Abstract
Honeysuckle, derived from the dried flower buds or blossoms of Lonicera japonica Thunb, is a traditional Chinese medicine known for its properties in eliminating heat and toxins, reducing inflammation, and alleviating swelling. In this study, we investigated the potential therapeutic and preventive benefits of L. japonica extract on inflammatory diseases induced by lipopolysaccharide (LPS) using Misgurnus anguillicaudatus as a model organism. The fish were fed a diet supplemented with L. japonica extract, followed by LPS injection to induce inflammation. We then analyzed the transcriptional profile to identify differentially expressed genes (DEGs). A total of 6611 DEGs were identified through comprehensive analysis, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. Our results revealed significant enrichment of DEGs in pathways associated with proteasome function, immune system regulation, and infectious disease response. These findings suggest a strong correlation between L. japonica and immune defense mechanisms, providing valuable insights into the potential anti-inflammatory effects of this plant, particularly in the context of LPS-induced inflammation. This study highlights the potential use of L. japonica in treating inflammatory diseases and underscores its role in immune regulation.
Key Contribution:
This study provides novel insights into the anti-inflammatory mechanisms of Lonicera japonica extract by identifying differentially expressed genes and enriched pathways in Misgurnus anguillicaudatus subjected to LPS-induced inflammation, highlighting its potential therapeutic value for inflammatory diseases and immune regulation.
1. Introduction
Honeysuckle, scientifically known as the dried flower buds or blossoms of Lonicera japonica Thunb, is a traditional Chinese medicine with extensive applications. It is a dual-purpose plant, valued both for its medicinal and edible properties. Honeysuckle contains numerous active constituents, including volatile oils, flavonoids, and organic acids, which contribute to its therapeutic effects [1,2]. It is primarily used to clear heat and toxins, disperse wind-heat, and treat conditions such as wind-heat cold, carbuncles, heat toxicity, and dysentery. Recent studies have further demonstrated its anti-inflammatory [3,4], antipyretic, antibacterial, antiviral, anti-tumor, immunomodulatory, and intestinal microenvironment-regulating properties [5]. Given its significant contributions to both the medical and food industries, the demand for L. japonica Thunb is increasing as health awareness grows [6,7].
Misgurnus anguillicaudatus, a member of the Cyprinidae family, is a typical freshwater benthic fish in China that possesses intestinal respiration capabilities enabling survival in hypoxic environments. As an important economic species, the application value of M. anguillicaudatus is manifested in multiple dimensions. Its flesh is tender and delicious, rich in unsaturated fatty acids and various trace elements, aligning with modern healthy dietary concepts [8]. Traditional medicine recognizes its efficacy in replenishing qi, invigorating the spleen, dispelling dampness, and detoxifying, endowing it with both edible and medicinal properties [9,10]. Additionally, this species demonstrates strong adaptability to water quality, tolerating harsh environments such as high temperatures and hypoxia. With a short reproductive cycle, high fecundity, low farming cost, and rapid growth [11], it has become a distinctive variety with high promotion value in China’s freshwater aquaculture industry. M. anguillicaudatus exhibits dual attributes as both an “economic fish” and a “model organism” [12]. Its robust environmental adaptability reduces the difficulty of experimental environment control, while the short reproductive cycle facilitates the rapid establishment of infection-treatment research systems. Meanwhile, as a high-protein and low-fat aquaculture variety, its meat quality and health characteristics can be linked to anti-inflammatory research, providing a new perspective for disease prevention under the framework of “food as medicine”.
Inflammation is a protective response of the body to various stimuli; however, an inappropriate response, either too low or too high, can lead to disease [13]. When the inflammatory response is excessive, such as the persistent high expression of pro-inflammatory factors (IL-1β, TNF-α) exceeding the demand for tissue repair, it often leads to excessive immunity in the body, thereby causing tissue damage. When the response is too weak, such as the sluggish response of immune cells that fail to eliminate pathogens or abnormal cells, it often results in the failure of immune surveillance and an increased susceptibility to infection. Moreover, we have also learned that numerous studies have highlighted the link between intestinal flora imbalances and inflammatory diseases, and these findings have multiple implications for our study [14].
The liver serves as a pivotal immunometabolic organ during inflammatory responses and represents a critical target site for LPS action. From the perspective of honeysuckle’s pharmacological properties, existing studies confirm that its bioactive components can target the liver to exert anti-inflammatory, antioxidant, and hepatoprotective effects [15]. Furthermore, as the primary organ for drug metabolism, L. japonica extracts achieve higher accumulation concentrations in the liver, enabling more direct therapeutic efficacy. Additionally, liver tissue samples are readily accessible for collection and analysis, where inflammatory biomarkers and associated gene expression changes provide reliable indicators for evaluating inflammatory severity and pharmaceutical intervention outcomes.
Lipopolysaccharide (LPS), a bacterial endotoxin, is widely used in experimental models to induce inflammation [16]. Upon entering the body, LPS binds to LPS-binding protein (LBP) on the host cell surface, forming a complex that subsequently binds to CD14 receptors to produce a ternary complex. This complex activates Toll-like receptor 4 (TLR4), triggering an inflammatory response [17]. This study investigates the effects of honeysuckle extract on LPS-induced inflammation and immunomodulation in M. anguillicaudatus. Through previous studies, we also know that in addition to M. anguillicaudatus, some researchers choose zebrafish to receive LPS injection to obtain an inflammatory model [18]. In addition, fish organisms such as Turbot (Scophthalmus maximus L.) and lampreys can all be induced by LPS injection to become the inflammatory models required for experiments [19,20].
In this study, M. anguillicaudatus was used as a model organism to systematically explore the preventive and therapeutic effects of L. japonica extract on LPS-induced inflammation. A total of 6611 DEGs were obtained by constructing an inflammation model and combining transcriptome sequencing analysis. Gene function enrichment analysis showed that these genes were significantly enriched in immune response, energy metabolism, autophagy, and other pathways. The study revealed that L. japonica might exert anti-inflammatory effects by regulating proteasome activity, remodeling energy metabolism and activating intestinal immunity, it provides a transcriptome-level scientific basis for clarifying the anti-inflammatory material basis of L. japonica and developing natural anti-inflammatory drugs, and also provides a new idea for the application of traditional Chinese medicine in aquaculture and the prevention and treatment of inflammatory diseases.
2. Materials and Methods
2.1. Material and Experimental Procedure
Three hundred M. anguillicaudatus were purchased from an aquatic animal breeding base in Zhejiang province and evenly distributed into six basins, with fifty fish per basin. M. anguillicaudatus had an average body weight of 13 g, an average standard length of 9 cm, and an average total length of 10 cm. M. anguillicaudatus used in the experiment were 6-month-old juveniles from the same batch and in an immature stage. The daily feeding amount of the basic diet was approximately 4% of the total body weight of the M. anguillicaudatus. Each culture container measured 40 cm × 28 cm × 12 cm, with 4 L of water per pot. The water source was tap water dechlorinated by aeration for 48 h. The basic water quality before stocking was as follows: DO > 6.0 mg/L, pH 7–8, TAN < 0.1 mg/L, NO2-N < 0.05 mg/L, and water temperature 25.0 ± 0.5 °C.
Over a 3-day period, each basin was fed 20 g of feed daily. The basal diet contained crude protein ≥38%, crude fat 8%, crude fiber ≤5%, ash ≤12%, phosphorus 1.2%, and lysine 2.1%. On the fourth day, three basins were fed 20 g of feed supplemented with 10 g of L. japonica extract powder, while the remaining three basins received only 20 g of feed as a control. After 12 h, all fish were injected with 0.1 mL of LPS. The activity and condition of the fish were monitored at 3, 6, 12, and 24 h post-injection. These four time points correspond to different stages of inflammation. Studies on intestinal inflammation in M. anguillicaudatus infected with LPS have shown that this time interval can effectively reflect the temporal characteristics of the immune response in fish. From each M. anguillicaudatus, 100 mg of liver tissue samples were immediately collected, rapidly frozen in liquid nitrogen, and stored at −80 °C. M. anguillicaudatus were euthanized following AVMA Guidelines and institutional animal care standards. A portion of the samples from each group was sent to a testing company for transcriptomic sequencing.
2.2. RNA Sequencing
RNA extraction from M. anguillicaudatus was conducted using the OMEGA kit according to the manufacturer’s instructions. The quality and concentration of the extracted RNA were assessed using Nanodrop photometers. Subsequently, the first strand of cDNA was synthesized using template RNA, arbitrary primers, and reverse transcriptase. This template was then used to generate the second strand of cDNA.
For Next-Generation Sequencing (NGS), libraries were constructed using the Illumina NovaSeq 6000 (Illumina Inc., San Diego, CA, USA) Sequencing platform and subjected to double-end sequencing. To ensure high-quality data, initial offline data were filtered to remove reads with adaptors, those shorter than 50 base pairs, and sequences with an average quality score below Q20. Based on the assembly principle of De Bruijn Graph (DBG), high-quality sequences were assembled de novo using Trinity software v2.15.1 to obtain transcripts, and the longest transcript from each cluster was designated as the Unigene.
The Unigene sequences were annotated using various databases, including GO, KEGG, eggNOG, SwissProt, and Pfam. Additionally, the filtered sequences were aligned to the Unigene database to determine the read counts for each Unigene.
2.3. Transcript Splicing and Analysis of DEGs
Given that the transcriptome was sequenced without a reference genome, Trinity software was employed to splice clean reads and obtain transcripts for subsequent analysis. Differential expression analysis was performed using DESeq and genes with a log2 fold change greater than 1 and a p-value less than 0.05 were identified as differentially expressed genes (DEGs).
2.4. Analysis by Quantitative Real-Time PCR
To validate the transcriptome data, 11 DEGs were randomly selected and their expression levels were assessed using Quantitative Real-Time PCR (qPCR). Primer sequences for each DEG are provided in Table S1.
