
Bio-Based Polyurethane Composites from Macauba Kernel Oil: Part 1, Matrix Synthesis from Glycerol-Based Polyol


 ,
, 
 and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
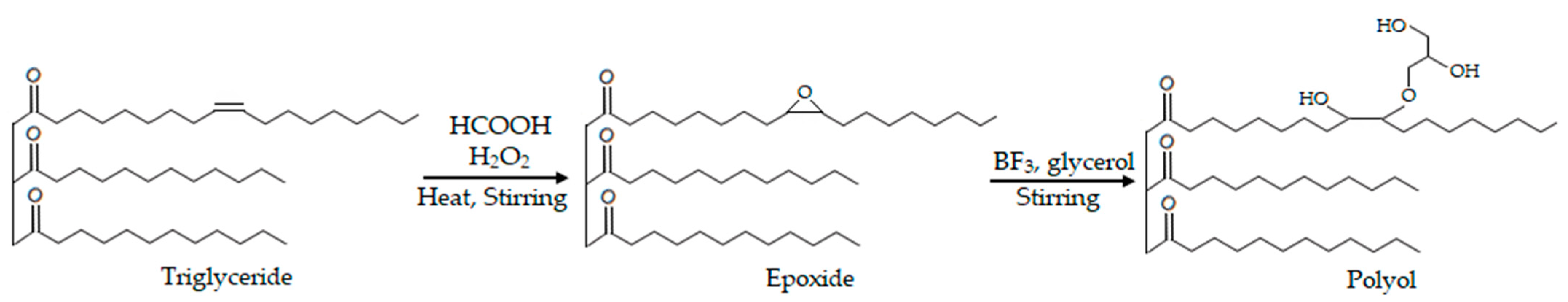
2.2. Epoxide Synthesis
2.3. Polyol Synthesis
2.4. Polyol Hydroxyl Index
2.5. Polyurethane Synthesis
2.6. Fourier-Transform Infrared Spectroscopy
2.7. Thermogravimetric Analysis
2.8. Dynamic Mechanical Analysis
3. Results and Discussion
3.1. Vegetable Oil Molar Mass and Degree of Unsaturation
3.2. Hydroxyl Index
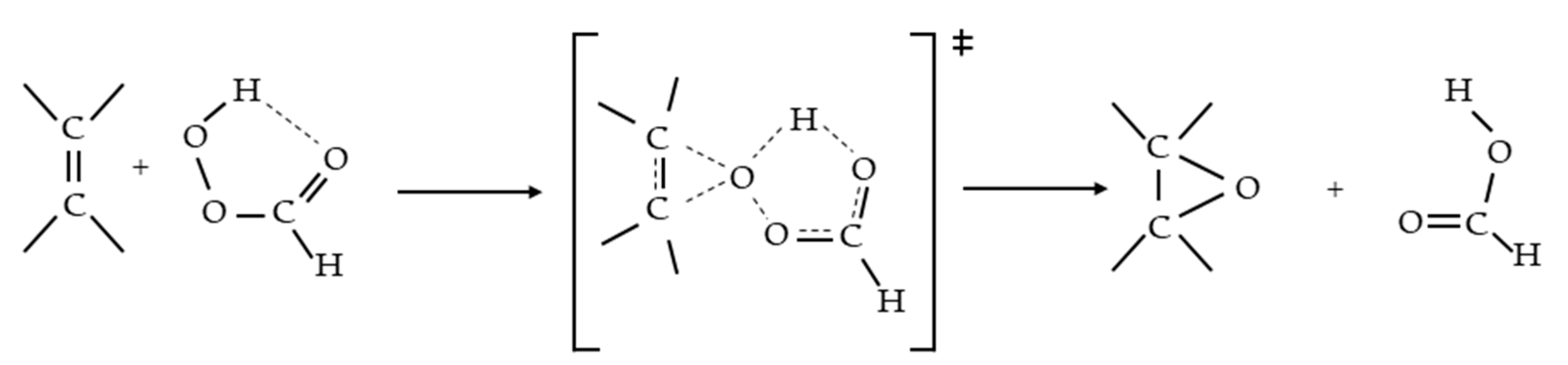
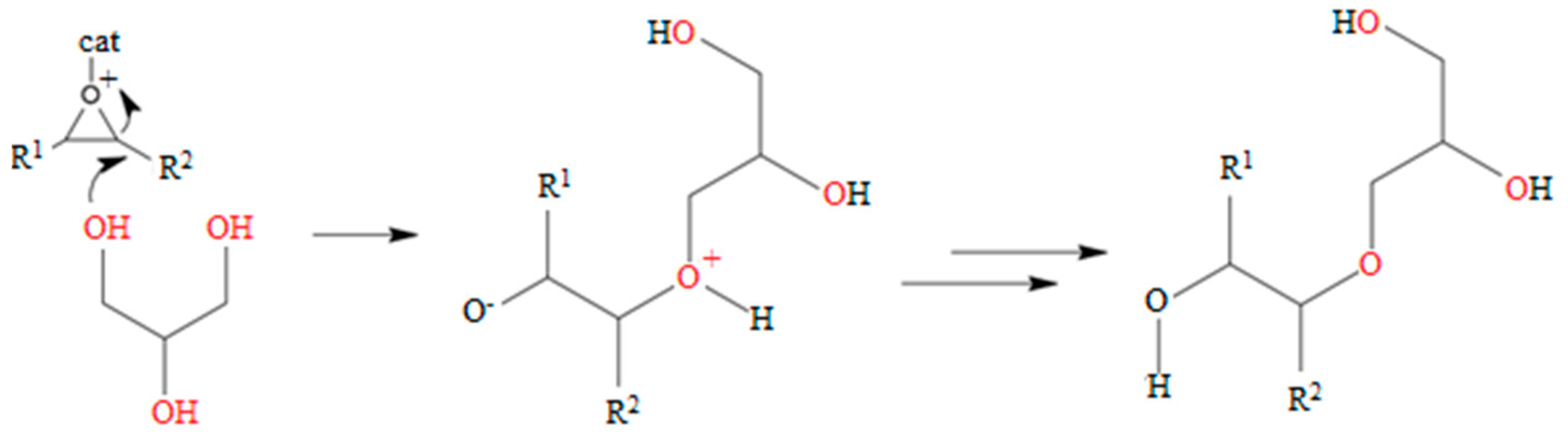
3.3. Reaction Mechanism
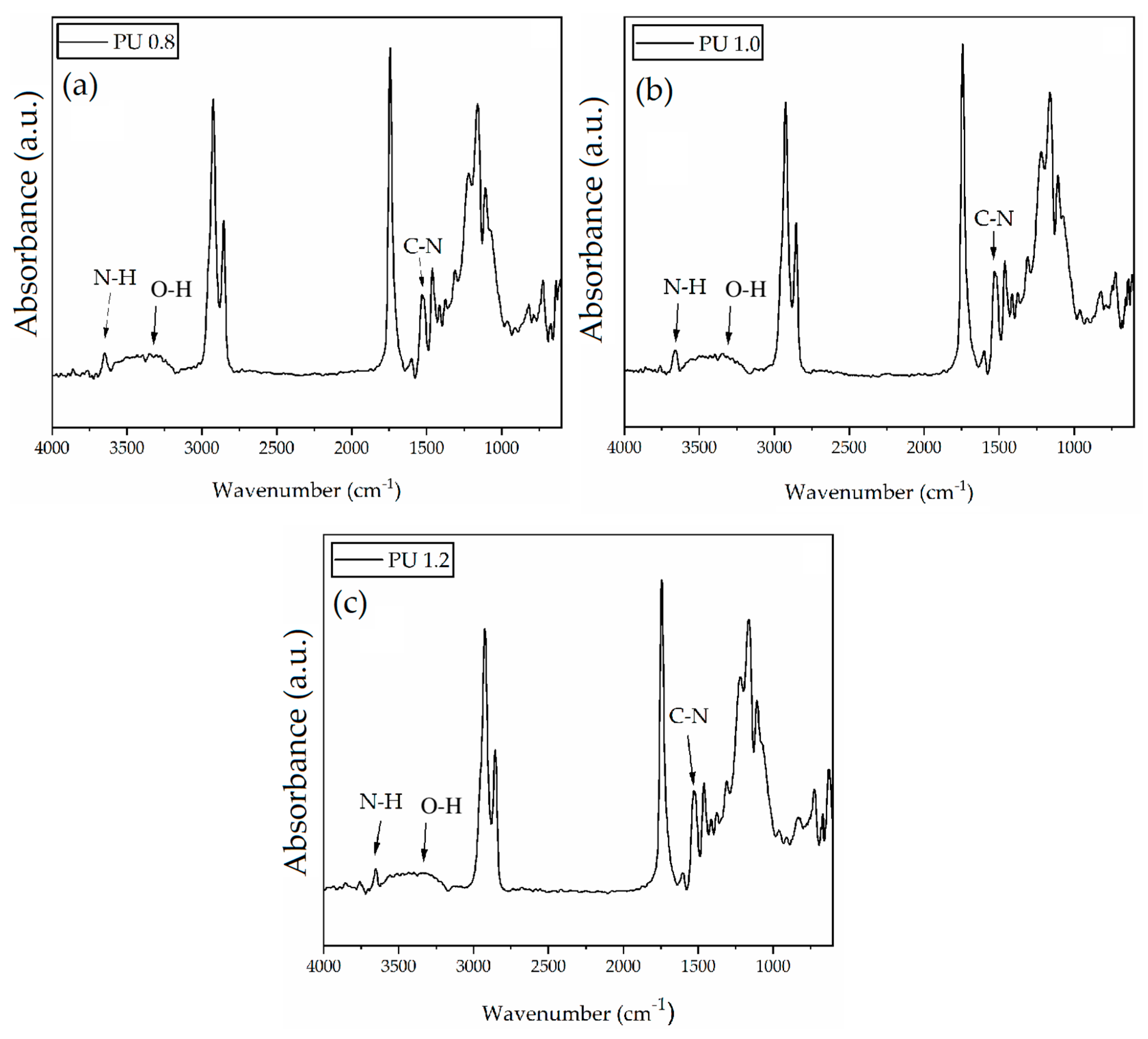
3.4. Fourier-Transform Infrared Spectroscopy
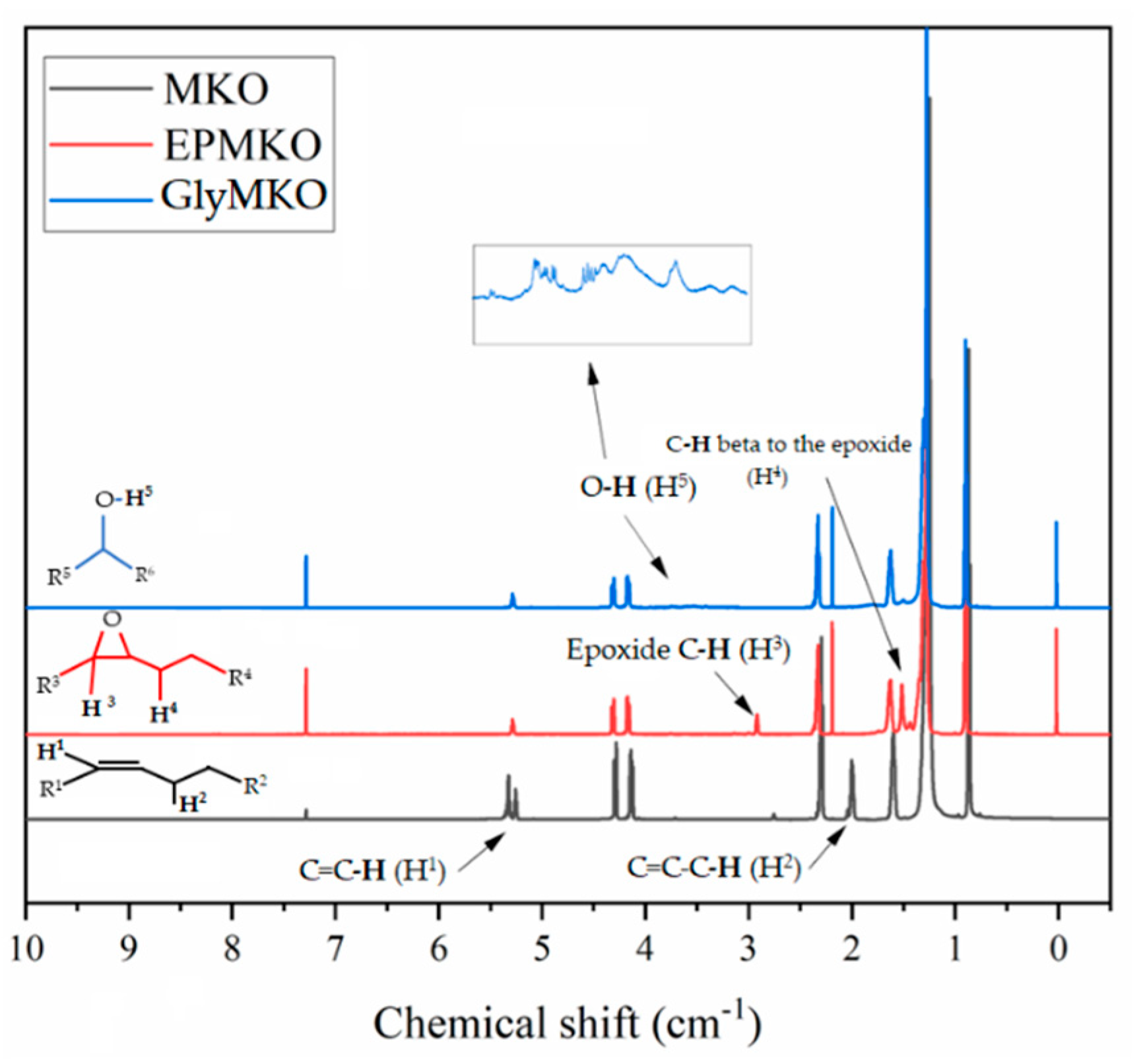
3.5. Proton Nuclear Magnetic Resonance
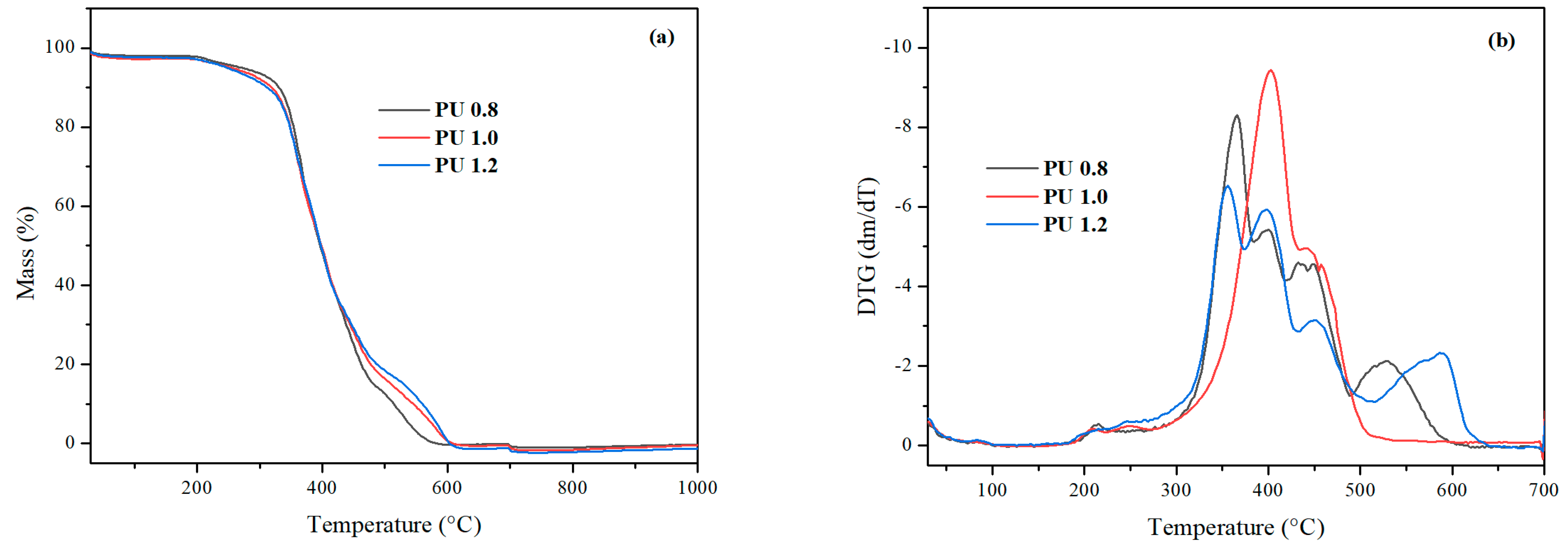
3.6. Thermogravimetric Analysis
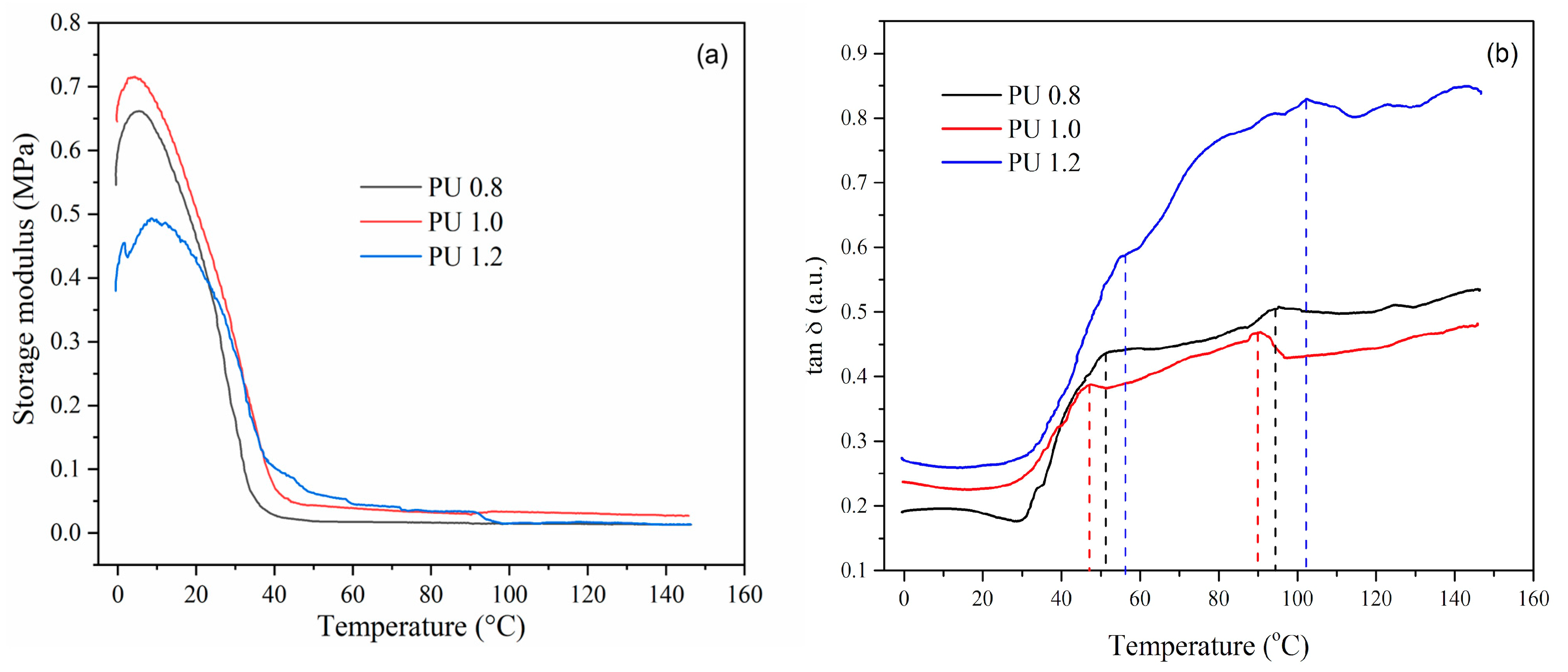
3.7. Dynamical Mechanical Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendiburu-Valor, E.; Calvo-Correas, T.; Martin, L.; Harismendy, I.; Peña-Rodriguez, C.; Eceiza, A. Synthesis and Characterization of Sustainable Polyurethanes from Renewable and Recycled Feedstocks. J. Clean. Prod. 2023, 400, 136749. [Google Scholar] [CrossRef]
- Bishop, G.; Styles, D.; Lens, P.N.L. Environmental Performance Comparison of Bioplastics and Petrochemical Plastics: A Review of Life Cycle Assessment (LCA) Methodological Decisions. Resour. Conserv. Recycl. 2021, 168, 105451. [Google Scholar] [CrossRef]
