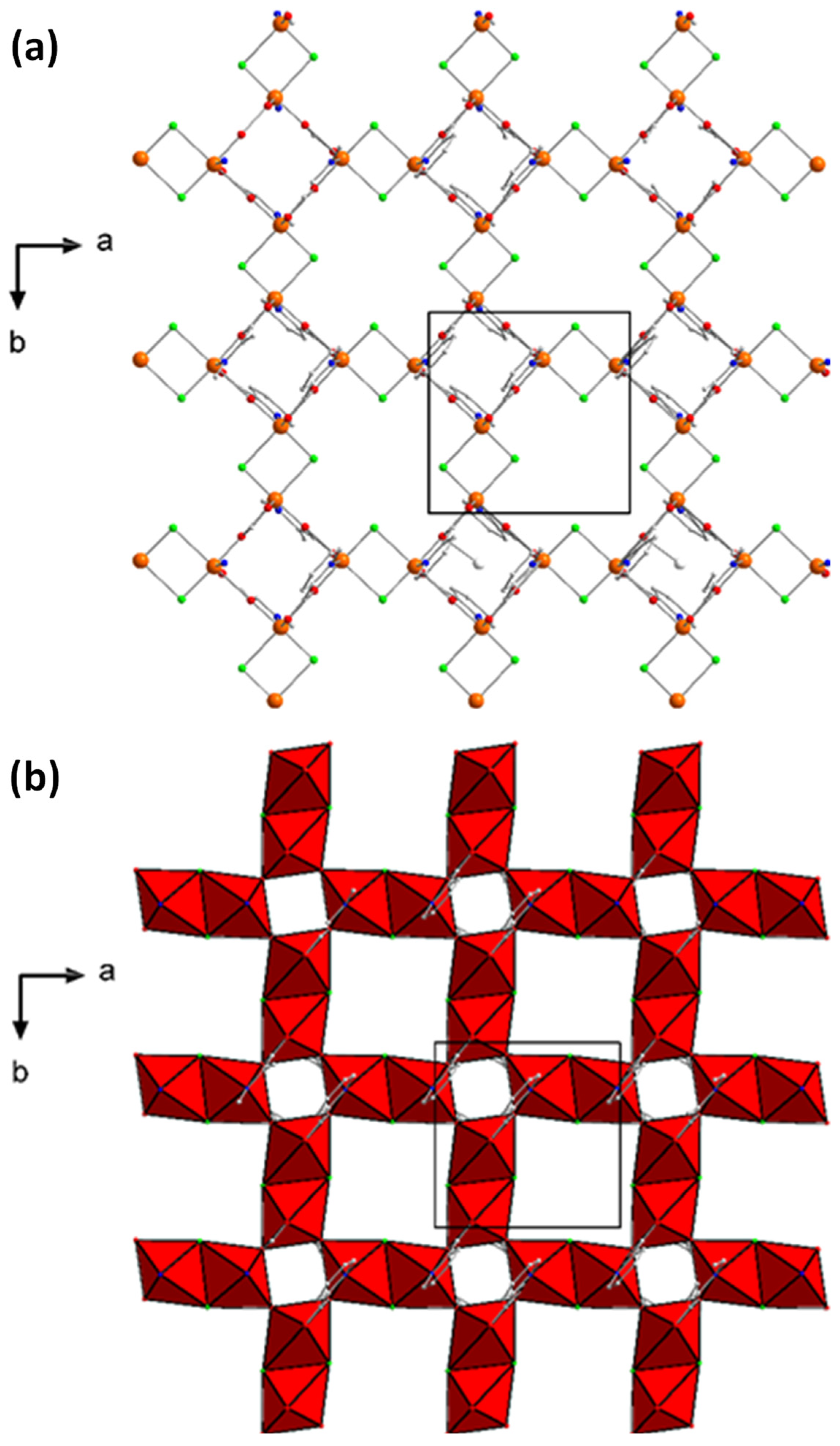
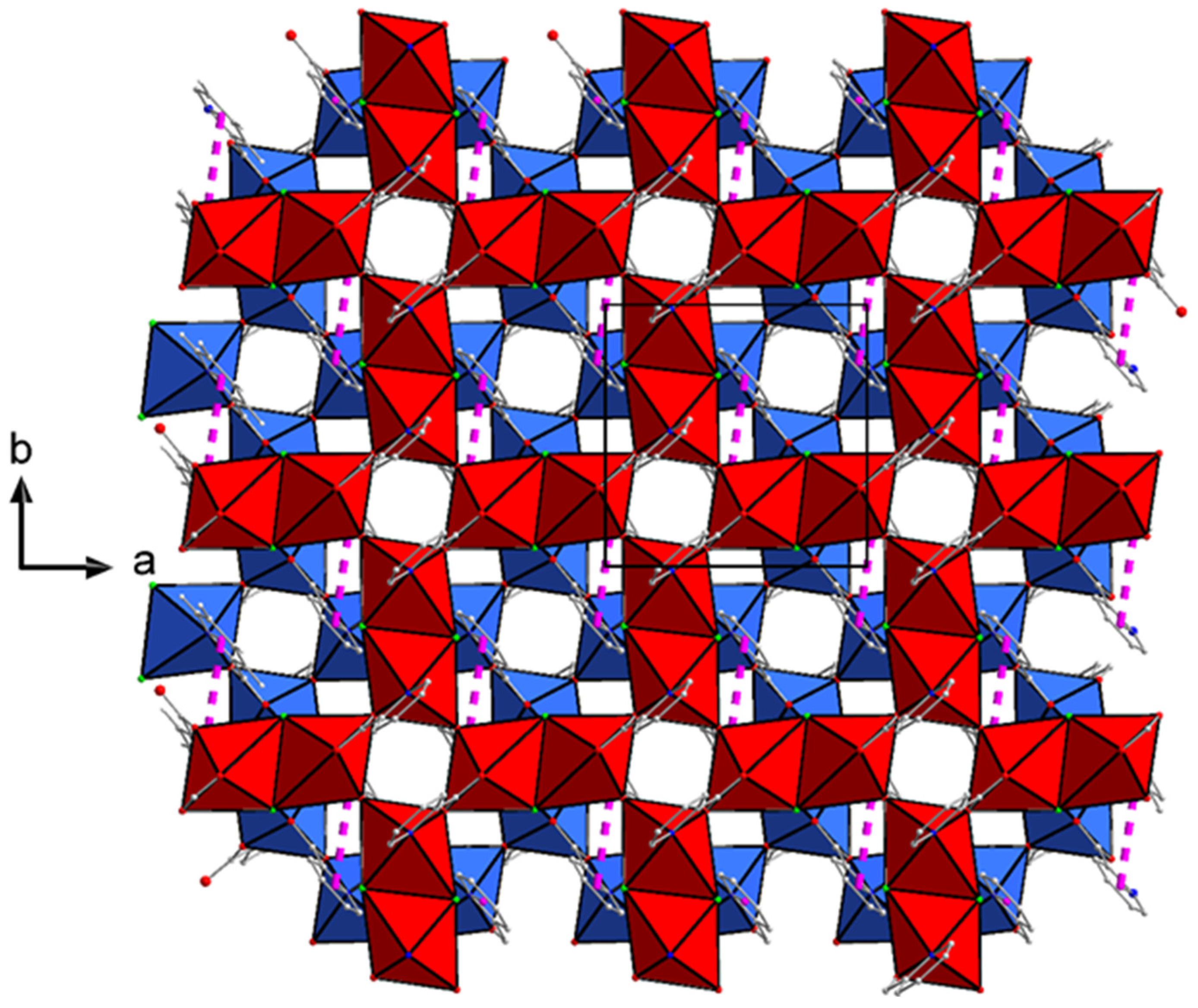
2.3. Description of Structures
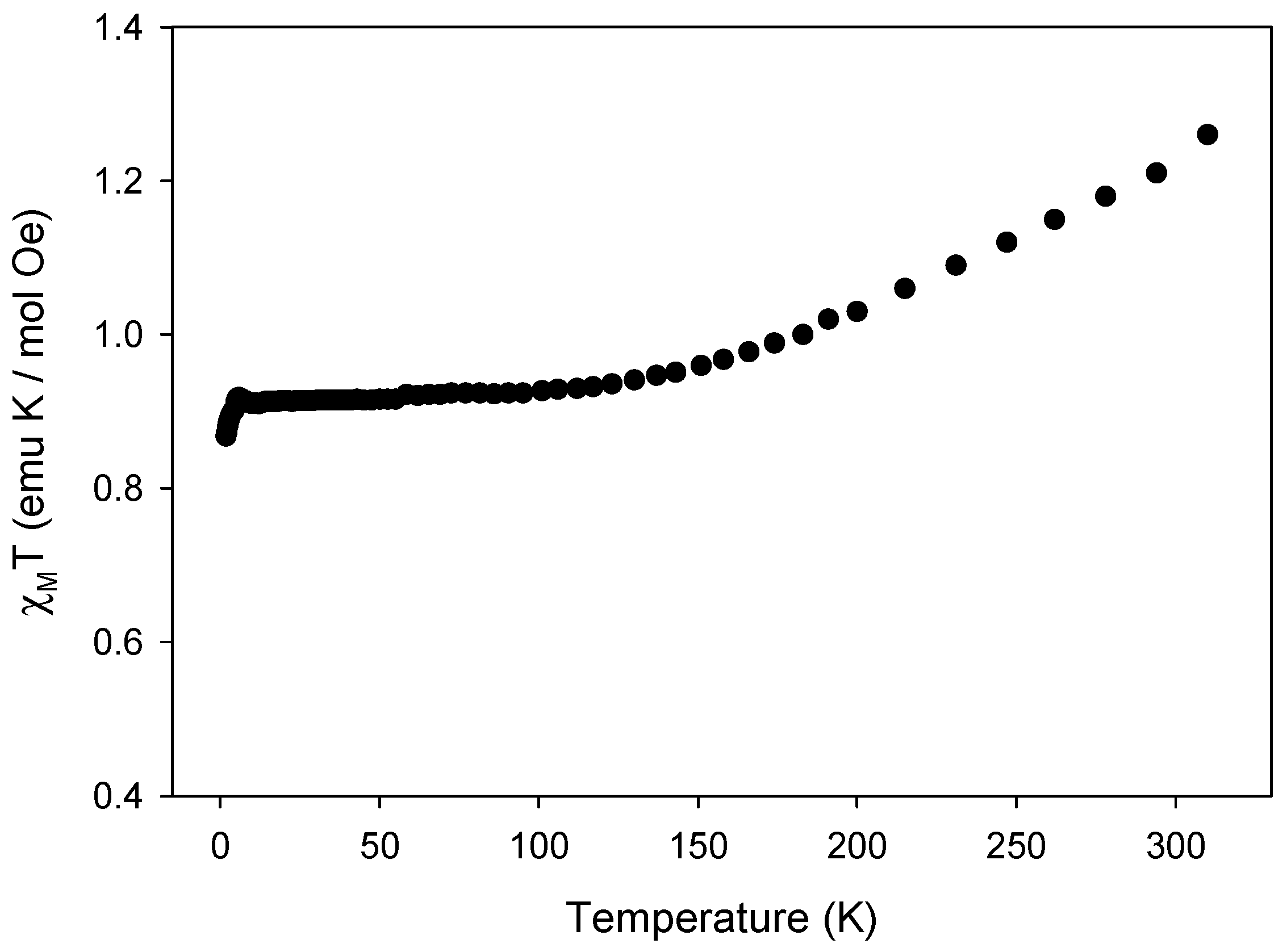
2.4. Magnetic Properties
3. Experimental Section
3.1. Materials and Instrumentation
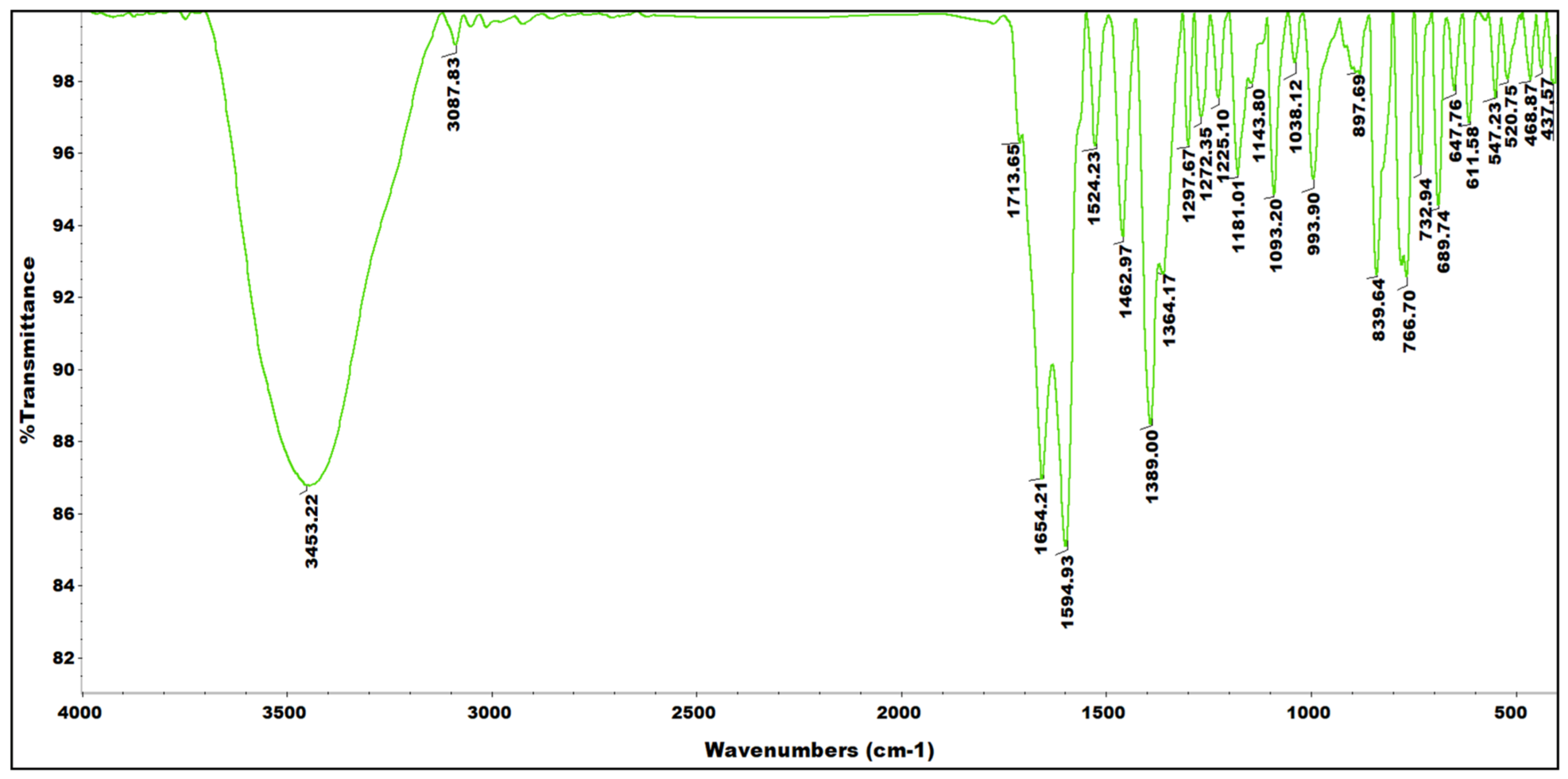
3.2. Synthesis of 2-Hydroxy-1,2-di(pyridin-2-yl)ethanone Oxime (α-pyroxH2)
3.3. Preparation of the Complexes
3.4. Single-Crystal X-Ray Crystallography
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blackman, A. Reactions of Coordinated Ligands. Adv. Heterocycl. Chem. 1993, 58, 123–170. [Google Scholar]
- Pombeiro, A.J.L.; Kukushkin, V.Y. Ligand Reactivity: General Introduction. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 585–594. [Google Scholar]
- Constable, E.C. Metals and Ligand Reactivity; VCH: Weinheim, Germany, 1996. [Google Scholar]
- Jones, M.M. Activation of Small Molecules by Coordination. J. Chem. Educ. 1964, 41, 493–500. [Google Scholar] [CrossRef]
- Miessler, G.L.; Fischer, P.J.; Tarr, D.A. Inorganic Chemistry, 5th ed.; Pearson: Boston, MA, USA, 1994; pp. 468–470. [Google Scholar]
- Lippard, S.J.; Berg, J.M. Principles of Bioinorganic Chemistry; University Science Books: Mill Valley, CA, USA, 1994; pp. 35–37, 257, 258. [Google Scholar]
- Pombeiro, A.J.L.; Kukushkin, V.Y. Reactivity of Coordinated Nitriles. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 639–660. [Google Scholar]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Oxime and oximate metal complexes: Unconventional synthesis and reactivity. Coord. Chem. Rev. 1999, 181, 147–175. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Tudela, D.; Pombeiro, A.J.L. Metal-ion assisted reactions of oximes and reactivity of oxime-containing metal complexes. Coord. Chem. Rev. 1996, 156, 333–362. [Google Scholar] [CrossRef]
