



Pharmaceuticals 2023, 16(5), 640; https://doi.org/10.3390/ph16050640 - 23 Apr 2023
Cited by 13 | Viewed by 5683
Abstract
►
Show Figures
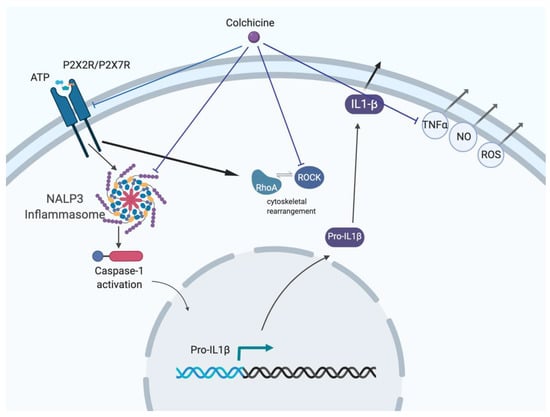
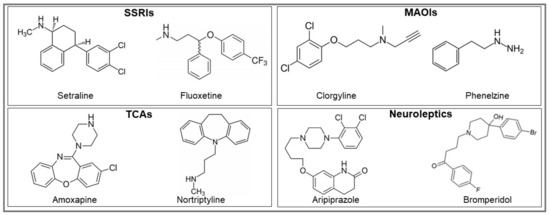
Checkpoint inhibitors can be a highly effective antitumor therapy but only to a subset of patients, presumably due to immunotherapy resistance. Fluoxetine was recently revealed to inhibit the NLRP3 inflammasome, and NLRP3 inhibition could serve as a target for immunotherapy resistance. Therefore, we
[...] Read more.
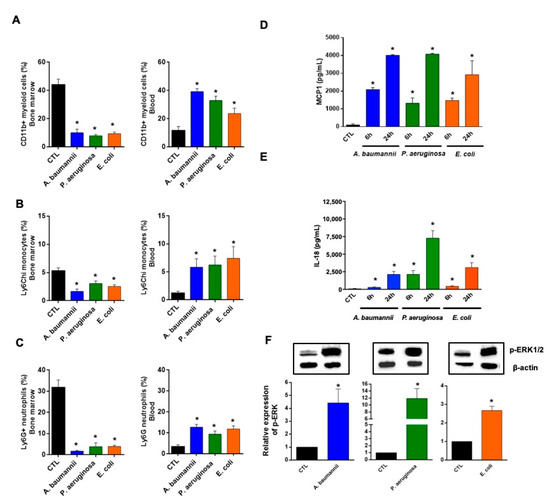
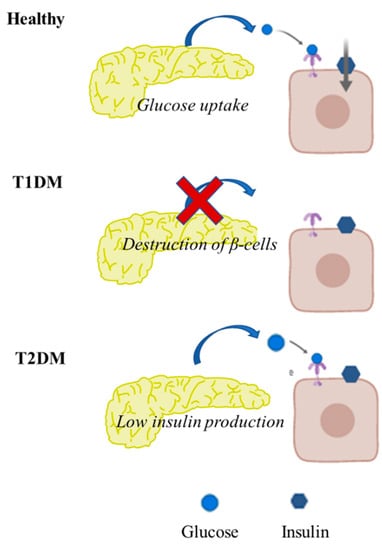
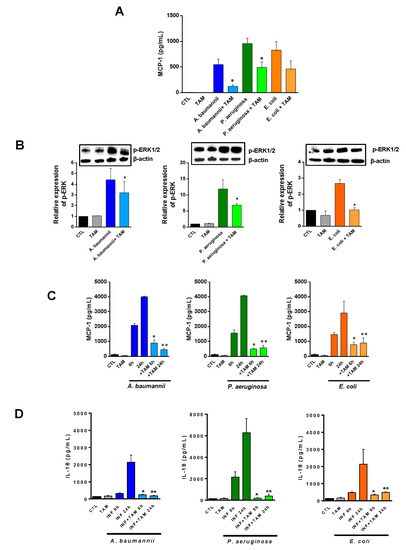
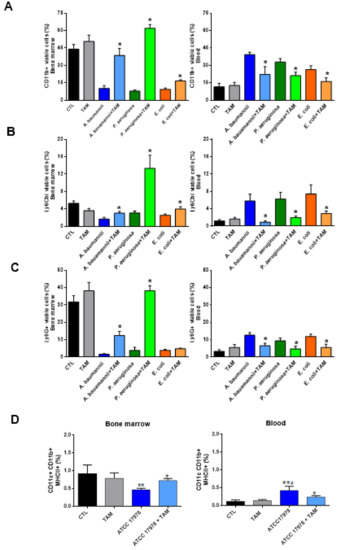
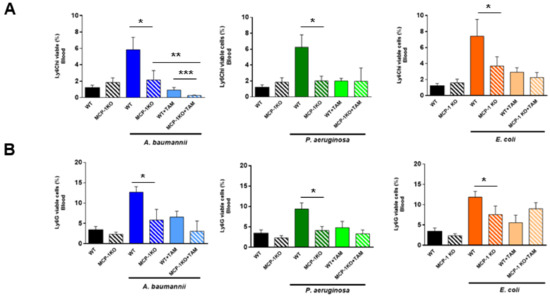
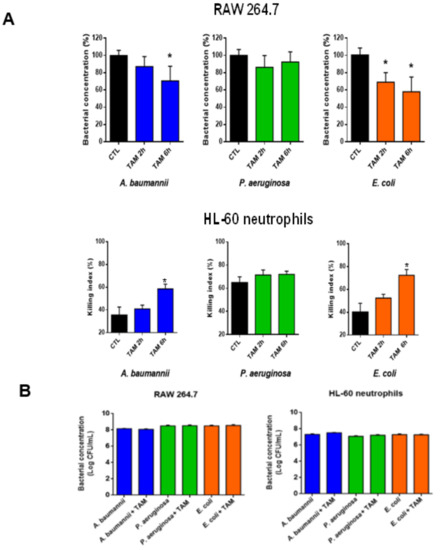
Checkpoint inhibitors can be a highly effective antitumor therapy but only to a subset of patients, presumably due to immunotherapy resistance. Fluoxetine was recently revealed to inhibit the NLRP3 inflammasome, and NLRP3 inhibition could serve as a target for immunotherapy resistance. Therefore, we evaluated the overall survival (OS) in patients with cancer receiving checkpoint inhibitors combined with fluoxetine. A cohort study was conducted among patients diagnosed with lung, throat (pharynx or larynx), skin, or kidney/urinary cancer treated with checkpoint inhibitor therapy. Utilizing the Veterans Affairs Informatics and Computing Infrastructure, patients were retrospectively evaluated during the period from October 2015 to June 2021. The primary outcome was overall survival (OS). Patients were followed until death or the end of the study period. There were 2316 patients evaluated, including 34 patients who were exposed to checkpoint inhibitors and fluoxetine. Propensity score weighted Cox proportional hazards demonstrated a better OS in fluoxetine-exposed patients than unexposed (HR: 0.59, 95% CI 0.371–0.936). This cohort study among cancer patients treated with checkpoint inhibitor therapy showed a significant improvement in the OS when fluoxetine was used. Because of this study’s potential for selection bias, randomized trials are needed to assess the efficacy of the association of fluoxetine or another anti-NLRP3 drug to checkpoint inhibitor therapy.