3. Results
3.1. Transcript Splicing Result
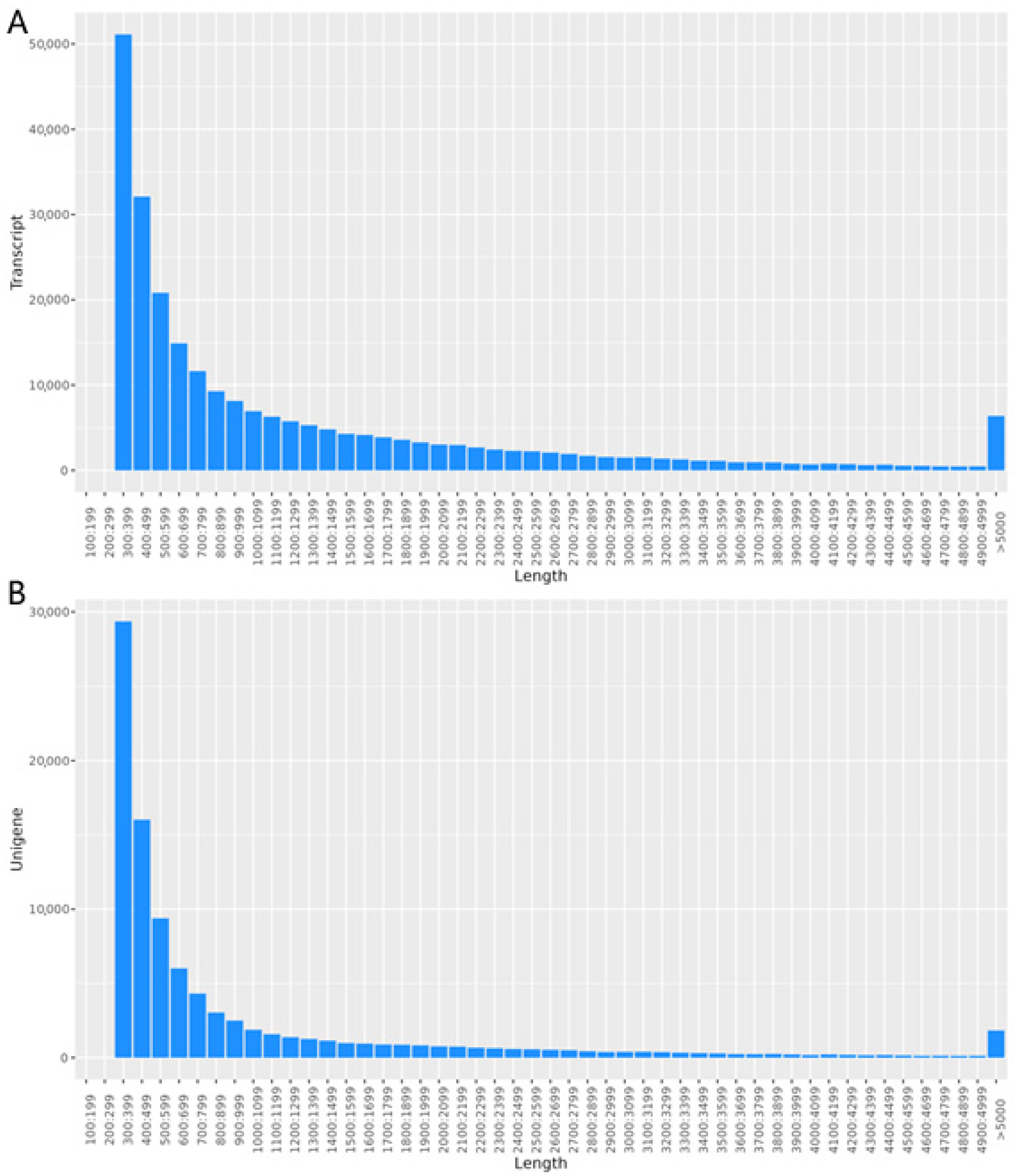
The quantity of high-quality reads was determined through the filtration of raw data, a step crucial for influencing subsequent analyses (Table S2). Upon splicing the transcripts, a total transcript length of 315,438,213 bp and a Unigene length of 96,247,359 bp were obtained. The maximum length observed for both transcripts and Unigenes was 48,863 bp. The average lengths of the transcript and Unigene sequences were 1295.26 bp and 1015.57 bp, respectively. The N50 values were 2179 bp for transcripts and 496 bp for Unigenes. The N90 values, which indicate the length at which 90% of the assembly is covered, were 1816 bp for transcripts and 393 bp for Unigenes. In addition, in transcripts and Unigenes, the total number of sequences longer than N50 is 41,839 and 13,964, respectively, while the total number of sequences longer than N90 is 161,226 and 66,653, respectively. The GC content was 42.21% for transcripts and 41.37% for Unigenes (Table S3, Figure 1).
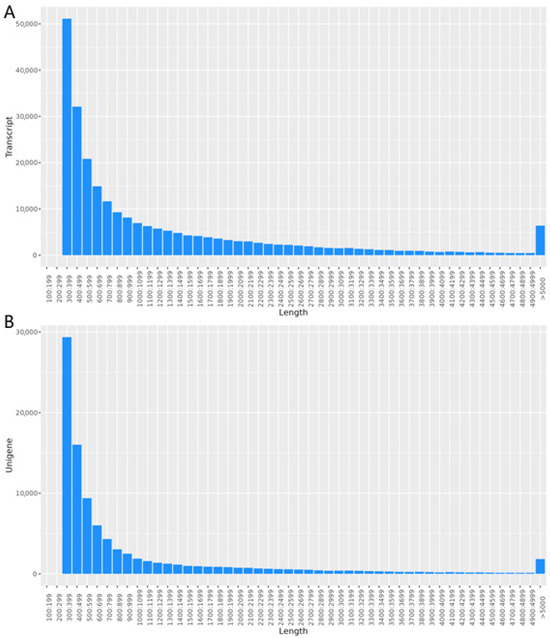
Figure 1.
Length distribution. (A) The length distribution of transcript; (B) the length distribution of Unigenes.
3.2. Function Annotation
3.2.1. Result of Annotation
Functional annotation of Unigenes yielded the following results: the NR database provided the highest number of annotations, with 40,696 Unigenes (42.94%) successfully annotated. The eggNOG database followed, with 37,193 annotations (39.24%). The SwissProt database annotated 29,701 Unigenes (31.34%), while the GO database annotated 26,825 Unigenes (28.30%). The Pfam and KEGG databases annotated 23,668 (24.97%) and 10,748 (11.34%) Unigenes, respectively. Notably, 7151 Unigenes (7.55%) were annotated across all databases (Table S4).
3.2.2. NR Annotation
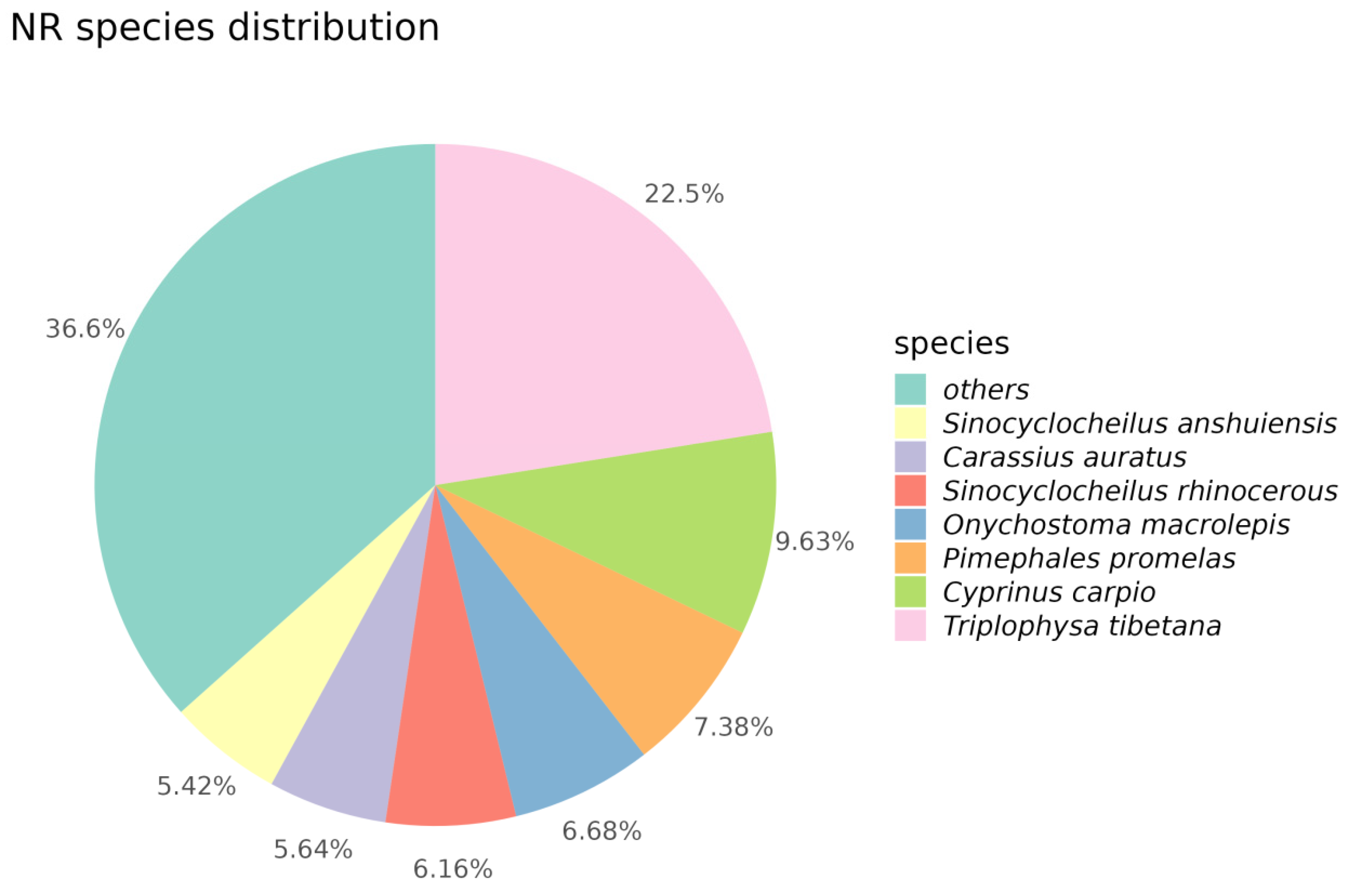
Through alignment and annotation of Unigenes with the NR database, we discovered that the majority of genes exhibited high similarity to various species, including Triplophysa tibetana, Cyprinus carpio, Pimephales promelas, Onychostoma macrolepis, Sinocyclocheilus rhinocerous, Carassius auratus, and Sinocyclocheilus anshuiensis. Specifically, 22.5% of Unigene sequences showed high similarity with Triplophysa tibetana, while 7.38% of Unigene sequences exhibited similarity to Pimephales promelas. Apart from these seven species, approximately 36.6% of Unigene sequences demonstrated similarity to other species (Figure 2).
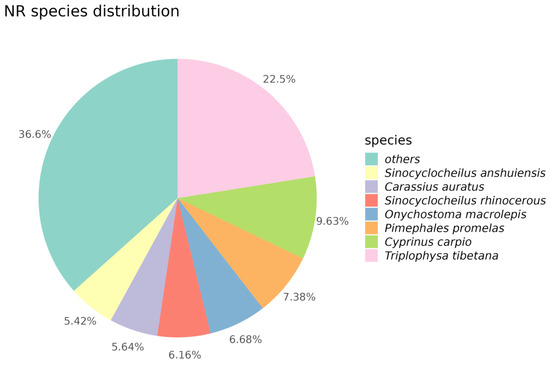
Figure 2.
Pie chart of NR species distribution. In this pie chart, different colors correspond to different species.
