An Efficient Synthesis of Pyridoxal Oxime Derivatives under Microwave Irradiation


Abstract
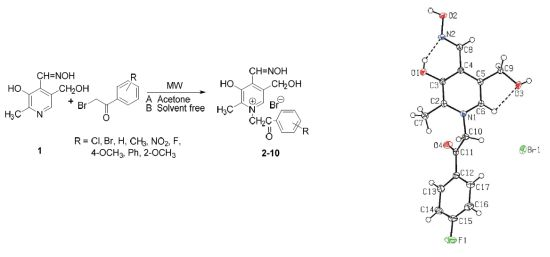
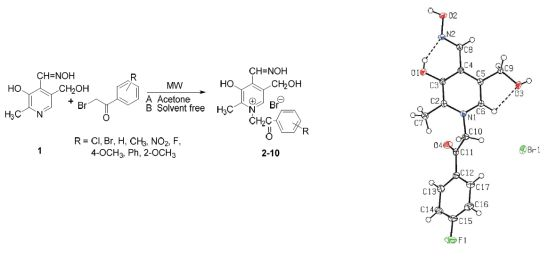
:
1. Introduction
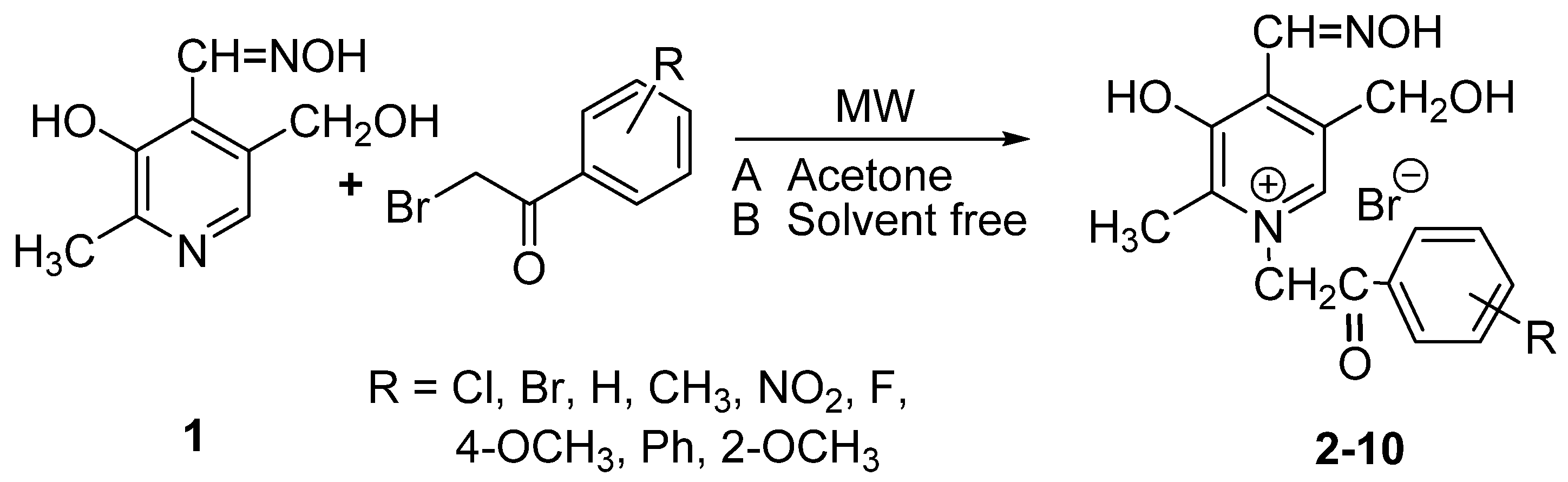
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | MW (A) | MW (B) | Conventional [15] | |||
|---|---|---|---|---|---|---|---|
| t/min | Yield (%) | t/min | Yield (%) | t/weeks | Yield (%) | ||
| 2 | 4'-Cl | 5 | 75 | 10 | 63 | 3 | 66 |
| 3 | 4'-Br | 4.5 | 90 | 10 | 74 | 3 | 44 |
| 4 | 4'-H | 5 | 74 | 7 | 53 | 1 | 38 |
| 5 | 4'-CH3 | 4 | 94 | 8 | 76 | 1 | 62 |
| 6 | 4'-NO2 | 4.5 | 80 | 10 | 79 | 1 | 60 |
| 7 | 4'-F | 3 | 70 | 10 | 48 | 3 | 36 |
| 8 | 4'-OCH3 | 5 | 90 | 10 | 71 | 3 | 67 |
| 9 | 4'-Ph | 5 | 79 | 10 | 46 | 1 | 46 |
| 10 | 2'-OCH3 | 5 | 58 | 10 | 42 | 12 | 12 |


3. Experimental Section
3.1. General Information
3.2. General Procedure for the Synthesis of Compounds 2–10 with MW Procedure A
3.3. General Procedure for the Solvent-Free Synthesis of Compounds 2–10 with MW Procedure B
3.4. Characterization Data
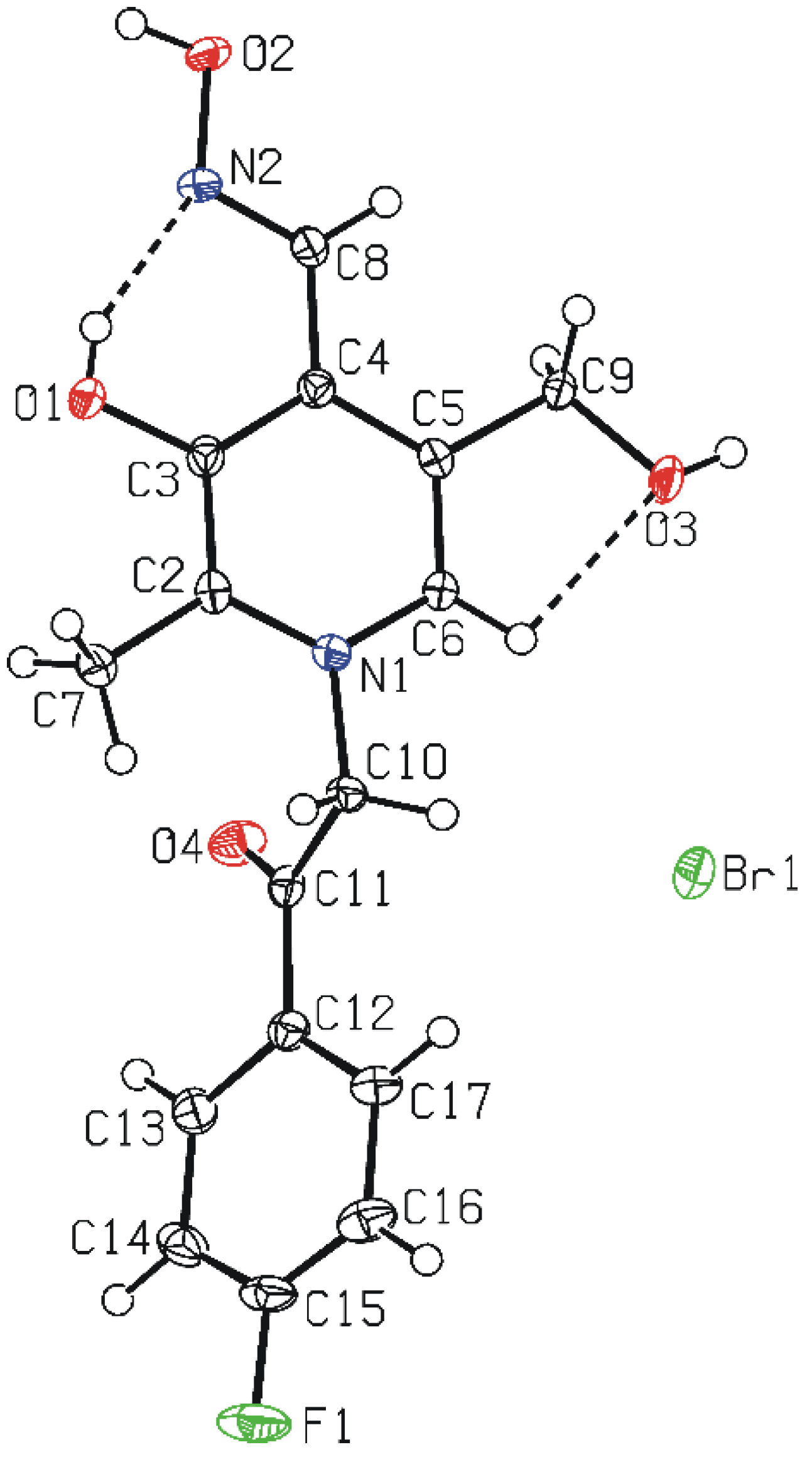
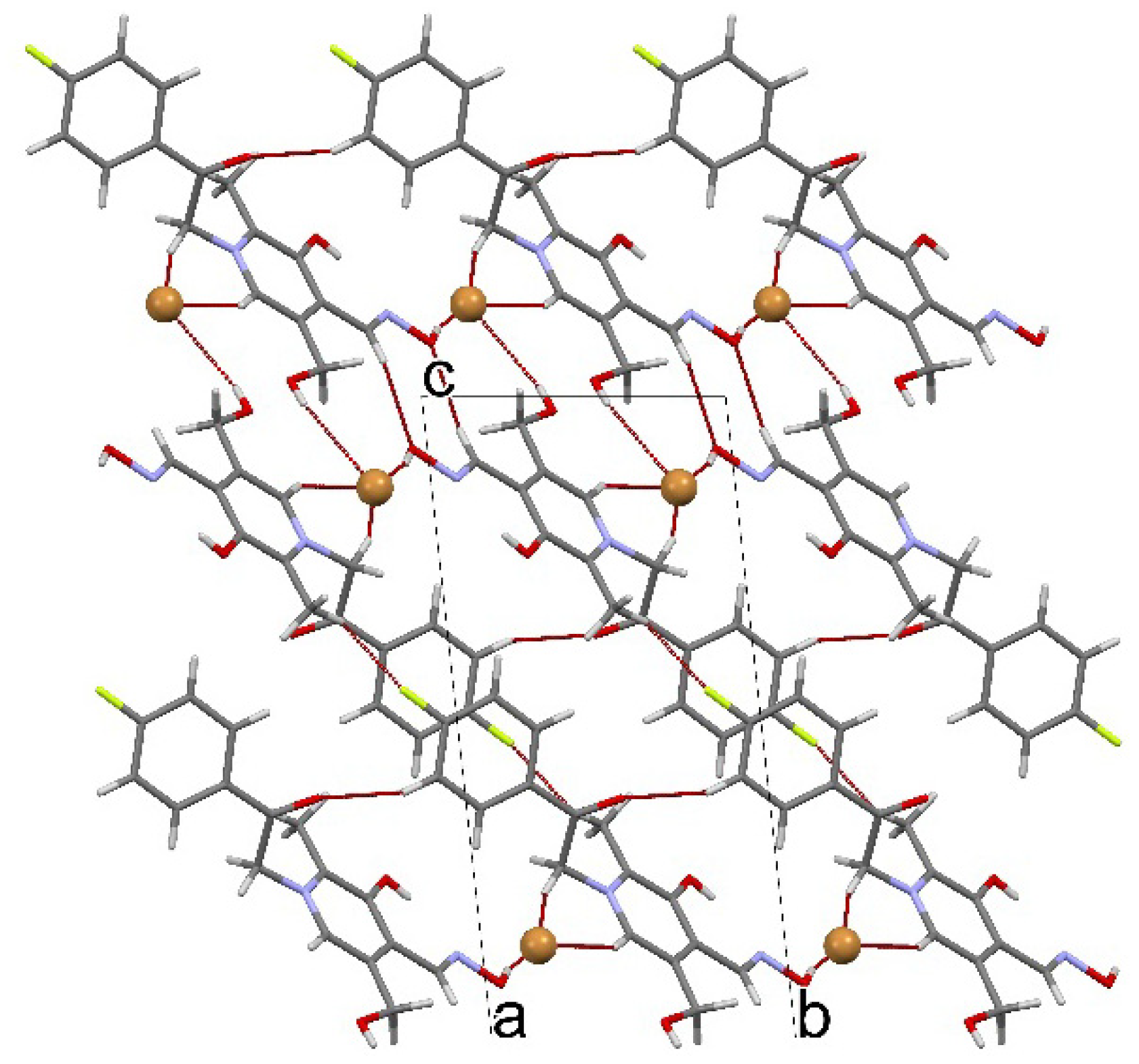
3.5. Crystal Structure Determination