- Pham, T.Q.; Longing, S.; Siebecker, M.G. Consumption and Degradation of Different Consumer Plastics by Mealworms (Tenebrio Molitor): Effects of Plastic Type, Time, and Mealworm Origin. J. Clean. Prod. 2023, 403, 136842. [Google Scholar] [CrossRef]
- Borowicz, M.; Paciorek-Sadowska, J.; Isbrandt, M. Synthesis and Application of New Bio-Polyols Based on Mustard Oil for the Production of Selected Polyurethane Materials. Ind. Crops Prod. 2020, 155, 112831. [Google Scholar] [CrossRef]
- Datta, J.; Głowińska, E. Chemical Modifications of Natural Oils and Examples of Their Usage for Polyurethane Synthesis. J. Elastomers Plast. 2014, 46, 33–42. [Google Scholar] [CrossRef]
- Narine, S.; Kong, X.; Bouzidi, L.; Sporns, P. Physical Properties of Polyurethanes Produced from Polyols from Seed Oils: I. Elastomers. JAOCS J. Am. Oil Chem. Soc. 2007, 84, 55–63. [Google Scholar] [CrossRef]
- Garrison, T.F.; Kessler, M.R. 3—Plant Oil-Based Polyurethanes. In Bio-Based Plant Oil Polymers and Composites; Madbouly, S.A., Zhang, C., Kessler, M.R., Eds.; William Andrew Publishing: Norwich, NY, USA, 2016; pp. 37–54. ISBN 978-0-323-35833-0. [Google Scholar]
- da Silva César, A.; de Azedias Almeida, F.; de Souza, R.P.; Silva, G.C.; Atabani, A.E. The Prospects of Using Acrocomia Aculeata (Macaúba) a Non-Edible Biodiesel Feedstock in Brazil. Renew. Sustain. Energy Rev. 2015, 49, 1213–1220. [Google Scholar] [CrossRef]