3.2.3. GO Annotation
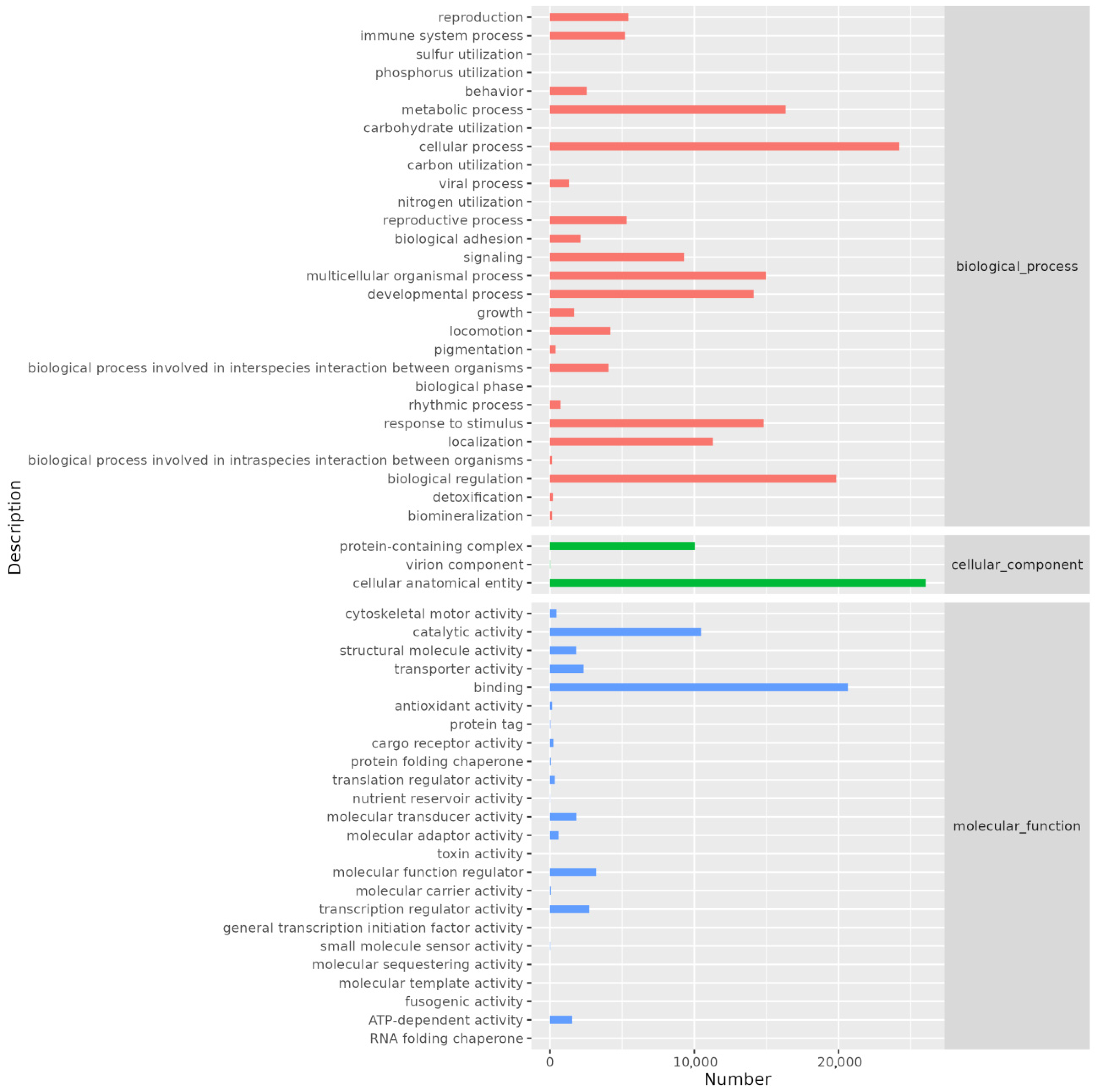
GO annotation revealed that genes were distributed across three main categories: cellular component, biological process, and molecular function. The biological process category contained the highest number of annotated genes. Specifically, the cellular anatomical entity category had the most annotations (26,046), followed by the cellular process category (24,221). Other notable annotations included binding (20,641), biological regulation (19,834), metabolic process (16,337), multicellular organismal process (14,960), response to stimulus (14,815), developmental process (14,116), localization (11,287), and catalytic activity (10,467) (Figure 3).
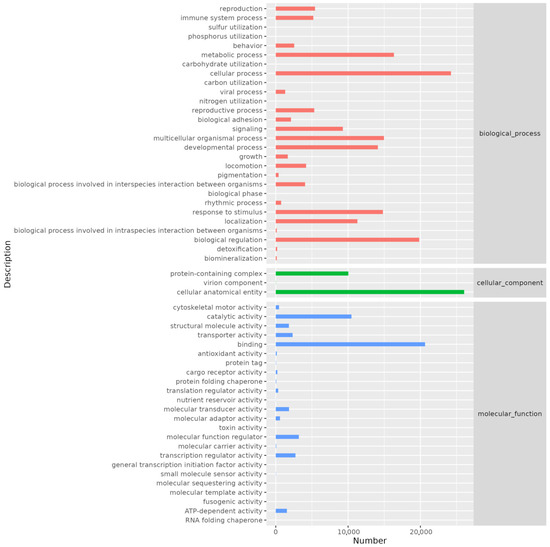
Figure 3.
Histogram of GO annotation. The abscissa is the number of genes annotated on secondary classification of GO term and the ordinate is the name of secondary classification.
3.2.4. KEGG Annotation
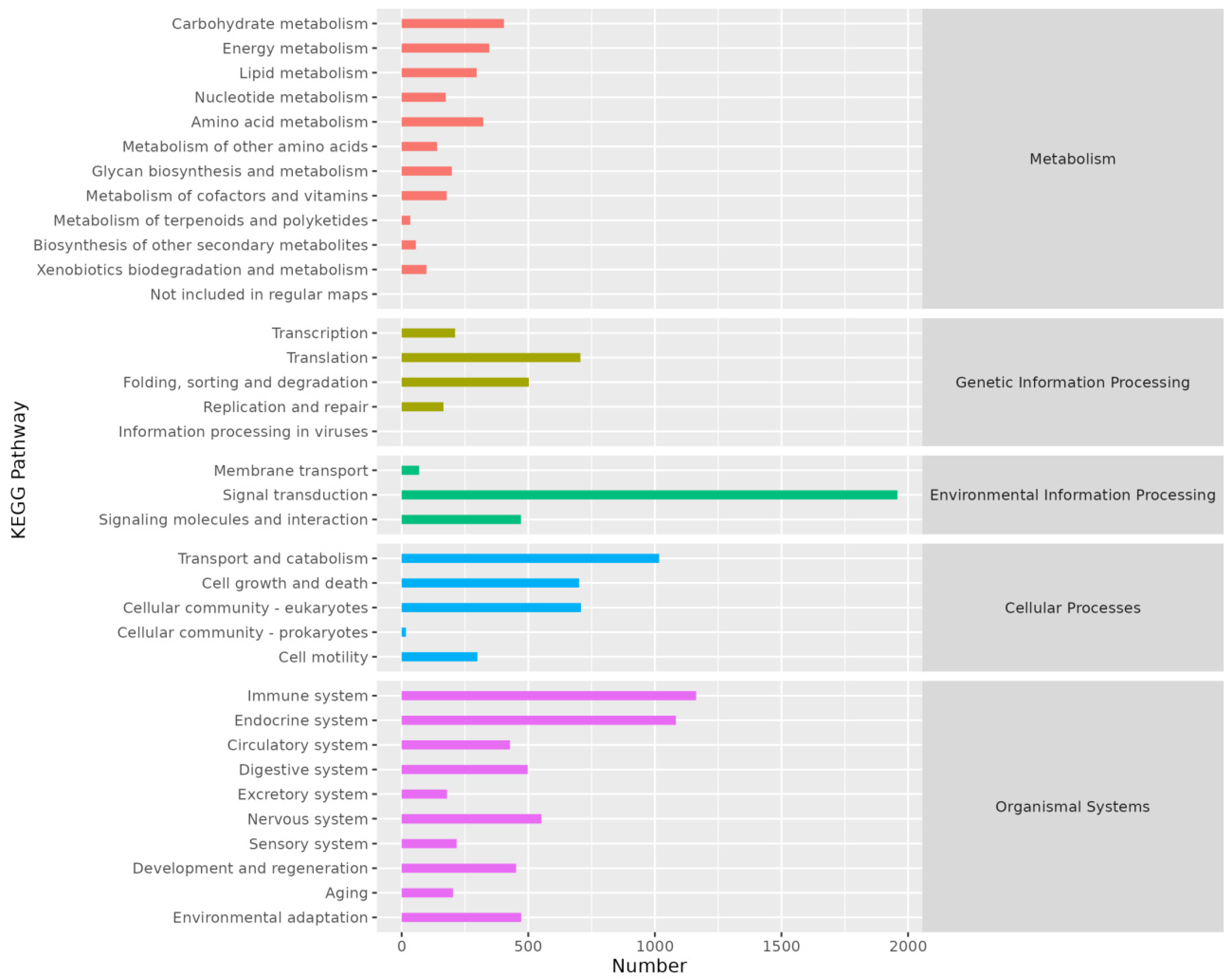
KEGG annotation results indicated that genes were primarily annotated in five pathways: Metabolism, Genetic Information Processing, Environmental Information Processing, Cellular Processes, and Organismal Systems (Figure 4).
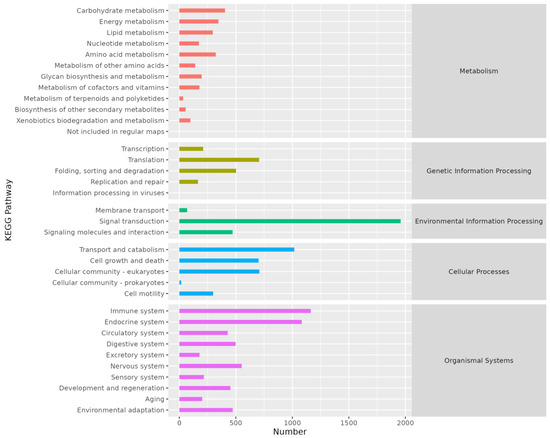
Figure 4.
Histogram of KEGG annotation. The abscissa is the number of genes annotated on KEGG pathway and the ordinate is the name of KEGG pathway.
3.2.5. EggNOG Annotation
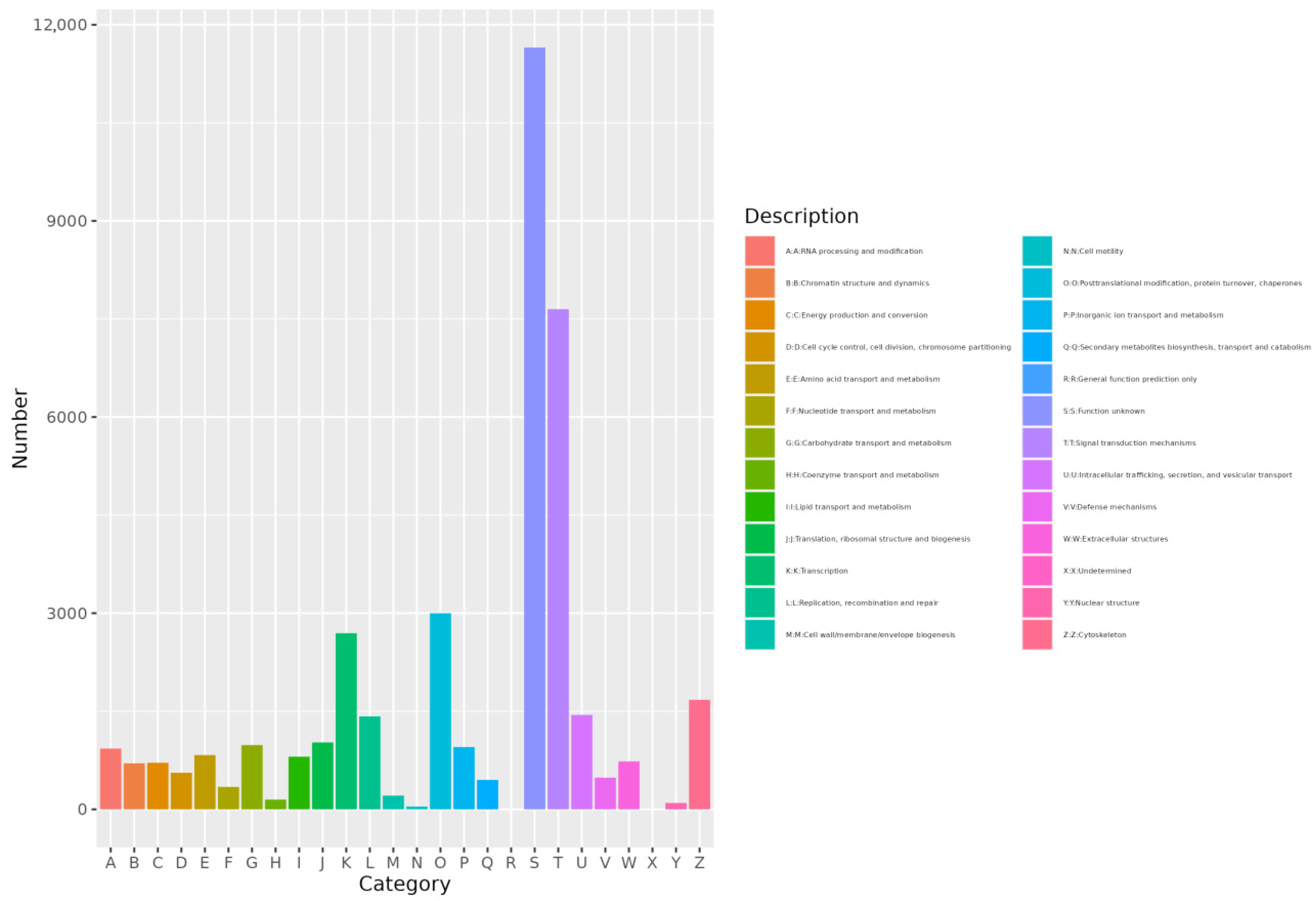
EggNOG annotation results showed that gene annotations were distributed across 24 categories. The category with the highest number of annotated genes was Signal transduction mechanisms (category T). Other notable categories included Posttranslational modification, protein turnover, and chaperones (category O) with 2999 annotations, and transcription (category K) with 2694 annotations (Figure 5).