 (No. 2); a = 6.7128(3), b = 8.1316(4), c = 15.9969(8) Å; α = 94.964(4), β = 92.224(4), γ = 111.877(5)°; V = 804.89(7) Å3; Z = 2; dx = 1.647 g cm−3; θmax = 27.0°; RInt = 0.0290; S = 1.004; R[I ≥ 2σ(I)] = 0.0357, wR[all data] = 0.0991; 0.698 < ∆ρ < −0.760 eA−3.
(No. 2); a = 6.7128(3), b = 8.1316(4), c = 15.9969(8) Å; α = 94.964(4), β = 92.224(4), γ = 111.877(5)°; V = 804.89(7) Å3; Z = 2; dx = 1.647 g cm−3; θmax = 27.0°; RInt = 0.0290; S = 1.004; R[I ≥ 2σ(I)] = 0.0357, wR[all data] = 0.0991; 0.698 < ∆ρ < −0.760 eA−3.4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Varma, R.S.; Namboodiri, V.V. Solvent-free preparation of ionic liquids using a household microwave oven. Pure. Appl. Chem. 2001, 73, 1309–1313. [Google Scholar]
- Surati, M.A.; Jauhari, S.; Desai, K.R. A brief review: Microwave assisted organic reaction. Arch. Appl. Sci. Res. 2012, 4, 645–661. [Google Scholar]
- Bogdal, D.; Pielichowski, J.; Jaskott, K. Fast N-Alkylation of azaheterocycles under microwave irradiation in dry media. Heterocycles 1997, 45, 715–722. [Google Scholar] [CrossRef]
- Bogdal, D.; Pielichowski, J.; Jaskott, K. New method of N-alkylation of carbazole under microwave irradiation in dry media. Synth. Commun. 1997, 27, 1553–1560. [Google Scholar] [CrossRef]