- Breves, A.B. Modificação Química Dos Óleos da Polpa e da Amêndoa da Macaúba (Acrocomia aculeata (Jacq.) Lood. ex Mart) Para a Obtenção de Epóxidos. Master’s Thesis, University of Brasília, Brasília, Brazil, 2018. [Google Scholar]
- do Amaral, F.P.; Broetto, F.; Batistella, C.B.; Jorge, S.M.A. Extração e caracterização qualitativa do óleo da polpa e amendoas de frutos de macaúba [Acrocomia aculeata (Jacq) Lodd. ex Mart] coletada na região de botucatu—SP. Energ. NA Agric. 2011, 26, 12–20. [Google Scholar] [CrossRef]
- Coimbra, M.C.; Jorge, N. Proximate Composition of Guariroba (Syagrus Oleracea), Jerivá (Syagrus Romanzoffiana) and Macaúba (Acrocomia Aculeata) Palm Fruits. Food Res. Int. 2011, 44, 2139–2142. [Google Scholar] [CrossRef]
- Hiane, P.A.; Baldasso, P.A.; Marangoni, S.; Macedo, M.L.R. Chemical and Nutritional Evaluation of Kernels of Bocaiuva, Acrocomia Aculeata (Jacq.) Lodd. Food Sci. Technol. 2006, 26, 683–689. [Google Scholar] [CrossRef]
- Silveira, A.L.M. Aproveitamento da Torta Residual Proveniente da Extração do óleo da Amêndoa de Macaúba (Acrocomia aculeata) para Produção de Farinha Destinada à Alimentação Humana. Master’s Thesis, UFMG, Belo Horizonte, Brazil, 30 October 2012. [Google Scholar]