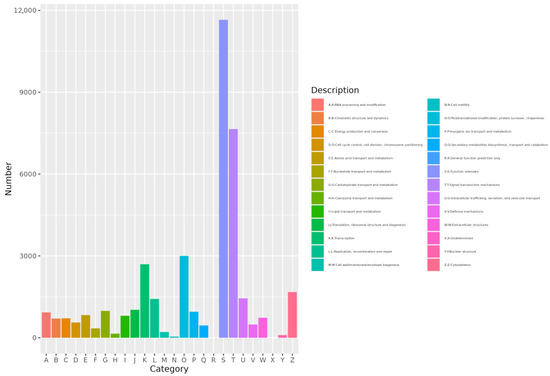
Figure 5.
Histogram of eggNOG. The abscissa is category ID of eggNOG and the ordinate is the number of eggNOG category.
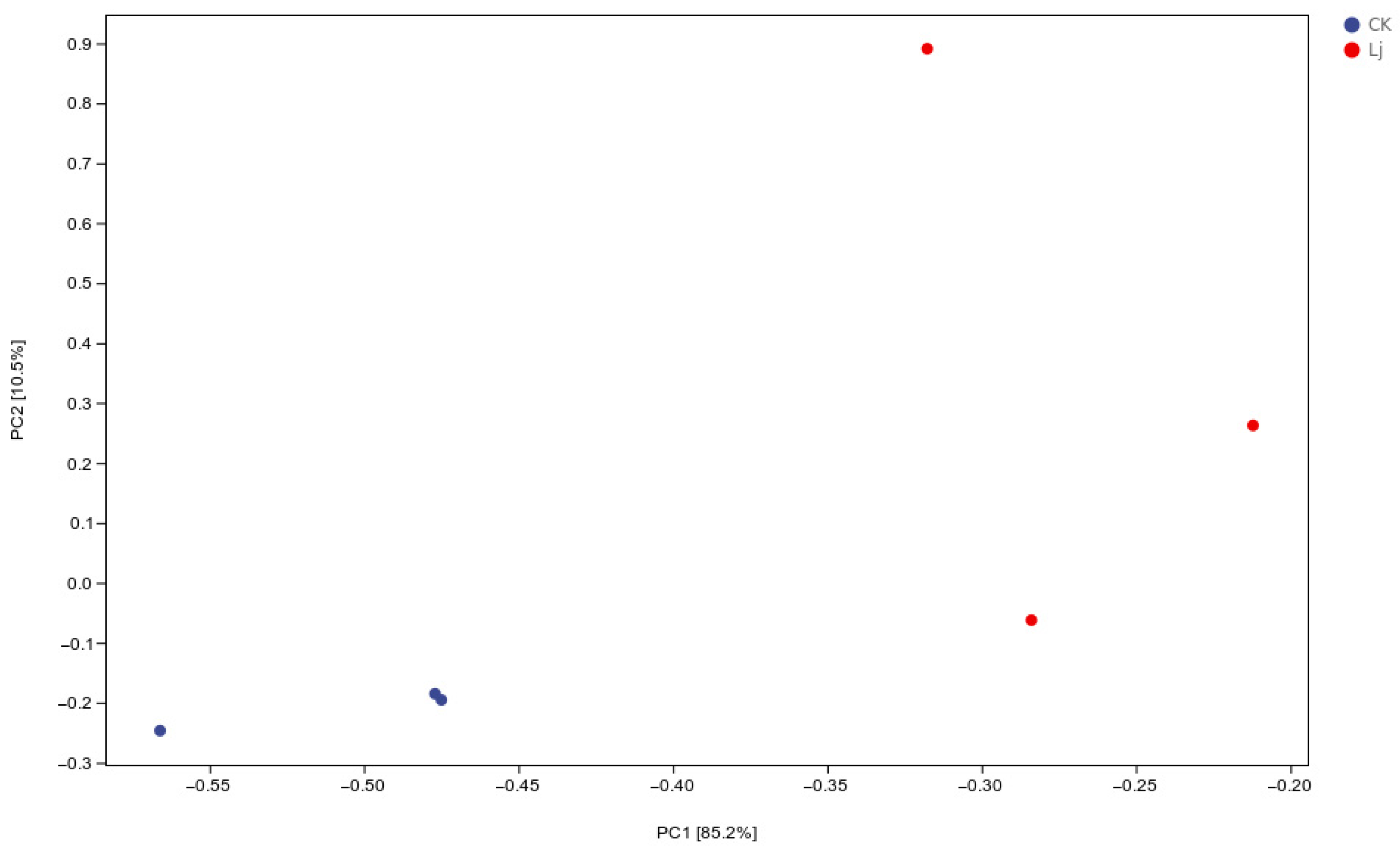
3.3. Analysis of PCA
Principal Component Analysis (PCA) was employed to group similar samples, with closer distances indicating higher similarity. The treatment and control groups were distinctly separated, demonstrating that the early administration of L. japonica extract significantly impacted inflammation (Figure 6).
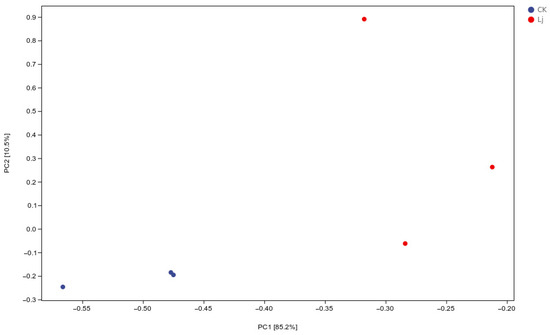
Figure 6.
Scatter plot of PCA analysis. In the two-dimensional scatter plot, the horizontal and vertical coordinates are represent principal component 1 (PC1) and principal component 2 (PC2), respectively. Every dot symbolized a sample, while the various hues of the dots stand for different groups. The closer the dots were, the greater the similarity between the samples.
3.4. Analysis of Difference
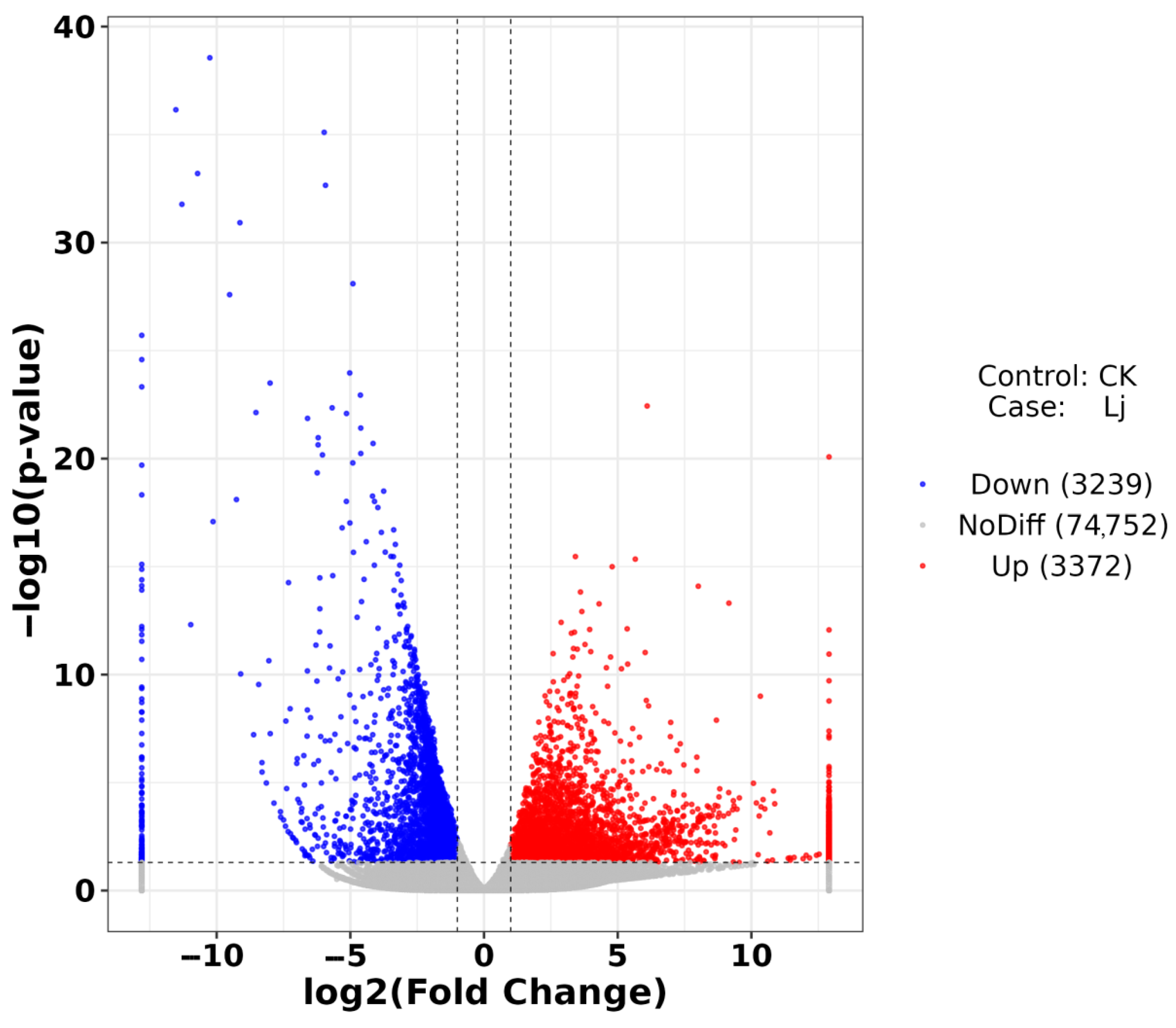
A total of 6611 DEGs were identified, comprising 3372 up-regulated and 3239 down-regulated genes (Figure 7). Up-regulated genes exhibited increased expression levels, while down-regulated genes showed decreased expression levels. Based on these findings, enrichment analysis was conducted to elucidate the functions and interactions of these DEGs.
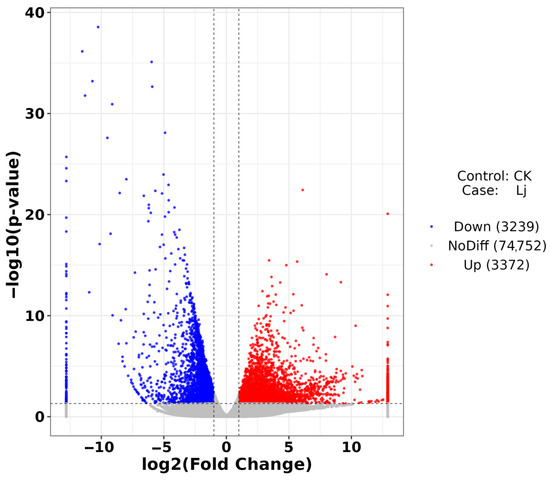
Figure 7.
Volcano plot of DEGs. The abscissa is log2FoldChange and the ordinate is −log10 (p-value). The two vertical dotted lines are two times the difference threshold and the horizontal dotted lines are p-value = 0.05. Red dots indicate up-regulated genes, blue dots indicate down-regulated genes, and gray dots indicate non-significant DEGs.
3.5. Enrichment Analysis of DEGs
3.5.1. GO Enrichment Analysis
GO enrichment analysis was conducted to assess whether differentially regulated genes were significantly clustered within three main categories: cellular component, molecular function, and biological process.
