




Toxics 2025, 13(8), 631; https://doi.org/10.3390/toxics13080631 - 27 Jul 2025
Cited by 1 | Viewed by 1345
Abstract
►
Show Figures
Human milk has been used for over 70 years to monitor pollutants such as polychlorinated biphenyls (PCBs) and organochlorine pesticides (OCPs). Despite the growing body of data, our understanding of the pollutant exposome, particularly co-exposure patterns and their interactions, remains limited. Artificial intelligence
[...] Read more.
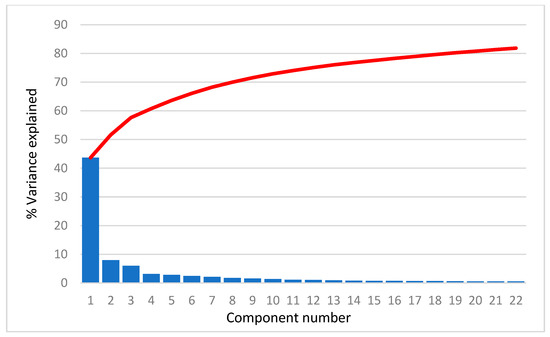
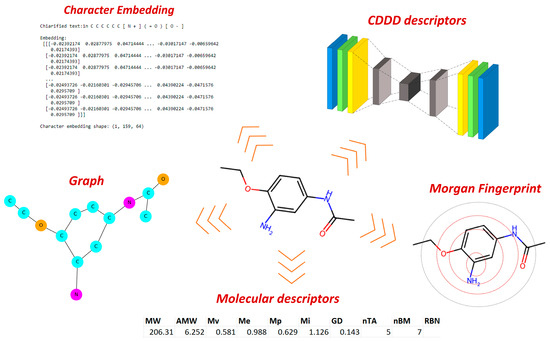
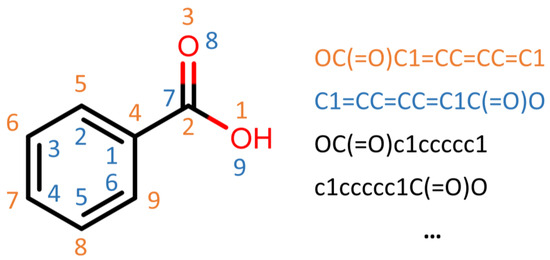
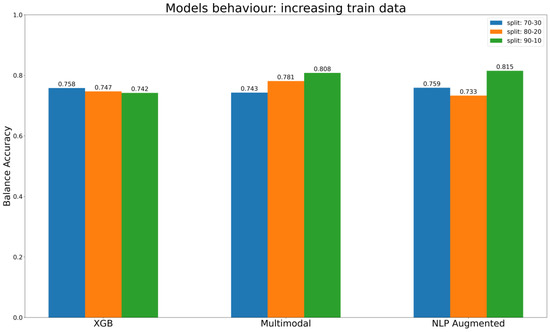
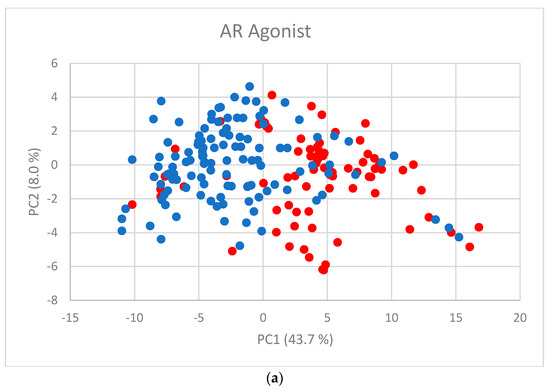
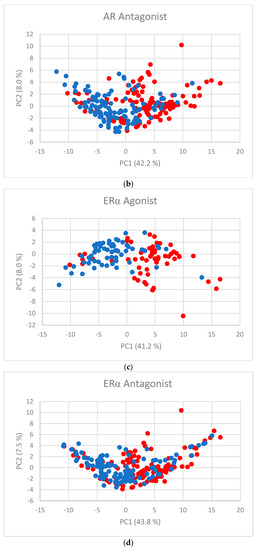
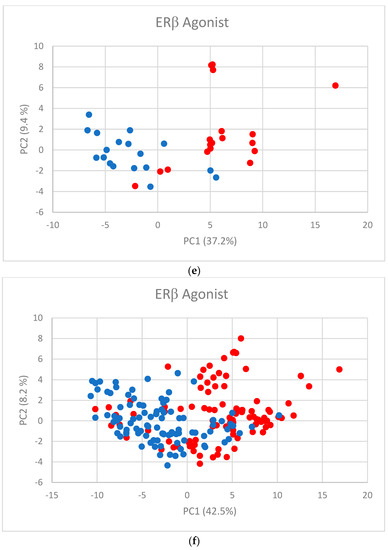
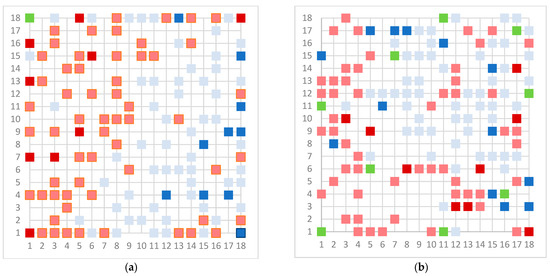
Human milk has been used for over 70 years to monitor pollutants such as polychlorinated biphenyls (PCBs) and organochlorine pesticides (OCPs). Despite the growing body of data, our understanding of the pollutant exposome, particularly co-exposure patterns and their interactions, remains limited. Artificial intelligence (AI) offers considerable potential to enhance biomonitoring efforts through advanced data modelling, yet its application to pollutant dynamics in complex biological matrices such as human milk remains underutilized. This study applied an AI-based framework, integrating machine learning, metaheuristic hyperparameter optimization, explainable AI, and postprocessing, to analyze PCB-170 levels in breast milk samples from 186 mothers in Zadar, Croatia. Among 24 analyzed POPs, the most influential predictors of PCB-170 concentrations were hexa- and hepta-chlorinated PCBs (PCB-180, -153, and -138), alongside p,p’-DDE. Maternal age and other POPs exhibited negligible global influence. SHAP-based interaction analysis revealed pronounced co-behavior among highly chlorinated congeners, especially PCB-138–PCB-153, PCB-138–PCB-180, and PCB-180–PCB-153. These findings highlight the importance of examining pollutant interactions rather than individual contributions alone. They also advocate for the revision of current monitoring strategies to prioritize multi-pollutant assessment and focus on toxicologically relevant PCB groups, improving risk evaluation in real-world exposure scenarios.
Full article

Figure 1








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}