- da Costa Júnior, M.B.; Arouca, C.L.C.; Maciel, M.P.; Aiura, F.S.; de Oliveira Fontes, D.; Rosa, B.O.; de Araújo Lima, C.; Fernandes, I.S. Torta da polpa da macaúba para suínos em terminação. Rev. Bras. Saúde E Produção Anim. 2015, 16, 325–336. [Google Scholar] [CrossRef]
- Magalhães, K.T.; De Sousa Tavares, T.; Nunes, C.A. The Chemical, Thermal and Textural Characterization of Fractions from Macauba Kernel Oil. Food Res. Int. 2020, 130, 108925. [Google Scholar] [CrossRef] [PubMed]
- Ben, Z.Y.; Samsudin, H.; Yhaya, M.F. Glycerol: Its Properties, Polymer Synthesis, and Applications in Starch Based Films. Eur. Polym. J. 2022, 175, 111377. [Google Scholar] [CrossRef]
- Attarbachi, T.; Kingsley, M.D.; Spallina, V. New Trends on Crude Glycerol Purification: A Review. Fuel 2023, 340, 127485. [Google Scholar] [CrossRef]
- Calderon, M.J.P.; Dumancas, G.G.; Gutierrez, C.S.; Lubguban, A.A.; Alguno, A.C.; Malaluan, R.M.; Lubguban, A.A. Producing Polyglycerol Polyester Polyol for Thermoplastic Polyurethane Application: A Novel Valorization of Glycerol, a by-Product of Biodiesel Production. Heliyon 2023, 9, e19491. [Google Scholar] [CrossRef]
- Coutinho, F.M.B.; Delpech, M.C. Degradation Profile of Films Cast from Aqueous Polyurethane Dispersions. Polym. Degrad. Stab. 2000, 70, 49–57. [Google Scholar] [CrossRef]
- Miyake, Y.; Yokomizo, K.; Matsuzaki, N. Rapid Determination of Iodine Value by 1H Nuclear Magnetic Resonance Spectroscopy. J. Am. Oil Chem. Soc. 1998, 75, 15–19. [Google Scholar] [CrossRef]