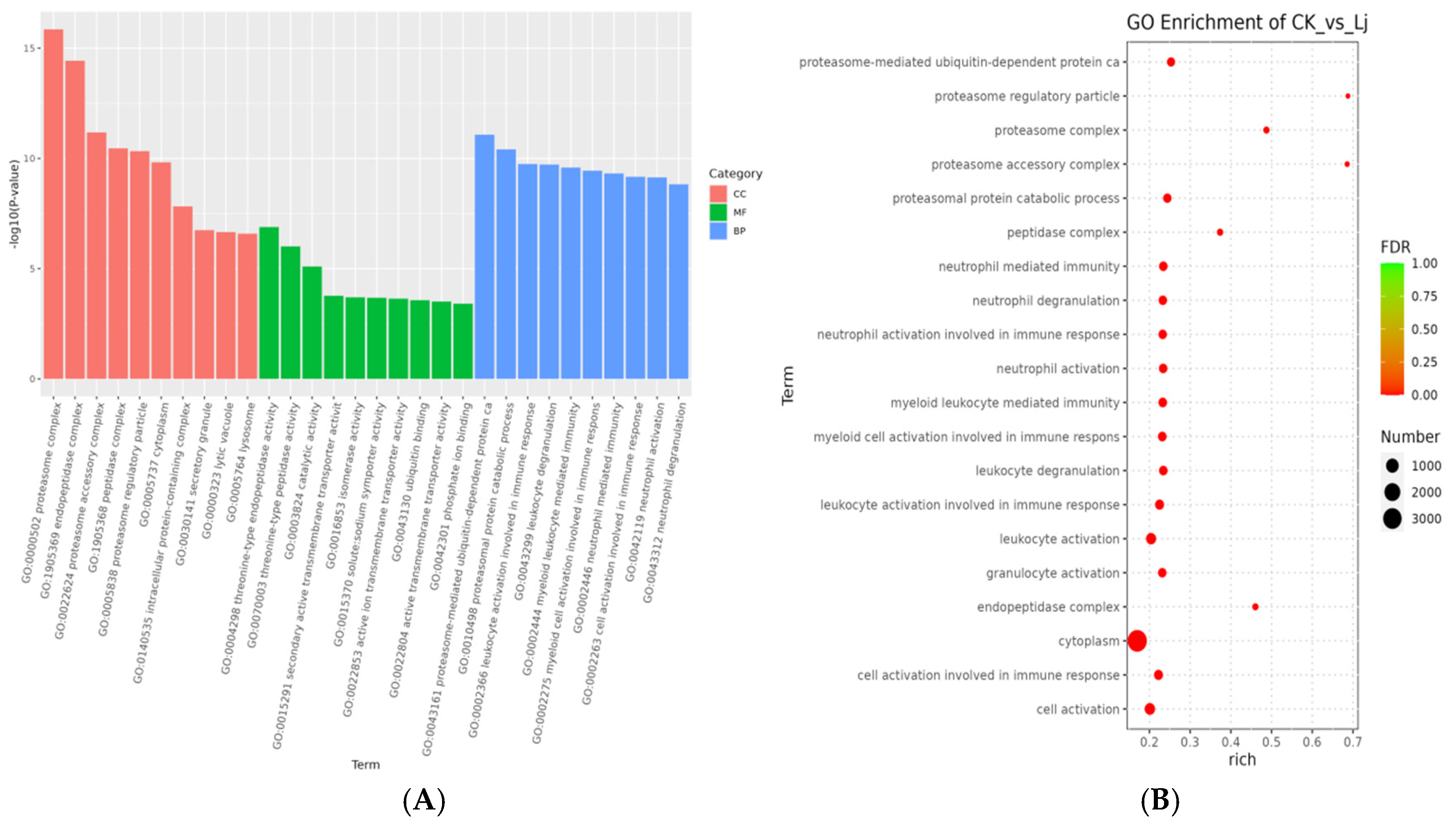
The top ten GO terms with the most significant enrichment in CC included proteasome complex (GO:0000502), endopeptidase complex (GO:1905369), proteasome accessory complex (GO:0022624), peptidase complex (GO:1905368), proteasome regulatory particle (GO:0005838), cytoplasm (GO:0005737), intracellular protein-containing complex (GO:0140535), secretory granule (GO:0030141), lytic vacuole (GO:0000323), and lysosome (GO:0005764). In MF, the top ten terms were threonine-type endopeptidase activity (GO:0004298), threonine-type peptidase activity (GO:0070003), catalytic activity (GO:0003824), secondary active transmembrane transporter activity (GO:0015291), isomerase activity (GO:0016853), solute:sodium symporter activity (GO:0015370), active ion transmembrane transporter activity (GO:0022853), ubiquitin binding (GO:0043130), active transmembrane transporter activity (GO:0022804), and phosphate ion binding (GO:0042301). In BP, the top ten terms were proteasome-mediated ubiquitin-dependent protein catabolism (GO:0043161), proteasomal protein catabolic process (GO:0010498), leukocyte activation involved in immune response (GO:0002366), leukocyte degranulation (GO:0043299), myeloid leukocyte-mediated immunity (GO:0002444), myeloid cell activation involved in immune response (GO:0002275), neutrophil-mediated immunity (GO:0002446), cell activation involved in immune response (GO:0002263), neutrophil activation (GO:0042119), and neutrophil degranulation (GO:0043312) (Figure 8).
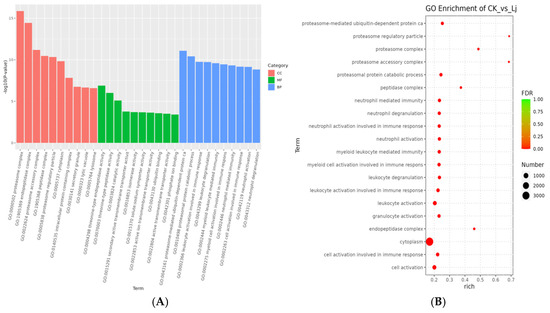
Figure 8.
GO enrichment analysis. (A) Histogram of GO enrichment analysis. The abscissa is term of GO level2 and the ordinate is −log10 (p-value) of each term is enriched. (B) Bubble diagram of GO enrichment analysis. Rich factor refers to the ratio of the number of DEGs enriched in the GO term to the number of DEGs annotated. The larger the Rich factor, the greater the degree of enrichment. FDR values generally range from 0 to 1, the closer to zero, the more significant enrichment.
Combining the three classifications, the top ten GO terms identified were proteasome complex, endopeptidase complex, proteasome accessory complex, proteasome-mediated ubiquitin-dependent protein catabolism, peptidase complex, proteasomal protein catabolic process, proteasome regulatory particle, cytoplasm, leukocyte activation involved in immune response, and leukocyte degranulation.
The top 10 pathways with significant enrichment of up-regulated genes in the GO database were primarily categorized under Biological Process (BP) and Cellular Component (CC). These pathways, listed in descending order of significance, are cell activation (GO:0001775), leukocyte activation (GO:0045321), leukocyte activation involved in immune response (GO:0002366), cell activation involved in immune response (GO:0002263), myeloid leukocyte-mediated immunity (GO:0002444), myeloid cell activation involved in immune response (GO:0002275), leukocyte degranulation (GO:0043299), apical plasma membrane (GO:0016324), neutrophil-mediated immunity (GO:0002446), and regulated exocytosis (GO:0045055).
Conversely, the top 10 pathways with significant enrichment of down-regulated genes, in descending order, are protein modification by small protein conjugation or removal (GO:0070647), proteasomal protein catabolic process (GO:0010498), proteasome-mediated ubiquitin-dependent protein catabolic process (GO:0043161), intracellular protein-containing complex (GO:0140535), proteasome complex (GO:0000502), endopeptidase complex (GO:1905369), ubiquitin-dependent protein catabolic process (GO:0006511), modification-dependent protein catabolic process (GO:0019941), proteolysis involved in protein catabolic process (GO:0051603), and modification-dependent macromolecule catabolic process (GO:0043632).
3.5.2. KEGG Enrichment Analysis
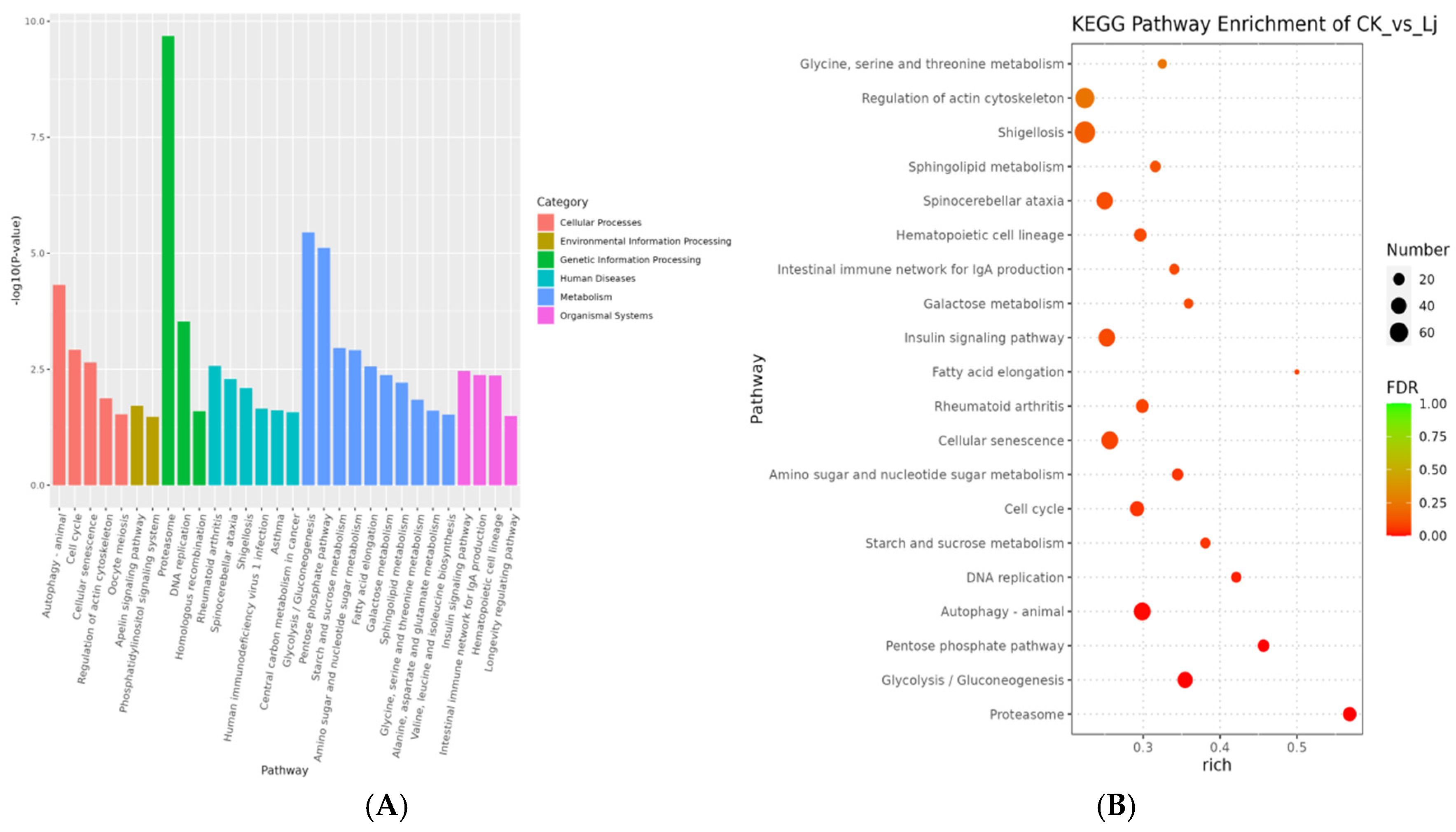
DEGs were primarily enriched in six KEGG categories: cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and organismal systems.