- Garnovskii, D.A.; Kukushkin, V.Y. Metal-mediated reactions of oximes. Russ. Chem. Rev. 2006, 75, 111–124. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Kukushkin, V.Y. Reactivity of Coordinated Oximes. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 631–637. [Google Scholar]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-Involving Synthesis and Reactions of Oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef]
- Tschugaeff, L. Uber ein neues, empfindliches reagens auf nickel. Ber. Dtsch. Chem. Ges. 1905, 38, 2520–2522. [Google Scholar] [CrossRef]
- Smith, A.G.; Tasker, P.A.; White, D.J. The structures of phenolic oximes and their complexes. Coord. Chem. Rev. 2003, 241, 61–85. [Google Scholar] [CrossRef]
- Tasker, P.A.; Tong, C.C.; Westra, A.N. Co-extraction of cations and anions in base metal recovery. Coord. Chem. Rev. 2007, 251, 1868–1877. [Google Scholar] [CrossRef]
- Gerasimchuk, N.; Maher, T.; Durham, P.; Domasevitch, K.V.; Wilking, J.; Mokhir, A. Tin(IV) cyanoximates: Synthesis, Characterization, and Cytotoxicity. Inorg. Chem. 2007, 46, 7268–7284. [Google Scholar] [CrossRef]
- Tarai, A.; Nath, B. A review on oxime functionality: An ordinary functional group with significant impacts in supramolecular chemistry. Chem. Commun. 2024, 60, 7266–7287. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-J.; Zhang, Z.-Z.; Lin, S.-B. A review of manganese-based molecular magnets and supramolecular architectures from phenolic oximes. Coord. Chem. Rev. 2015, 289–290, 289–314. [Google Scholar] [CrossRef]
- Alonso, D.A.; Nájera, C. Oxime-derived palladacycles as source of palladium nanoparticles. Chem. Soc. Rev. 2010, 39, 2891–2902. [Google Scholar] [CrossRef] [PubMed]
- Lada, Z.G.; Polyzou, C.D.; Nika, V.; Stamatatos, T.C.; Konidaris, K.F.; Perlepes, S.P. Adventures in the coordination chemistry of 2-pyridyl oximes: On the way to 3d/4f-metal coordination clusters. Inorg. Chim. Acta 2022, 539, 120954. [Google Scholar] [CrossRef]
- Stamatatos, T.C.; Foguet-Albiol, D.; Lee, S.-C.; Stoumpos, C.C.; Raptopoulou, C.P.; Terzis, A.; Wernsdorfer, W.; Hill, S.O.; Perlepes, S.P.; Christou, G. “Switching on” the Properties of Single-Molecule Magnetism in Triangular Manganese(III) Complexes. J. Am. Chem. Soc. 2007, 129, 9484–9499. [Google Scholar] [CrossRef]