- Bogdal, D. Fast solvent-free alkylation of amides and lactams under microwave irradiation. Molecules 1999, 4, 333–337. [Google Scholar] [CrossRef]
- Deshayes, S.; Liagre, M.; Loupy, A.; Luche, J.L.; Petit, A. Microwave activation in phase transfer catalysis. Tetrahedron 1999, 55, 10851–10870. [Google Scholar] [CrossRef]
- Perez, E.; Sotelo, E.; Loupy, A.; Mocelo, R.; Suarez, M.; Perez, R.; Autie, M. An easy and efficient microwave-assisted method to obtain 1-(4-bromophenacyl)azoles in dry media. Heterocycles 1996, 43, 539–543. [Google Scholar] [CrossRef]
- Somani, S.M.; Romano, J.A. Chemical Warfare Agents: Toxicity at Low Levels; CRS Press: LLC Boca Raton, FL, USA, 2001. [Google Scholar]
- Bajgar, J. Organophosphates/nerve agent poisoning: Mechanism of action, diagnosis, prophylaxis, and treatment. J. Adv. Clin. Chem. 2004, 38, 151–216. [Google Scholar] [CrossRef]
- Kassa, J.; Cabal, J.; Kuča, K. A comparison of the efficacy of currently available oximes against tabun in rats. Biologia 2005, 60, 77–79. [Google Scholar]
- Kuča, K.; Bartošovál, D.; Patočka, J.; Cabal, J.; Kassa, J.; Kunešová, G. New quaternary pyridine aldoximes as causal antidotes against nerve agents intoxications. Biomed. Papers 2005, 149, 75–82. [Google Scholar] [CrossRef]