Full article
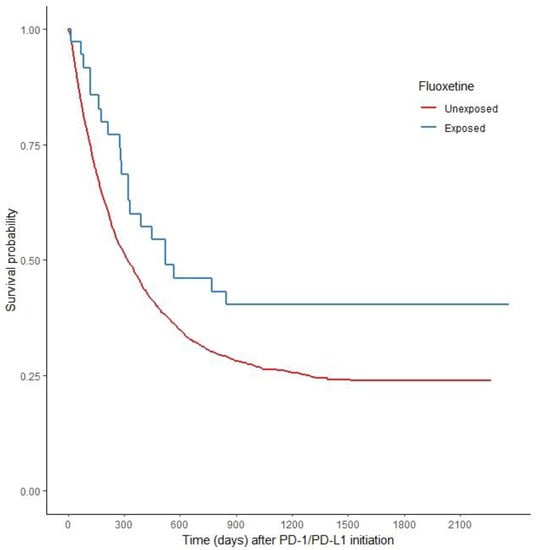
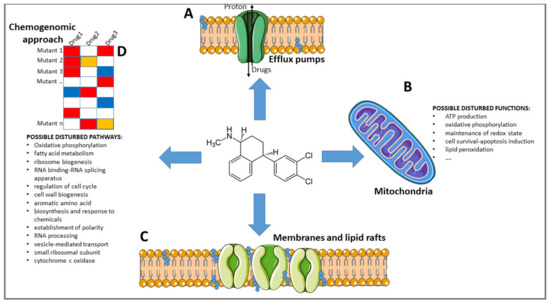
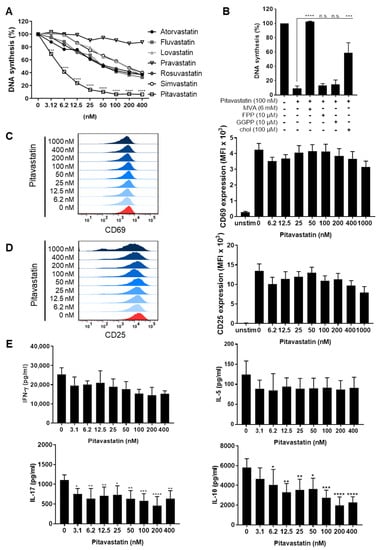
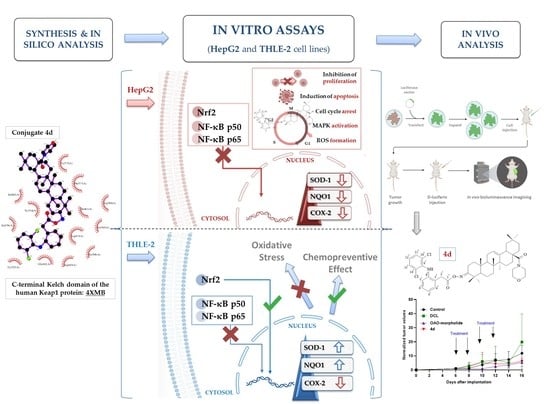
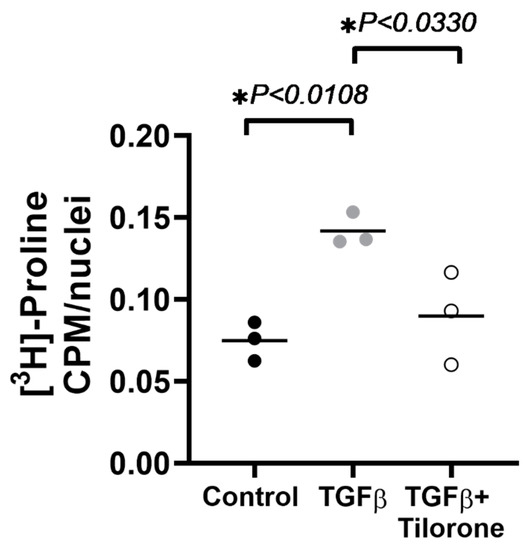
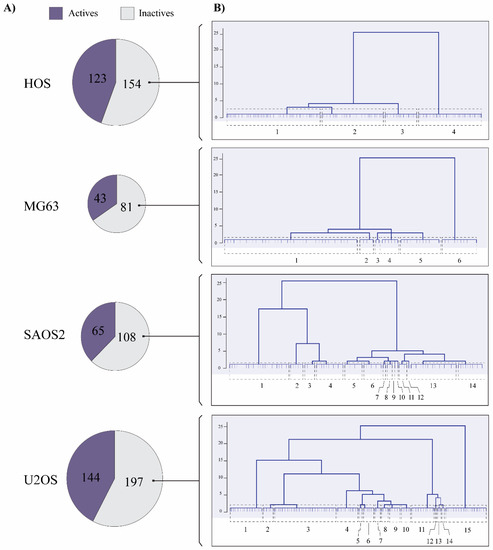