- Milios, C.J.; Stamatatos, T.C.; Perlepes, S.P. The coordination chemistry of pyridyl oximes. Polyhedron 2006, 25, 134–194. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Bekiari, V.; Tzimopoulos, D.I.; Raptopoulou, C.P.; Psycharis, V.; Tsipis, A.; Perlepes, S.P. Reactivity of Coordinated 2-Pyridyl Oximes: Synthesis, Structure, Spectroscopic Characterization and Theoretical Studies of Dichlorodi{(2-pyridyl)Furoxan}Zinc(II) Obtained from the Reaction between Zinc(II) Nitrate and Pyridine-2-Chloroxime. Inorganics 2020, 8, 47. [Google Scholar] [CrossRef]
- Deville, C.; Padamati, S.K.; Sundberg, J.; McKee, V.; Browne, W.R.; McKenzie, C.J. O2 Activation and Double C–H Oxidation by a Mononuclear Manganese(II) Complex. Angew. Chem. 2015, 128, 555–559. [Google Scholar] [CrossRef]
- Deville, C.; McKee, V.; McKenzie, C.J. Copper-promoted methylene C-H oxidation to a ketone derivative by O2. Dalton Trans. 2017, 46, 709–719. [Google Scholar] [CrossRef]
- Dolai, M.; Saha, U. A simple Cu(II) complex of phenolic oxime: Synthesis, crystal structure, supramolecular interactions, DFT calculation and catecholase activity study. Heliyon 2020, 6, e04942. [Google Scholar] [CrossRef]
- Hassan, E.A.; Ebrahium, M.M.; Ebrahium, A.M. Metal complexes of hydrazone-oxime derivative as promising in vitro antimicrobial agents against some fungal and bacterial strains. Appl. Organomet. Chem. 2022, 36, e6663. [Google Scholar] [CrossRef]
- Luo, S.; Tu, T.; Chen, Z.; Zhong, Y.; Xu, J.; Lu, S.; Sun, X. Magneto-structural correlations of oximato-bridged dinuclear copper(II) complex: A theoretical perspective. Int. J. Quantum Chem. 2023, 123, e27142. [Google Scholar] [CrossRef]
- Hensel, H.R. Über eine Benzoin-Kondensation ohne Cyan-Ionen. Angew. Chem. 1953, 65, 491. [Google Scholar] [CrossRef]
