1. Introduction
The use of controllable therapeutic agents and the blockage of their therapeutic effect after treatment can significantly reduce drug-induced side effects. Over the past 25 years, aptamers have gained clinical interest due to their several unique characteristics. Aptamers are short single-stranded oligonucleotides which can fold into three-dimensional structures. These molecules can bind with a high affinity and specificity to the target of interest [
1]. The modifications of aptamers at defined positions enable the fine-tuning of their stability and bioavailability, and allow their application as theranostics [
2].
The important criteria for the optimal application of drugs are the dosing accuracy and the controllability of their effect. Therefore, so-called antidotes (ADs) provide excellent possibilities, which can turn off the effect of administered drugs by competitive blocking, altering, binding, or general inactivation of the agents.
The binding of an aptamer to its target can be easily prevented by changing the three-dimensional structure of the aptamer, for example by addition of a complementary oligonucleotide which hybridizes to the aptamer. Thus, a further important characteristic of aptamers is that the binding ability of a specific aptamer can be abrogated by the addition of an oligonucleotide with a complementary sequence to the aptamer [
3,
4,
5].
In this study, we analyzed the required time for the complexation of NU172 [
6] and R10-60 [
7,
8] aptamers by their ADs in human serum. Against the concerns that the 3D folding of the aptamers and the existence of several proteins in biological fluids could impede the specific and quick recognition and binding of the ADs to the aptamers, here, we demonstrated using human serum the rapid binding of respective ADs to the NU172 or R10-60 aptamer and their complexation. Furthermore, the abolition of each aptamer´s effect was demonstrated in human blood or cell culture medium.
2. Materials and Methods
2.1. Oligonucleotides
The experiments were performed with two different DNA aptamers, the Toll-like receptor 9 (TLR 9) binding R10-60 aptamer and the thrombin binding NU172 aptamer, and their complementary DNA sequences, called ADs (
Table 1). Furthermore, nonsense (NS) AD and nonsense aptamers were used as negative controls. All oligonucleotides were ordered as HPLC-purified from Ella Biotech GmbH (Martinsried, Germany).
2.2. Ethics Statement
The Ethics Committee of the University of Tuebingen approved the blood sampling procedures and all subjects gave written informed consent.
2.3. Generation of Human Serum
Human blood samples (7.5 mL) were collected in tubes containing kaolin-coated granulate (Sarstedt AG & Co., Nümbrecht, Germany) as a clot activator and the blood was incubated for approximately 30 min at room temperature. Then, the tubes were centrifuged for 15 min at 2000× g. The liquid supernatant (serum) was collected and frozen at −20 °C.
2.4. Collection of Human Blood for Determining of ACT (Activated Clotting Time)
A total volume of 4.5 mL blood was collected from the antecubital vein of each healthy, non-medicated volunteer into neutral monovettes or 3 IU/mL sodium heparin-containing (Ratiopharm, Ulm, Germany) monovettes.
2.5. Cultivation of PMDC05 Cells
The leukemic plasmacytoid dendritic cell line (PMDC05) is similar to normal plasmacytoid dendritic cells (pDCs) in its surface expression of phenotypic markers and expression of TLRs. These cells were donated from the Laboratory of Hematology and Oncology of Niigata University in Japan. PMDC05 cells were cultured in suspension culture flasks in IMDM medium (Iscove’s Modified Dulbecco’s Medium) containing 10% FBS (fetal bovine serum), 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C, 5% CO2, and 20% O2. All cell culture reagents were obtained from Life Technologies (Darmstadt, Germany). A medium change was performed every 3 days. The number of cells was determined using a CASY® cell counter (Roche Innovatis AG, Mannheim, Germany) and the cultivation was performed with 30 × 106 cells/30 mL medium.