- Monteavaro, L.L.; da Silva, E.O.; Costa, A.P.O.; Samios, D.; Gerbase, A.E.; Petzhold, C.L. Polyurethane Networks from Formiated Soy Polyols: Synthesis and Mechanical Characterization. J. Am. Oil Chem. Soc. 2005, 82, 365–371. [Google Scholar] [CrossRef]
- ASTM D1193-06; Standard Specification for Reagent Water. ASTM International: West Conshohocken, PA, USA, 2018.
- ASTM D5155; Standard Test Methods for Polyurethane Raw Materials Determination of the Isocyanate Content of Aromatic Isocyanates. ASTM International: West Conshohocken, PA, USA, 2017.
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Ward, I.M.; Sweeney, J. Mechanical Properties of Solid Polymers, 3rd ed.; Wiley-Interscience: New Deli, India, 2013; ISBN 978-0-471-91995-7. [Google Scholar]
- Rubio, M.; Ramírez-Galicia, G.; López-Nava, L.J. Mechanism Formation of Peracids. J. Mol. Struct. THEOCHEM 2005, 726, 261–269. [Google Scholar] [CrossRef]
- Rocha, T.L.A.C.; Schuster, R.H.; Jacobi, M.M.; Samios, D. Estudo da modificação química de polidienos do tipo SBR e BR. Polímeros 2004, 14, 318–321. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Stuart, W. Organic Chemistry, 2nd ed.; Clayden, J., Greeves, N., Warren, S., Eds.