- Bueher, C.A.; Addleburg, J.W.; Glem, D.M. The Influence of Chelation on the Stability of Enediols. I. 2-Pyridoin, 1-Phenyl-2-(2-pyridyl)-1,2-ethenediol and 1-Phenyl-2-(4-pyridyl)-1,2-ethenediol. J. Org. Chem. 1955, 20, 1350–1355. [Google Scholar] [CrossRef]
- Inoue, H.; Matsumoto, M.; Kiyoi, S.; Yamanaka, M. The Solvent Effect on the Stability of α-Pyridoin. Bull. Chem. Soc. Jpn. 1973, 46, 3900–3901. [Google Scholar] [CrossRef]
- Hatanaka, M.; Takahashi, K.; Nakamura, S.; Mashino, T. Preparation and antioxidant activity of α-pyridoin and its derivatives. Bioorg. Med. Chem. 2005, 13, 6763–6770. [Google Scholar] [CrossRef]
- Ashida, T.; Hirokawa, S.; Okaya, Y. The Crystal Structure of α-Pyridoin, 1,2-Di-2-pyridylethenediol-1,2. Acta Crystallogr. 1965, 18, 122–127. [Google Scholar] [CrossRef]
- Routzoumani, A.; Lada, Z.G.; Angelidou, V.; Raptopoulou, C.P.; Psycharis, V.; Konidaris, K.F.; Chasapis, C.T.; Perlepes, S.P. Confirming the Molecular Basis of the Solvent Extraction of Cadmium(II) Using 2-Pyridyl Oximes through a Synthetic Inorganic Chemistry Approach and a Proposal for More Efficient Extractions. Molecules 2022, 27, 1619. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Romero, S.; Rodríguez-Barbarín, C.; Malendez-Bustamante, F.J. Synthesis of 1,2-dimethoxy-1,2-di(pyridin-2-yl)-1,2-ethanediol: Crystal and molecular structure determination. J. Chem. Crystallogr. 2006, 36, 303–308. [Google Scholar] [CrossRef]
- Sabaté, C.M.; Delalu, H. The Crystal Structure of (1E,2E)-2,2′-Dipyridylglyoxime. Heteratom. Chem. 2011, 23, 59–65. [Google Scholar] [CrossRef]
- Alsalim, T.A.; Hadi, J.S.; Ali, O.N.; Abbo, H.S.; Titinchi, S.J.J. Oxidation of Benzoin Catalyzed by Oxovanadium(IV) Schiff Base Complexes. Chem. Cent. J. 2013, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Depreux, P.; Bethegnies, G.; Marcimal-Lefeovre, A. Synthesis of Benzil from Benzoin with Copper(II) Acetate. J. Chem. Educ. 1988, 65, 553. [Google Scholar] [CrossRef]