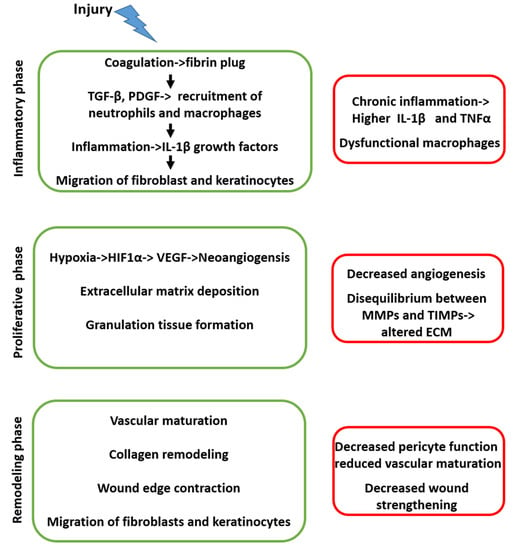
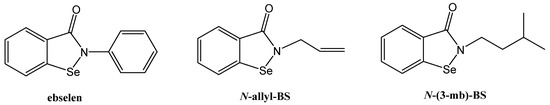
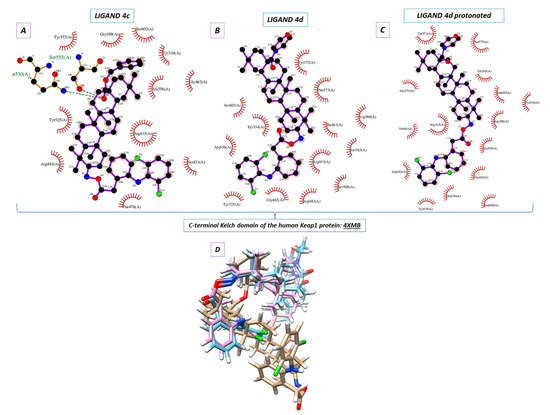
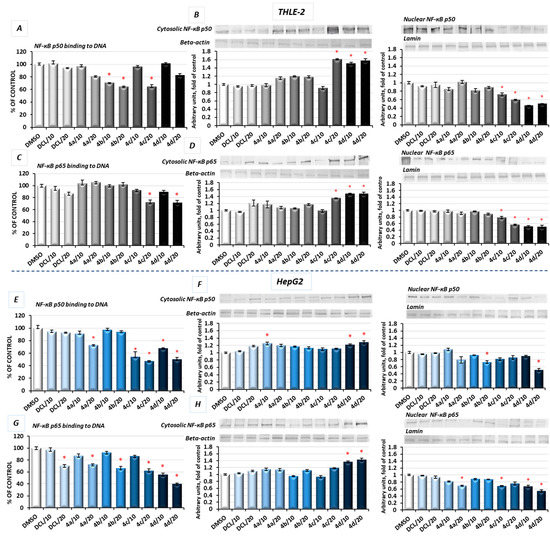
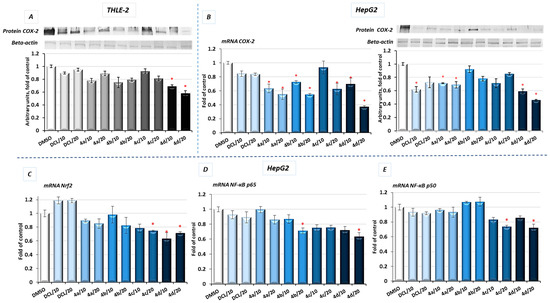
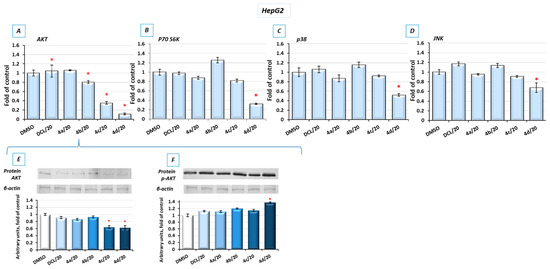
Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}