Given that lower p-values indicate more significant enrichment, the 30 pathways with the lowest p-values were selected. These pathways were distributed across the six categories as follows: In cellular processes, significant enrichment was observed in autophagy-animal, cell cycle, cellular senescence, regulation of the actin cytoskeleton, and oocyte meiosis. In environmental information processing, the Apelin signaling pathway and phosphatidylinositol signaling system showed significant enrichment. In genetic information processing, the proteasome, DNA replication, and homologous recombination pathways were significantly enriched. In the human diseases category, rheumatoid arthritis, spinocerebellar ataxia, shigellosis, Human immunodeficiency virus 1 infection, asthma, and central carbon metabolism in cancer exhibited high enrichment. In the metabolism category, significant enrichment was observed in Glycolysis/Gluconeogenesis, the pentose phosphate pathway, starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism, fatty acid elongation, Galactose metabolism, Sphingolipid metabolism, Glycine, serine and threonine metabolism, Alanine, aspartate and glutamate metabolism, and Valine, leucine and isoleucine biosynthesis. Finally, in organismal systems, the insulin signaling pathway, intestinal immune network for IgA production, hematopoietic cell lineage, and longevity regulating pathway showed significant enrichment (Figure 9).
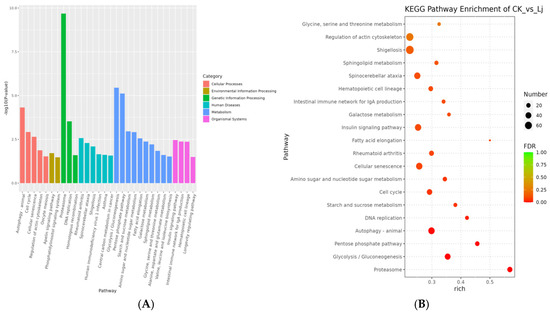
Figure 9.
KEGG pathway enrichment results. (A) Histogram of KEGG pathway enrichment results. The smaller the p-value value, the more significant the enrichment. The abscissa is the name of pathway, and the ordinate is −log10 (p-value) of each path enrichment. (B) Bubble diagram of KEGG enrichment analysis. Rich factor refers to the ratio of the number of DEGs enriched in the KEGG pathway to the number of DEGs annotated. The larger the Rich factor, the greater the degree of enrichment. FDR values generally range from 0 to 1, the closer to zero, the more significant enrichment.
Based on the Rich factor and FDR values, we have further identified the top 20 pathways in the KEGG database that exhibit notable enrichment. These pathways include the proteasome (ko03050), Glycolysis/Gluconeogenesis (ko00010), pentose phosphate pathway (ko00030), autophagy-animal (ko04140), DNA replication (ko03030), starch and sucrose metabolism (ko00500), cell cycle (ko04110), amino sugar and nucleotide sugar metabolism (ko00520), cellular senescence (ko04218), rheumatoid arthritis (ko05323), fatty acid elongation (ko00062), insulin signaling pathway (ko04910), galactose metabolism (ko00052), intestinal immune network for IgA production (ko04672), hematopoietic cell lineage (ko04640), spinocerebellar ataxia (ko05017), sphingolipid metabolism (ko00600), Shigellosis (ko05131), regulation of the actin cytoskeleton (ko04810), and glycine, serine, and threonine metabolism (Figure 10). Furthermore, we have summarized the top 10 KEGG pathway genes that exhibit significant enrichment among both up-regulated and down-regulated genes (Table 1).
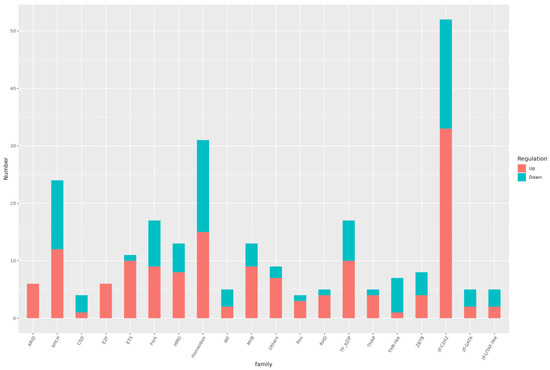
Figure 10.
Histogram of differential TFs. The abscissa is different TF families and the ordinate the number of genes in the TF family.

Table 1.
(A). Up-regulated genes were significantly enriched in the top ten pathways in KEGG. (B). Down-regulated genes were significantly enriched in the top ten pathways in KEGG.
In summary, our analysis revealed several pathways that exhibited significant enrichment and were closely correlated with immune system function, immunological diseases, and infectious diseases. These pathways include rheumatoid arthritis, the intestinal immune network for IgA production, hematopoietic cell lineage, spinocerebellar ataxia, Shigellosis, human immunodeficiency virus 1 infection, and asthma.
3.5.3. KO Analysis
Further analysis of the top 20 KEGG pathways with significant enrichment revealed that several genes were significantly upregulated in the treatment group. These included phosphoglucomutase, glucose-6-phosphatase, hexokinase, ADP-dependent glucokinase, aldose 1-epimerase, glyceraldehyde 3-phosphate dehydrogenase (phosphorylating), multiple inositol-polyphosphate phosphatase/2,3-bisphosphoglycerate 3-phosphatase, phosphoenolpyruvate carboxykinase (GTP), aldehyde dehydrogenase (NAD+), glucose-6-phosphate 1-dehydrogenase, transketolase, ribulose-phosphate 3-epimerase, ribose 5-phosphate isomerase A, phosphopentomutase, phosphoglucomutase, and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha/beta/delta, among others.
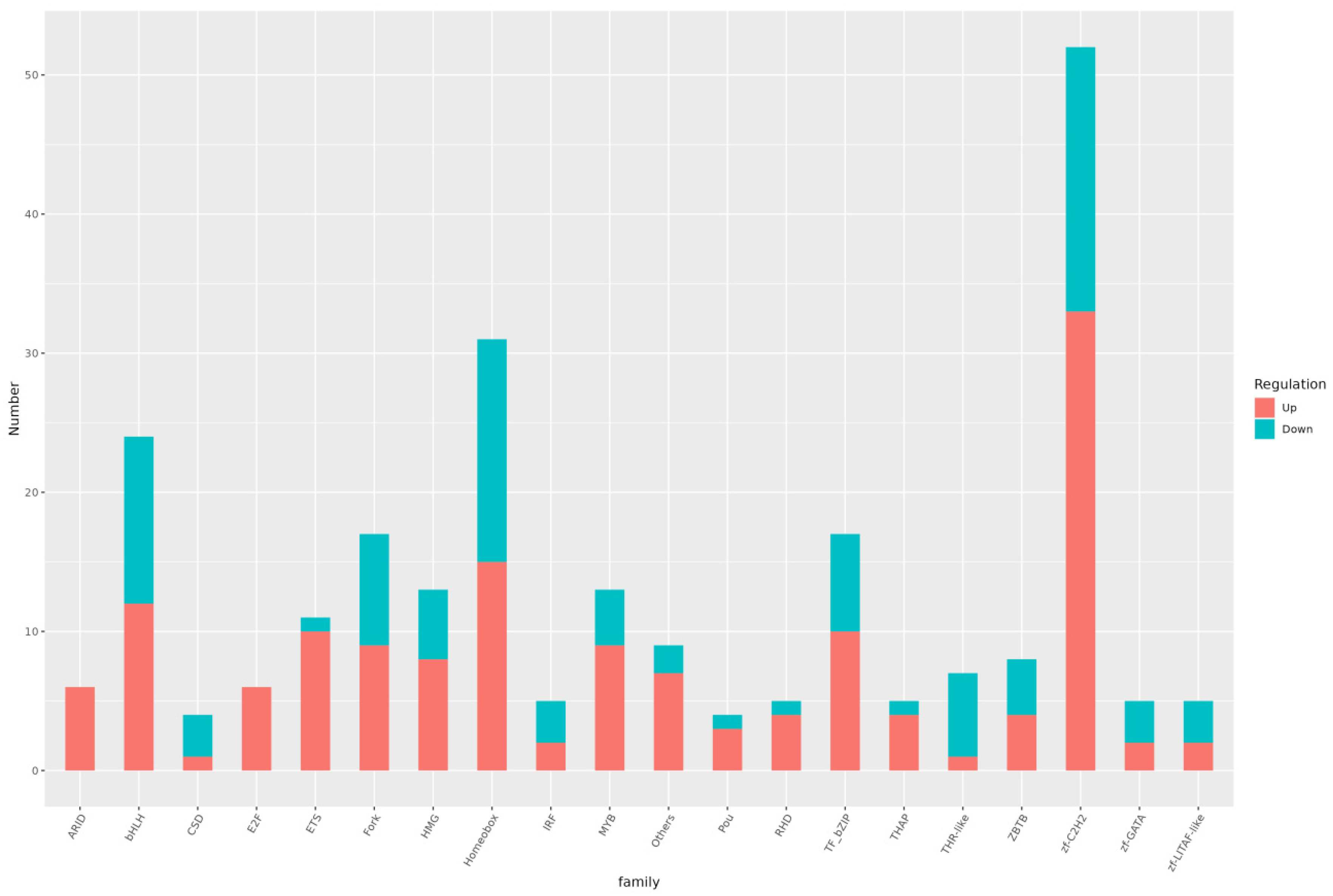
3.6. Differential TF Analysis
Analysis of transcription factors in both the treatment and control groups identified 51 differential transcription factor families. The top 10 families, ranked by the number of transcription factors in descending order, were zf-C2H2, Homeobox, bHLH, TF_bZIP, Fork, MYB, HMG, ETS, Others, and ZBTB (Figure 10).
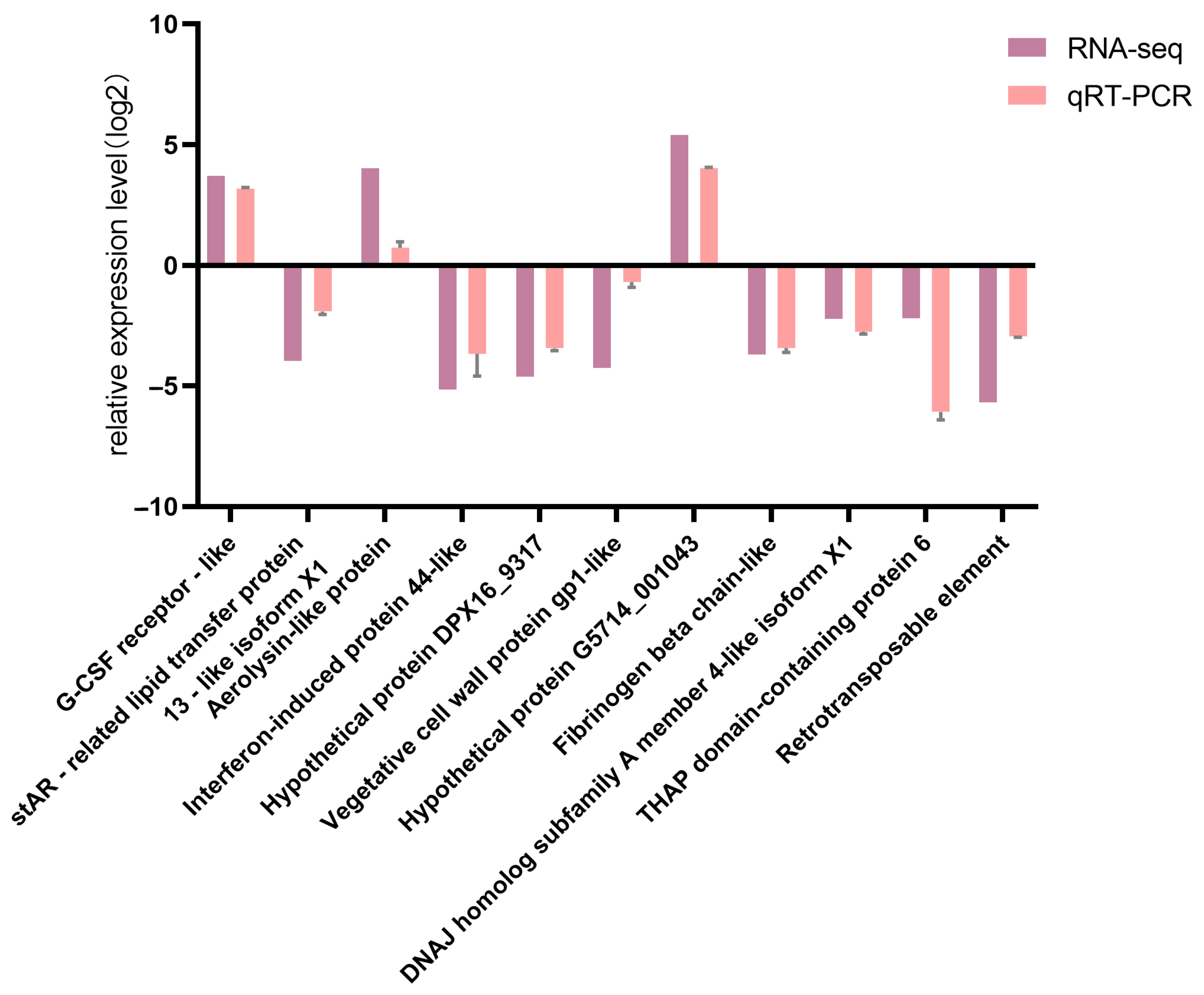
3.7. Validation of DEGs by qRT-PCR
Comparison of qRT-PCR results for 11 DEGs with corresponding RNA-seq data confirmed the high reliability of the RNA-seq sequencing results (Figure 11).