- Goher, M.A.S.; Hafez, A.K.; Abu-Youssef, M.A.M.; Popitsch, A.; Fritzer, H.P.; Mautner, F.A. Synthesis and Spectral and Structural Characterization of a Bridging Chloropicolinatocopper(II) Complex, Cu(C5H4NCOO)Cl. Monatshefte Chem. 1994, 125, 833–840. [Google Scholar] [CrossRef]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Krawczyk, M.K.; Krawczyk, M.S.; Siczek, M.; Lis, T. 2,2′-[((1E,2E)-1,2-Bis(hydroxyimino)ethane-1,2-diyl]dipyridinium hexachloridorhenate(IV). Acta Crystallogr. 2012, E68, m1174–m1175. [Google Scholar] [CrossRef]
- Papaefstathiou, G.S.; Raptopoulou, C.P.; Tsohos, A.; Terzis, A.; Bakalbassis, E.G.; Perlepes, S.P. Alcoholysis of 2,2′-Pyridyl, (2-C5H4N)C(O)C(O)(2-C5H4N), in the Presence of Copper(II): A Family of Planar Pentanuclear Copper(II) Complexes Stabilized by [(2-C5H4N)C(O)(OR)C(O)(OR)(2-C5H4N)]2− and Carboxylate Ligands. Inorg. Chem. 2000, 39, 4658–4662. [Google Scholar] [CrossRef]
- Ali, B.; David, G.; Gendron, F.; Li, X.-L.; Cador, O.; Plass, W.; Le Guennic, B.; Tang, J. A Cu12 Metallacycle Assembled from Four C3-Symmetric Spin Frustrated Triangular Units. Magnetochemistry 2023, 9, 122. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Tzeng, Y.-W.; Yang, C.-I.; Nakano, M.; Wan, W.-L.; Lai, L.-L.; Lee, G.-H. The synthesis of three new Cu5, Cu8 and Cu12 clusters via the use of a semi-flexible aminotriazine-based bis-methylpyridine ligand. Dalton Trans. 2017, 46, 1237–1248. [Google Scholar] [CrossRef]
- Chandrasekhar, V.; Kingsley, S. A Dodecanuclear Copper(II) Cage Containing Phosphonate and Pyrazole Ligands. Angew. Chem. Int. Ed. 2000, 39, 2320–2322. [Google Scholar] [CrossRef]
- Strotmeyer, K.P.; Fritsky, I.O.; Ott, R.; Pritzkow, H.; Krämer, R. Evaluating the Conformational Role of an Allosteric CuII Ion in Anion Recognition and Catalysis by a Tricopper Complex. Supramol. Chem. 2003, 15, 529–547. [Google Scholar] [CrossRef]
- Dodds, C.A.; Garner, M.; Reglinski, J.; Spicer, M.D. Coinage Metal Complexes of a Boron-Substituted Scorpionate Ligand. Inorg. Chem. 2006, 45, 2733–2741. [Google Scholar] [CrossRef]