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Arjunan, V.; Anitha, R.; Thirunarayanan, S.; Mohan, S. Simulations on the Structure, Vibrations and Electronic Properties of 1,2-Epoxy-3-Phenoxy Propane and 1,2-Epoxy-3-(p-Tolyloxy)Propane by FT–IR, FT–Raman, FT–NMR and DFT Methods. Chem. Data Collect. 2017, 11–12, 139–167. [Google Scholar] [CrossRef]
- Dai, Z.; Jiang, P.; Lou, W.; Zhang, P.; Bao, Y.; Gao, X.; Xia, J.; Haryono, A. Preparation of Degradable Vegetable Oil-Based Waterborne Polyurethane with Tunable Mechanical and Thermal Properties. Eur. Polym. J. 2020, 139, 109994. [Google Scholar] [CrossRef]
- Durand, S.; D’Orlando, A.; Mougnard, L.; Bourmaud, A.; Beaugrand, J. Combining Infrared and Raman Spectra to Assess MDI Localization in Novel Flax-Reinforced Automotive Composites. Compos. Part C Open Access 2023, 12, 100382. [Google Scholar] [CrossRef]
- Erhan, S.; Sharma, B.; Liu, Z.; Adhvaryu, A. Lubricant Base Stock Potential of Chemically Modified Vegetable Oils. J. Agric. Food Chem. 2008, 56, 8919–8925. [Google Scholar] [CrossRef]
- Popescu, R.; Diana, C.; Dinca, O.R.; Marinescu, A.; Stefanescu, I.; Roxana, I. Discrimination of Vegetable Oils Using NMR Spectroscopy and Chemometrics. Food Control 2015, 48, 84–90. [Google Scholar] [CrossRef]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.R. Introdução à Espectroscopia. Tradução Da Quarta Edição Norte Americana, 4th ed.; Cengage Learning: Boston, MA, USA, 2010. [Google Scholar]