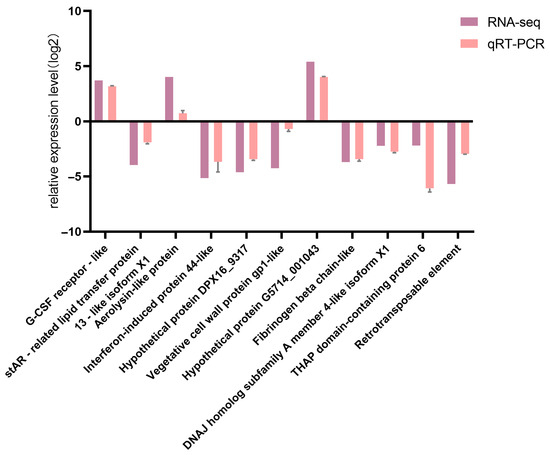
Figure 11.
Comparison between qPCR verification results and RNA-seq results.
4. Discussion
Proteasomes play a crucial role in maintaining intracellular stability and are involved in various cellular processes, including protein quality control, DNA repair, transcriptional regulation, cell cycle regulation, and signal transduction. These processes significantly impact cell proliferation, differentiation, and immune responses [21]. The proteasome pathway is primarily responsible for degrading proteins within cells, making it a key mechanism for the degradation of most target proteins [22,23]. Additionally, the ubiquitin-dependent protein degradation pathway mediated by proteasomes is recognized as a critical mechanism for selective intracellular protein degradation [24]. Numerous studies have demonstrated that proteasomes and their related pathways are essential for transcriptional regulation, protein degradation, and cell cycle progression.
Glycolysis/Gluconeogenesis and the pentose phosphate pathway are essential components of sugar metabolism. These pathways are responsible for metabolizing sugars to generate energy. Based on KEGG pathway analysis, DEGs were highly abundant in the Glycolysis/Gluconeogenesis pathway, indicating that our experimental treatment had a significant impact on energy metabolism.
Autophagy, a fundamental self-defense mechanism, helps maintain cellular homeostasis by promoting the breakdown and recycling of cellular components, thereby enabling cells to tolerate stressful conditions [25]. Furthermore, autophagy is crucial for the immune system’s defense against infectious diseases by regulating and processing invasive pathogens. It also plays an essential role in the onset and treatment of various diseases, including neurological and cardiovascular diseases [26].
Cell growth and development are closely linked to the cell cycle and DNA replication pathways. DNA replication is a fundamental cellular function that ensures the accurate replication of genetic material during cell division, influencing the growth, multiplication, and development of organisms. Many studies have shown that the cell cycle and DNA replication pathways are closely related to cell integrity and normal activity [27,28].
The insulin signaling system is a complex intracellular signal transduction mechanism primarily involved in cell metabolism, division, and survival, as well as other physiological processes [29]. Its main function is to facilitate the absorption and storage of nutrients by tissues and organs, such as glucose uptake. Additionally, the insulin signaling system is involved in the development of some cancers and plays a crucial role in maintaining metabolic balance, initiating anti-apoptotic pathways, and protecting cells from damaging stress.
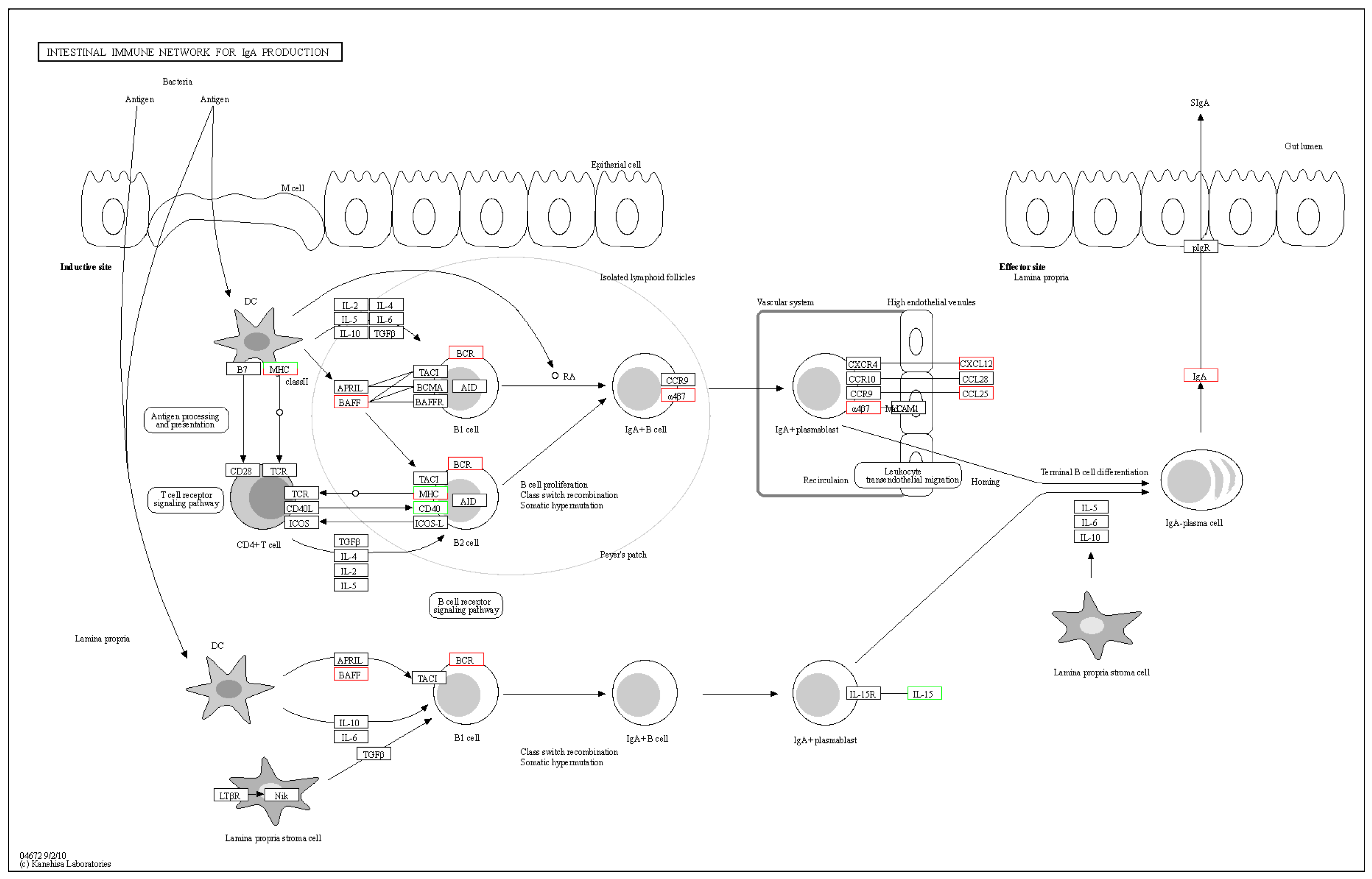
The intestinal immune network for IgA production is a crucial component of the immune system, playing a key role in maintaining intestinal stability. It protects the integrity of the intestinal system and prevents inflammation [30]. Inhibiting this pathway may impair the normal functioning of the intestinal immune system, increasing the risk of intestinal problems and associated diseases [31]. Understanding the intricate mechanisms of the intestinal immune network for IgA production can aid in maintaining intestinal health, preventing and treating intestinal inflammatory diseases, and improving quality of life. As shown in Figure 12, when bacteria invade the body, dendritic cells first identify them and initiate a cascade of immunological reactions. Ultimately, Secretory IgA (SIgA) is found on the surface of the intestinal mucosa. SIgA, a significant antibody of mucosal immunity, primarily prevents pathogenic microorganisms from adhering, protects the mucosa, neutralizes viruses, and stimulates bacterial lysis.
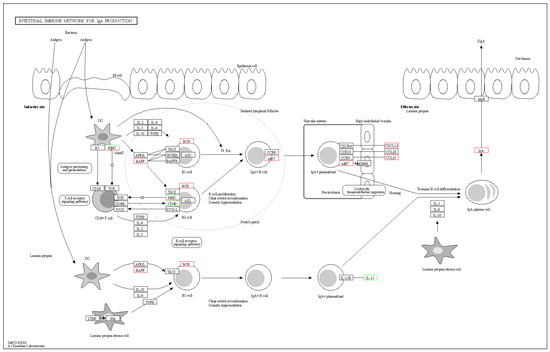
Figure 12.
The pathway of intestinal immune network for IgA production.
ZF-C2H2 is the largest transcription factor family in the animal kingdom, widely distributed across various DNA-binding domains [32]. It is essential for eukaryotic growth, development, and normal function, participating in multiple biological processes [33,34]. ZF-C2H2 transcription factors are involved in numerous biological processes, including gene regulation, cell differentiation, embryonic development, cell homeostasis maintenance, and stress response. These transcription factors play a crucial role in directing cell differentiation and ensuring the proper formation of tissues and organs during critical growth and development phases. Understanding the function and regulatory mechanisms of the ZF-C2H2 transcription factor family provides a scientific basis for exploring organismal growth and development, cell function, and environmental responses.
Evolutionarily conserved homeobox transcription factor families are widely distributed in many organisms and play a significant role in organismal growth and development, hematopoiesis, and tumorigenesis [35,36,37]. Hemapreethi Surendran et al. found that homeobox D10 (Hoxd10) plays a leading role in body development and tissue homeostasis, suggesting that elucidating its function could provide new signaling pathway targets for tumor therapy [38]. Additionally, Cui et al. found that HOXC8 is involved in the proliferation and differentiation of adipocytes, whose primary function is to store and metabolize fat. This study also identified several related genes, providing a basis for further exploration of HOXC8’s role in adipocyte regulation [39]. This research helps elucidate the complex molecular mechanisms of fat cell biology.
5. Conclusions
This study aimed to investigate the effects of L. japonica extract on inflammation through its immunomodulatory properties. We administered L. japonica extract to M. anguillicaudatus to induce an immunological response and subsequently assessed its impact on LPS-induced inflammation. Through transcriptome sequencing, we identified numerous differentially expressed genes (DEGs) and various pathways associated with the immune system, infectious diseases, and other significant biological processes. These findings provide key transcriptomic evidence for the subsequent functional verification of the immune regulatory mechanism of L. japonica and its potential applications in the prevention and treatment of inflammatory diseases. Additionally, this research enhances our understanding of the immune-related genes and pathways in L. japonica and their roles in anti-inflammatory processes.