- Shahbakhsh, M.; Saravani, H.; Dusek, M.; Poupon, M. Study of the one-step in situ growth synthesis of Cu-Pic coordination polymer and Cu-BTC MOF and their performance for detection of 4-Nitrophenol. Microchem. J. 2023, 189, 108557. [Google Scholar] [CrossRef]
- Żurowska, B.; Mroziński, J.; Ślepokura, K. Structure and magnetic properties of a double out-of-plane carboxylate-bridged Cu(II) compound with pyridine-2-carboxylate. Polyhedron 2007, 26, 3379–3387. [Google Scholar] [CrossRef]
- Hallett, A.J.; O’Brien, T.M.; Carter, E.; Kariuki, B.M.; Murphy, D.M.; Ward, B.D. Copper(II) complexes of pyridine-oxazoline (Pyox) ligands: Coordination chemistry, ligand stability, and catalysis. Inorg. Chim. Acta 2016, 441, 86–94. [Google Scholar] [CrossRef]
- Biswas, C.; Mukherjee, P.; Drew, M.G.B.; Gómez-Garcia, C.J.; Clemente-Juan, J.M.; Ghosh, A. Anion-Directed Synthesis of Metal-Organic Frameworks Based on 2-Picolinate Cu(II) Complexes: A Ferromagnetic Alternating Chain and Two Unprecedented Ferromagnetic Fish Backbone Chains. Inorg. Chem. 2007, 46, 10771–10780. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.A. 1-D Hydrogen bonded water in Cu(II)-picolinate coordination polymer: Synthesis, crystal structure, and thermogravimetric analysis. J. Coord. Chem. 2012, 65, 4168–4176. [Google Scholar] [CrossRef]
- Tessarolo, J.; Venzo, A.; Bottaro, G.; Armelao, L.; Rancan, M. Hampered Subcomponent Self-Assembly Leads to an Aminal Ligand: Reactivity with Silver(I) and Copper(II). Eur. J. Inorg. Chem. 2017, 2017, 30–34. [Google Scholar] [CrossRef]
- Ay, B.; Şahin, O.; Yildiz, E. One-pot hydrothermal synthesis of 1D copper(II) coordination polymers involving in-situ decarboxylation. Solid. State Sci. 2019, 96, 105958. [Google Scholar] [CrossRef]
- Sarkar, S.; Dey, S.; Mukherjee, T.; Zangrando, E.; Drew, M.G.B.; Chattopadhyay, P. Coordination behavior of pyridylmethylthioether system with cupric chloride and cupric bromide: C–S bond cleavage and crystal structures. J. Mol. Struct. 2010, 980, 94–100. [Google Scholar] [CrossRef]