- Kassa, J. Review of oximes in the antidotal treatment of poisoning by organophosphorus nerve agents. J. Clin. Toxicol. 2002, 40, 803–816. [Google Scholar] [CrossRef]
- Jokanović, M.; Stojiljković, M.P. Current understanding of the application of pyridinium oximes as cholinesterase reactivators in treatment of organophosphate poisoning. Eur. J. Pharmacol. 2006, 553, 10–17. [Google Scholar] [CrossRef]
- Kovarik, Z.; Čalić, M.; Šinko, G.; Bosak, A.; Berend, S.; Lucić Vrdoljak, A.; Radić, B. Oximes: Reactivators of phosphorylated acetylcholinesterase and antidotes in therapy against tabun poisoning. Chem. Biol. Interact. 2008, 175, 173–179. [Google Scholar] [CrossRef]
- Gašo-Sokač, D.; Katalinić, M.; Kovarik, Z.; Bušić, V.; Kovač, S. Synthesis and evaluation of novel analogues of vitamin B6 as reactivators of tabun and paraoxon inhibited acetylcholinesterase. Chem. Biol. Interact. 2010, 187, 234–237. [Google Scholar] [CrossRef]
- Cetina, M.; Nagl, A.; Gašo-Sokač, D.; Kovač, S.; Bušić, V.; Saftić, D. Extensive intramolecular and intermolecular interactions in two quaternary salts of the pyridoxal oxime. J Chem. Christallogr. 2012, 42, 752–758. [Google Scholar] [CrossRef]
- Jukić, M.; Hergold-Brundić, A.; Cetina, M.; Nagl, A.; Vorkapić-Furač, J. The synthesis and structures of the novel pyridoxal oxime derivatives. Struct.Chem. 2003, 14, 597–604. [Google Scholar] [CrossRef]