- Crawford, D.M.; Bass, R.G.; Haas, T.W. Strain Effects on Thermal Transitions and Mechanical Properties of Thermoplastic Polyurethane Elastomers. Thermochim. Acta 1998, 323, 53–63. [Google Scholar] [CrossRef]
- Saidpour, H.; Razmara, M.; Arunachalam, S. DMA Investigation on Polyurethane. In Proceedings of the International Conference on Fascinating Advancement in Mechanical Engineering (FAME 2008), Mepco Schlenk Engineering College, Sivakasi, India, 11 December 2008. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MKO (g) | Formic Acid (mL) | H2O2 (mL) | |
|---|---|---|---|
| Quantity | 55.55 | 53.57 | 200.19 |
| NCO/OH Ratio | Polyol (g) | MDI (g) |
|---|---|---|
| 0.8 | 10 | 1.71 |
| 1.0 | 10 | 2.14 |
| 1.2 | 10 | 2.57 |
| Molar Mass | Unsaturation Index | |
|---|---|---|
| MKO | 660.03 g·mol−1 | 0.85 |
| Fatty Acid (Cx:y) a | Percentage (%) |
|---|---|
| Lauric acid (C12:0) | 32.58–44.14 |
| Oleic acid (C18:1) | 18.70–36.27 |
| Myristic acid (C14:0) | 8.45–13.60 |
| Palmitic acid (C16:0) | 6.57–9.20 |
| Sample | Tonset | T5 | T10 |
|---|---|---|---|
| PU 0.8 | 339.2 | 272.3 | 326.8 |
| PU 1.0 | 332.3 | 254.6 | 314.3 |
| PU 1.2 | 332.7 | 249.6 | 313.8 |
| Sample | E’ at 25 °C (MPa) | E’ at 100 °C (MPa) | ε (µmol·cm−3) | Tg 1 (°C) | Tg 2 (°C) |
|---|---|---|---|---|---|
| PU 0.8 | 0.30 | 0.012 | 3.87 | 51 | 96 |
| PU 1.0 | 0.36 | 0.030 | 9.67 | 48 | 91 |