- Koumousi, E.S.; Raptopoulou, C.P.; Perlepes, S.P.; Escuer, A.; Stamatatos, T.C. Strong antiferromagnetic coupling in doubly N,O oximato-bridged dinuclear copper(II) complexes. Polyhedron 2010, 29, 204–211. [Google Scholar] [CrossRef]
- Skorda, K.; Stamatatos, T.C.; Vafiadis, A.P.; Lithoxoidou, A.T.; Terzis, A.; Perlepes, S.P.; Mrozinski, J.; Raptopoulou, C.P.; Plakatouras, J.C.; Bakalbassis, E.G. Copper(II) chloride/1-methylbenzotriazole chemistry: Influence of various synthetic parameters on the product identity, structural and magnetic characterization, and quantum-chemical studies. Inorg. Chim. Acta 2005, 358, 565–582. [Google Scholar] [CrossRef]
- Marsh, W.E.; Patel, K.C.; Hatfield, W.F.; Hodgson, D.J. Magnetic Interactions in Chloro-Bridged Copper(II) Dimers. Structural and Magnetic Characterizations of Bis(μ-chloro)bis[chloro(N,N,N′-triethylethylenediamine)copper(II)], [Cu(Et3en)Cl2]2. Inorg. Chem. 1983, 23, 511–515. [Google Scholar] [CrossRef]
- Landee, C.P.; Turnbull, M.M. Review: A gentle introduction to magnetism: Units, fields, theory, and experiment. J. Coord. Chem. 2014, 67, 375–439. [Google Scholar] [CrossRef]
- Carlin, R.L. Magnetochemistry; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- APEX2. Data Collection Software, version 2012.4; Bruker AXS: Delft, The Netherlands, 2012.
- SAINT+. Data Integration Engine, version 7.23a; Bruker AXS: Madison, WI, USA, 2005.
- Sheldrick, G.M. SADABS, version 2.01; Bruker/Siemens Area Detector Absorption Correction Program; Bruker AXS: Madison, WI, USA, 1998.
- Sheldrick, G.M. SHELXL, version 2014; Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 2014.
- Spek, A.L. PLATON, An integrated tool for the analysis of the results of a single crystal structure determination. Acta Crystallogr. 1990, A46, C34. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Bradenburg, K. DIAMOND, Release 3.1f; Crystal Impact GbR: Bonn, Germany, 2008.