Simultaneously, this study, by deciphering the molecular basis of inflammatory regulation in M. anguillicaudatus, not only addresses practical issues such as diseases and reproduction in loach aquaculture but also provides a scientific technical pathway and theoretical support for global freshwater fish farming, combining applied value and academic innovation. The mechanistic analysis at the transcriptome level offers theoretical support for the green transformation of the entire aquaculture industry. In the future, the deep industrialization of research achievements can be promoted by integrating synthetic biology technologies to develop functional proteins with high-efficiency expression.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fishes10070333/s1, Table S1: Primer pairs for RT-PCR; Table S2: Sorting and filtering of sequencing data; Table S3: Transcript splicing result; Table S4: The success rate of gene annotation.
Author Contributions
Conceptualization, Q.L.; data curation, Q.L.; formal analysis, Y.Z. and C.W.; funding acquisition, Q.L.; investigation, C.W.; project administration, Q.L.; resources, Q.L.; software, Y.Z.; supervision, Q.L.; writing—original draft, Y.Z.; writing—review and editing, Q.L. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support of this work by the Natural Science Research Project of Anhui Educational Committee (2023AH052098).
Institutional Review Board Statement
The study was conducted approved by the School of Biological and Environmental Engineering, Chaohu University (Approval Code: 2023009; Approval Date: 3 December 2023).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lee, E.J.; Kim, J.S.; Kim, H.P.; Lee, J.-H.; Kang, S.S. Phenolic constituents from the flower buds of Lonicera japonica and their 5-lipoxygenase inhibitory activities. Food Chem. 2010, 120, 134–139. [Google Scholar] [CrossRef]
- Chen, X.; Guo, D.; Gong, X.; Wan, N.; Wu, Z. Optimization of Steam Distillation Process for Volatile Oils from Forsythia suspensa and Lonicera japonica according to the Concept of Quality by Design. Separations 2023, 10, 25. [Google Scholar] [CrossRef]
- Tang, X.; Liu, X.; Zhong, J.; Fang, R. Potential Application of Lonicera japonica Extracts in Animal Production: From the Perspective of Intestinal Health. Front. Microbiol. 2021, 12, 719877. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Zhu, Z.H.; Zhang, L.; Wang, Q.; Xu, M.M.; Lu, C.; Zhu, Y.; Zeng, J.; Duan, J.A.; Zhao, M. Anti-inflammatory property and functional substances of Lonicerae Japonicae Caulis. J. Ethnopharmacol. 2021, 267, 113502. [Google Scholar] [CrossRef]
- Liu, C.; Yin, Z.; Feng, T.; Zhang, M.; Zhou, Z.; Zhou, Y. An integrated network pharmacology and RNA-Seq approach for exploring the preventive effect of Lonicerae japonicae flos on LPS-induced acute lung injury. J. Ethnopharmacol. 2021, 264, 113364. [Google Scholar] [CrossRef]
- Xiao, O.; Li, M.; Chen, D.; Chen, J.; Simal-Gandara, J.; Dai, X.; Kong, Z. The dissipation, processing factors, metabolites, and risk assessment of pesticides in honeysuckle from field to table. J. Hazard. Mater. 2022, 431, 128519. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, Y.; Chao, Z. A Comprehensive Quality Analysis of Different Colors of Medicinal and Edible Honeysuckle. Foods 2023, 12, 3126. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, X.; Li, D.; Qian, K.; Liu, Y.; Xu, T.; Dai, L.; Cheng, J. The transcriptome sequencing analysis reveals immune mechanisms of soybean fermented powder on the loach (Misgurnus anguillicaudatus) in response to Lipopolysaccharide (LPS) infection. Front. Immunol. 2023, 14, 1247038. [Google Scholar] [CrossRef]
- Qin, C.G.; Huang, K.X.; Xu, H.B. Protective effect of polysaccharide from the loach on the in vitro and in vivo peroxidative damage of hepatocyte. J. Nutr. Biochem. 2002, 13, 592–597. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Gao, Z.X.; Luo, S.S.; Su, J.X.; Yi, S.K. Evidence for the Growth Superiority and Delayed Ovarian Development in Tetraploid Loach Misgurnus anguillicaudatus. Fishes 2022, 7, 322. [Google Scholar] [CrossRef]
- Ma, B.H.; Tao, Z.Y.; Wu, Z.B.; Wang, H.H.; Xu, X.D.; Chen, C.Y. Artificial Propagation and Embryonic Development in New Strain of Bigscale Loach Paramisgurnus dabryanus. Jiangxi Fish. Res. Inst. 2020, 6, 1003–1111. [Google Scholar]
- Mao, S.Q.; Yan, J.R.; Xu, P.; Zhang, Y.Y.; Song, L.P.; Hu, B.; Wu, J.; Wang, B.L. Comparative study on body index, nutrient composition, and digestive enzyme activity of Misgurnus anguillicaudatus, Paramisgurnus dabryanus, and Paramisgurnus dabryanus ssp. Isr. J. Aquac. 2022, 74, 1724798. [Google Scholar] [CrossRef]
- Guo, X.; Yu, X.; Zheng, B.; Zhang, L.; Zhang, F.; Zhang, Y.; Li, J.; Pu, G.; Zhang, L.; Wu, H. Network Pharmacology-Based Identification of Potential Targets of Lonicerae japonicae Flos Acting on Anti-Inflammatory Effects. BioMed Res. Int. 2021, 2021, 5507003. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Lin, T.L.; Chang, C.J.; Wu, T.R.; Lai, W.F.; Lu, C.C.; Lai, H.C. Probiotics, prebiotics and amelioration of diseases. J. Biomed. Sci. 2019, 26, 3. [Google Scholar] [CrossRef]
- Shang, X.; Pan, H.; Li, M.; Miao, X.; Ding, H. Lonicera japonica Thunb.: Ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2011, 138, 1–21. [Google Scholar] [CrossRef]
- Kwon, S.H.; Ma, S.X.; Hong, S.I.; Lee, S.Y.; Jang, C.G. Lonicera japonica THUNB. Extract Inhibits Lipopolysaccharide-Stimulated Inflammatory Responses by Suppressing NF-κB Signaling in BV-2 Microglial Cells. J. Med. Food 2015, 18, 762–775. [Google Scholar] [CrossRef]
- Malgorzata-Miller, G.; Heinbockel, L.; Brandenburg, K.; van der Meer, J.W.; Netea, M.G.; Joosten, L.A. Bartonella quintana lipopolysaccharide (LPS): Structure and characteristics of a potent TLR4 antagonist for in-vitro and in-vivo applications. Sci. Rep. 2016, 6, 34221. [Google Scholar] [CrossRef]
- Medina, C.; Royo, J.L. Zebrafish as a model organism to study host-pathogen interactions. Methods 2013, 62, 241–245. [Google Scholar] [CrossRef]
- Pan, M.; Liu, J.; Huang, D.; Guo, Y.; Luo, K.; Yang, M.; Gao, W.; Xu, Q.; Zhang, W.; Mai, K. FoxO3 Modulates LPS-Activated Hepatic Inflammation in Turbot (Scophthalmus maximus L.). Front. Immunol. 2021, 12, 679704. [Google Scholar] [CrossRef]
- Chai, M.; Liu, X.; Wei, L.; Li, J.; Gou, M.; Zhu, T.; Han, Y.; Liu, X. Molecular Characterization of a B Cell Adaptor for Phosphoinositide 3-Kinase Homolog in Lamprey (Lampetra japonica) and Its Function in the Immune Response. Int. J. Mol. Sci. 2022, 23, 14449. [Google Scholar] [CrossRef]
- Pickart, C.M.; Cohen, R.E. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004, 5, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Raffeiner, M.; Zhu, S.; González-Fuente, M.; Üstün, S. Interplay between autophagy and proteasome during protein turnover. Trends Plant Sci. 2023, 28, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, S. Crosstalk between autophagy and proteasome protein degradation systems: Possible implications for cancer therapy. Folia Histochem. Cytobiol. 2013, 51, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Russell, R.C.; Guan, K.L. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41, e110031. [Google Scholar] [CrossRef]
- Yue, A.C.; Zhou, X.D.; Song, H.P.; Liu, X.H.; Bi, M.J.; Han, W.; Li, Q. Effect and molecular mechanism of Sulforaphane alleviates brain damage caused by acute carbon monoxide poisoning: Network pharmacology analysis, molecular docking, and experimental evidence. Environ. Toxicol. 2023, 39, 1140–1162. [Google Scholar] [CrossRef]
- Siddiqui, K.; On, K.F.; Diffley, J.F. Regulating DNA replication in eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a012930. [Google Scholar] [CrossRef]
- Tanaka, S.; Araki, H. Helicase activation and establishment of replication forks at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010371. [Google Scholar] [CrossRef]
- Le, T.K.C.; Dao, X.D.; Nguyen, D.V.; Luu, D.H.; Bui, T.M.H.; Le, T.H.; Nguyen, H.T.; Le, T.N.; Hosaka, T.; Nguyen, T.T.T. Insulin signaling and its application. Front. Endocrinol. 2023, 14, 1226655. [Google Scholar] [CrossRef]
- Miao, Y.; Zhang, Q.; Yuan, Z.; Wang, J.; Xu, Y.; Chai, Y.; Du, M.; Yu, Q.; Zhang, L.; Jiang, Z. Proteomics analysis reveals novel insights into the mechanism of hepatotoxicity induced by Tripterygium wilfordii multiglycoside in mice. Front. Pharmacol. 2022, 13, 1032741. [Google Scholar] [CrossRef]
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Yao, Z.; Kong, J.; Zhang, X.; Li, H.; Chen, W.; Xie, Q. Transcriptomic analysis reveals the dynamic changes of transcription factors during early development of chicken embryo. BMC Genom. 2022, 23, 825. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Chen, Z.; Du, X.; Huang, Y.; Qin, J.; Wen, L.; Pan, X.; Lin, Y. SMRT Sequencing Reveals Candidate Genes and Pathways With Medicinal Value in Cipangopaludina chinensis. Front. Genet. 2022, 13, 881952. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, Y.; Peng, X.; Bo, L.; Wang, Z.; Song, Q. Characterization of cadmium-responsive transcription factors in wolf spider Pardosa pseudoannulata. Chemosphere 2021, 268, 129239. [Google Scholar] [CrossRef]
- Yu, M.; Zhan, J.; Zhang, H. HOX family transcription factors: Related signaling pathways and post-translational modifications in cancer. Cell Signal. 2020, 66, 109469. [Google Scholar] [CrossRef]
- Gorski, D.H.; Walsh, K. Control of vascular cell differentiation by homeobox transcription factors. Trends Cardiovasc. Med. 2003, 13, 213–220. [Google Scholar] [CrossRef]
- Parrillo, L.; Spinelli, R.; Longo, M.; Zatterale, F.; Santamaria, G.; Leone, A.; Campitelli, M.; Raciti, G.A.; Beguinot, F. The Transcription Factor HOXA5: Novel Insights into Metabolic Diseases and Adipose Tissue Dysfunction. Cells 2023, 12, 2090. [Google Scholar] [CrossRef]
- Surendran, H.; Palaniyandi, T.; Natarajan, S.; Hari, R.; Viwanathan, S.; Baskar, G.; Wahab, M.R.A.; Ravi, M.; Rajendran, B.K. Role of homeobox d10 gene targeted signaling pathways in cancers. Pathol. Res. Pract. 2023, 248, 154643. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Q.; Wang, H.; Zhang, X.; Tian, M.; Liu, D.; Yang, X. Effects of HOXC8 on the Proliferation and Differentiation of Porcine Preadipocytes. Animals 2023, 13, 2615. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).