| PU 1.2 | 0.39 | 0.024 | 7.74 | 56 | 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade Breves, R.; Ajiola, D.; de Vasconcelos Vieira Lopes, R.; Quirino, R.L.; Colin, B.; Petrissans, A.; Petrissans, M.; Sales, M.J.A. Bio-Based Polyurethane Composites from Macauba Kernel Oil: Part 1, Matrix Synthesis from Glycerol-Based Polyol. J. Compos. Sci. 2024, 8, 363. https://doi.org/10.3390/jcs8090363
Andrade Breves R, Ajiola D, de Vasconcelos Vieira Lopes R, Quirino RL, Colin B, Petrissans A, Petrissans M, Sales MJA. Bio-Based Polyurethane Composites from Macauba Kernel Oil: Part 1, Matrix Synthesis from Glycerol-Based Polyol. Journal of Composites Science. 2024; 8(9):363. https://doi.org/10.3390/jcs8090363
Chicago/Turabian StyleAndrade Breves, Rodolfo, Daniel Ajiola, Roseany de Vasconcelos Vieira Lopes, Rafael L. Quirino, Baptiste Colin, Anelie Petrissans, Mathieu Petrissans, and Maria José Araújo Sales. 2024. "Bio-Based Polyurethane Composites from Macauba Kernel Oil: Part 1, Matrix Synthesis from Glycerol-Based Polyol" Journal of Composites Science 8, no. 9: 363. https://doi.org/10.3390/jcs8090363
APA StyleAndrade Breves, R., Ajiola, D., de Vasconcelos Vieira Lopes, R., Quirino, R. L., Colin, B., Petrissans, A., Petrissans, M., & Sales, M. J. A. (2024). Bio-Based Polyurethane Composites from Macauba Kernel Oil: Part 1, Matrix Synthesis from Glycerol-Based Polyol. Journal of Composites Science, 8(9), 363. https://doi.org/10.3390/jcs8090363