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.K.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge structural database and visualizing crystal structures. Acta Crystallogr. 2002, B58, 389–397. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Diamond, Crystal and Molecular Structure Visualization, version 3.2; Crystal Impact: Bonn, Germany, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IUPAC Name | Abbreviation |
|---|---|
| 2-hydroxy-1,2-di(pyridin-2-yl)ethanone a | α-pyrH2 |
| (E)-2-hydroxy-1,2-di(pyridin-2-yl)ethanone oxime b | α-pyroxH2 |
| 1,2-dimethoxy-1,2-di(pyridin-2-yl)ethane-1,2-diol | mpydolH2 |
| (E,E)-1,2-di(pyridin-2-yl)ethanedione dioxime c | pydoxH2 |
| (Z)-1,2-di(pyridin-2-yl)ethanone oxime | dpeoH |
| 1,2-di(pyridin-2-yl)ethane-1,2-dione d | (py)2COCO |
| pyridine-2-carboxylic acid e | picH |
| Parameter | 1∙12MeOH | 2 |
|---|---|---|
| Empirical formula | C96H136Cl12Cu12N16O36 | C24H16Cl4Cu4N4O8 |
| Formula weight/g mol−1 | 3278.08 | 884.37 |
| Temperature/°C | −123 | −113 |
| Crystal system | triclinic | tetragonal |
| Space group | P42/n | |
| a/Å | 13.0613(6) | 9.7196(2) |
| b/Å | 16.5801(5) | 9.7196(2) |
| c/Å | 17.0534(7) | 14.9474(4) |
| α/° | 101.523(3) | 90.0 |
| β/° | 94.103(4) | 90.0 |
| γ/° | 112.393(4) | 90.0 |
| Volume/Å3 | 3301.0(2) | 1412.09(7) |
| Z | 1 | 2 |
| ρcalc/g cm−3 | 1.649 | 2.080 |
| μ/mm−1 | 2.21 | 3.41 |
| 2θmax/° | 52.8 | 54.0 |
| Radiation (wavelength/Å) | Mo Kα (λ = 0.71073) | Mo Kα (λ = 0.71073) |
| Reflections collected | 317430 | 17269 |
| Independent reflections (Rint) | 13464 (0.0547) | 1542 (0.025) |
| No. of parameters | 729 | 116 |
| Goodness of fit on F2 | 0.93 | 1.09 |
| R1 a [I ≥ 2σ(I)] | 0.0466 | 0.0208 |
| wR2 b [I ≥ 2σ(I)] | 0.1269 | 0.0543 |
| Δρmax/Δρmin (e Å−3) | 1.70/−1.26 | 0.38/−0.31 |
| CCDC | 2425425 | 2426248 |
| Complex | Coordination Mode(s) i of pic− | Dimensionality | Reference |
|---|---|---|---|
| {[Cu(pic)2]}n a,b | 2.111 | 1D | [48,49] |
| {[Cu4(pic)6(H2O)2](ClO4)2}n c | 2.111 | 2D | [50,51] |
| {[Cu(pic)2]∙2H2O}n d,e,f | 2.111 | 1D | [52,53,54] |
| {[CuCl(pic)(H2O)]∙H2O }n g | 1.101 | 1D | [55] |
| {[Cu4(pic)6(H2O)2](BF4)2}n | 2.111 | 2D | [51] |
| {[Cu2(pic)3(H2O)2(NO3)]}n | 2.111, 1.101 | 1D | [51] |
| {[Cu4Cl4(pic)4}n h | 2.211 | 2D | [39], this work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baka, K.H.; Cunha-Silva, L.; Raptopoulou, C.P.; Psycharis, V.; Papaioannou, D.; Turnbull, M.M.; Lada, Z.G.; Perlepes, S.P.; Stamatatos, T.C. Copper(II)-Promoted Reactions of α-Pyridoin Oxime: A Dodecanuclear Cluster and a 2D Coordination Polymer. Magnetochemistry 2025, 11, 35. https://doi.org/10.3390/magnetochemistry11040035
Baka KH, Cunha-Silva L, Raptopoulou CP, Psycharis V, Papaioannou D, Turnbull MM, Lada ZG, Perlepes SP, Stamatatos TC. Copper(II)-Promoted Reactions of α-Pyridoin Oxime: A Dodecanuclear Cluster and a 2D Coordination Polymer. Magnetochemistry. 2025; 11(4):35. https://doi.org/10.3390/magnetochemistry11040035
Chicago/Turabian StyleBaka, Konstantina H., Luís Cunha-Silva, Catherine P. Raptopoulou, Vassilis Psycharis, Dionissios Papaioannou, Mark M. Turnbull, Zoi G. Lada, Spyros P. Perlepes, and Theocharis C. Stamatatos. 2025. "Copper(II)-Promoted Reactions of α-Pyridoin Oxime: A Dodecanuclear Cluster and a 2D Coordination Polymer" Magnetochemistry 11, no. 4: 35. https://doi.org/10.3390/magnetochemistry11040035
APA StyleBaka, K. H., Cunha-Silva, L., Raptopoulou, C. P., Psycharis, V., Papaioannou, D., Turnbull, M. M., Lada, Z. G., Perlepes, S. P., & Stamatatos, T. C. (2025). Copper(II)-Promoted Reactions of α-Pyridoin Oxime: A Dodecanuclear Cluster and a 2D Coordination Polymer. Magnetochemistry, 11(4), 35. https://doi.org/10.3390/magnetochemistry11040035