- Oxford Diffraction. Xcalibur CCD System, CrysAlisPro, Oxford Diffraction Ltd: Abingdon, England, 2010.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; de Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar]
- Farrugia, L.J. WinGX and Ortep for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gašo-Sokač, D.; Bušić, V.; Cetina, M.; Jukić, M. An Efficient Synthesis of Pyridoxal Oxime Derivatives under Microwave Irradiation. Molecules 2014, 19, 7610-7620. https://doi.org/10.3390/molecules19067610
Gašo-Sokač D, Bušić V, Cetina M, Jukić M. An Efficient Synthesis of Pyridoxal Oxime Derivatives under Microwave Irradiation. Molecules. 2014; 19(6):7610-7620. https://doi.org/10.3390/molecules19067610
Chicago/Turabian StyleGašo-Sokač, Dajana, Valentina Bušić, Mario Cetina, and Marijana Jukić. 2014. "An Efficient Synthesis of Pyridoxal Oxime Derivatives under Microwave Irradiation" Molecules 19, no. 6: 7610-7620. https://doi.org/10.3390/molecules19067610
APA StyleGašo-Sokač, D., Bušić, V., Cetina, M., & Jukić, M. (2014). An Efficient Synthesis of Pyridoxal Oxime Derivatives under Microwave Irradiation. Molecules, 19(6), 7610-7620. https://doi.org/10.3390/molecules19067610