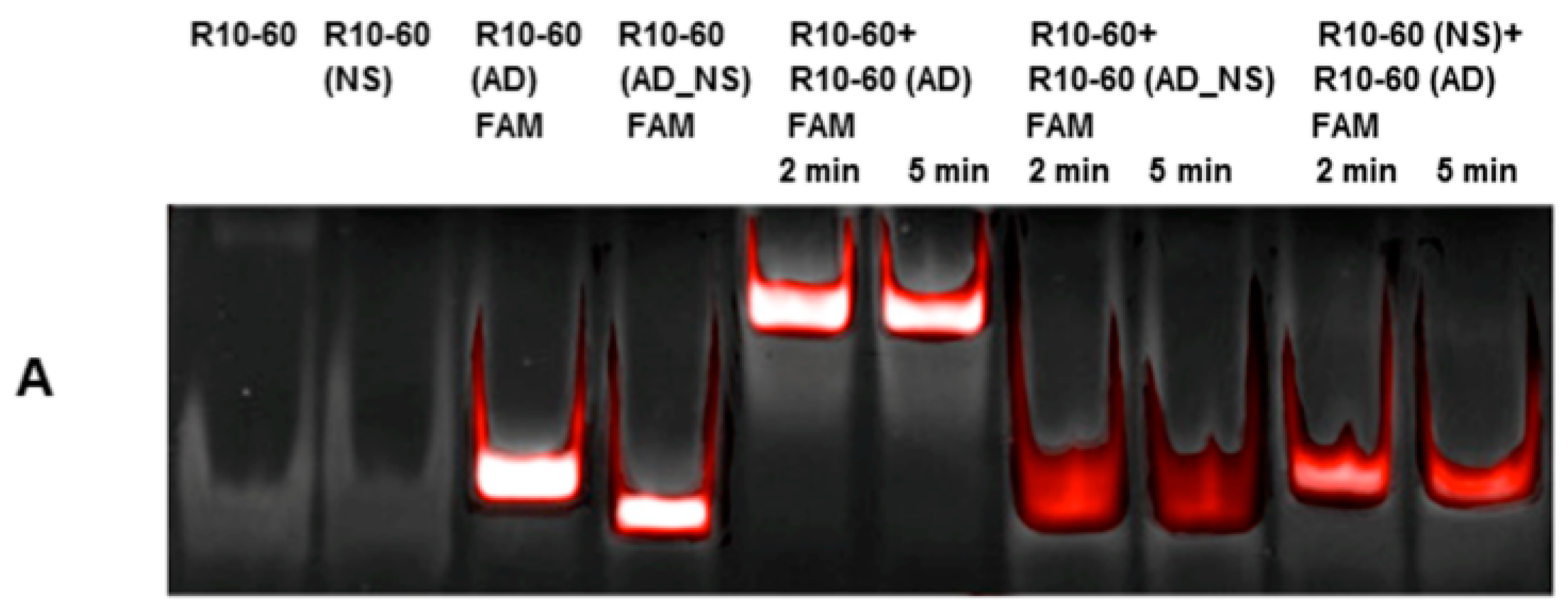
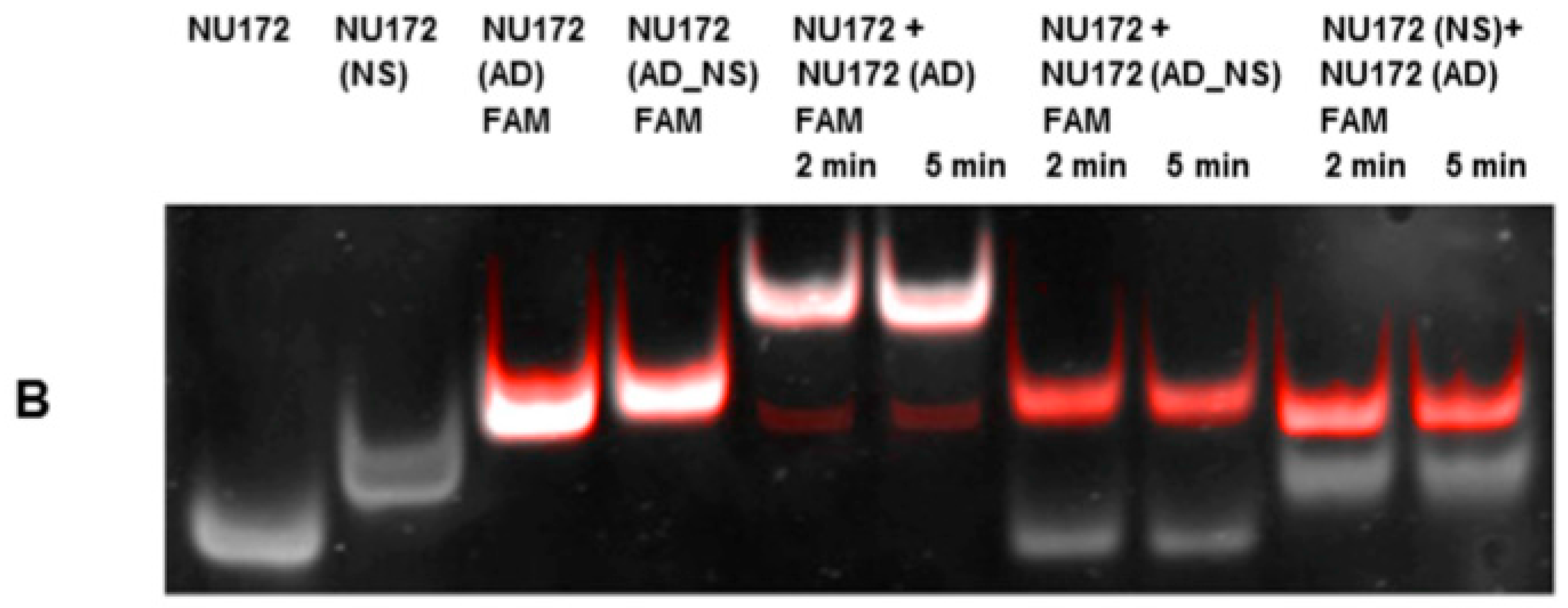
2.6. Required Time for Binding of AD to Aptamers R10-60 and NU172 in Human Serum
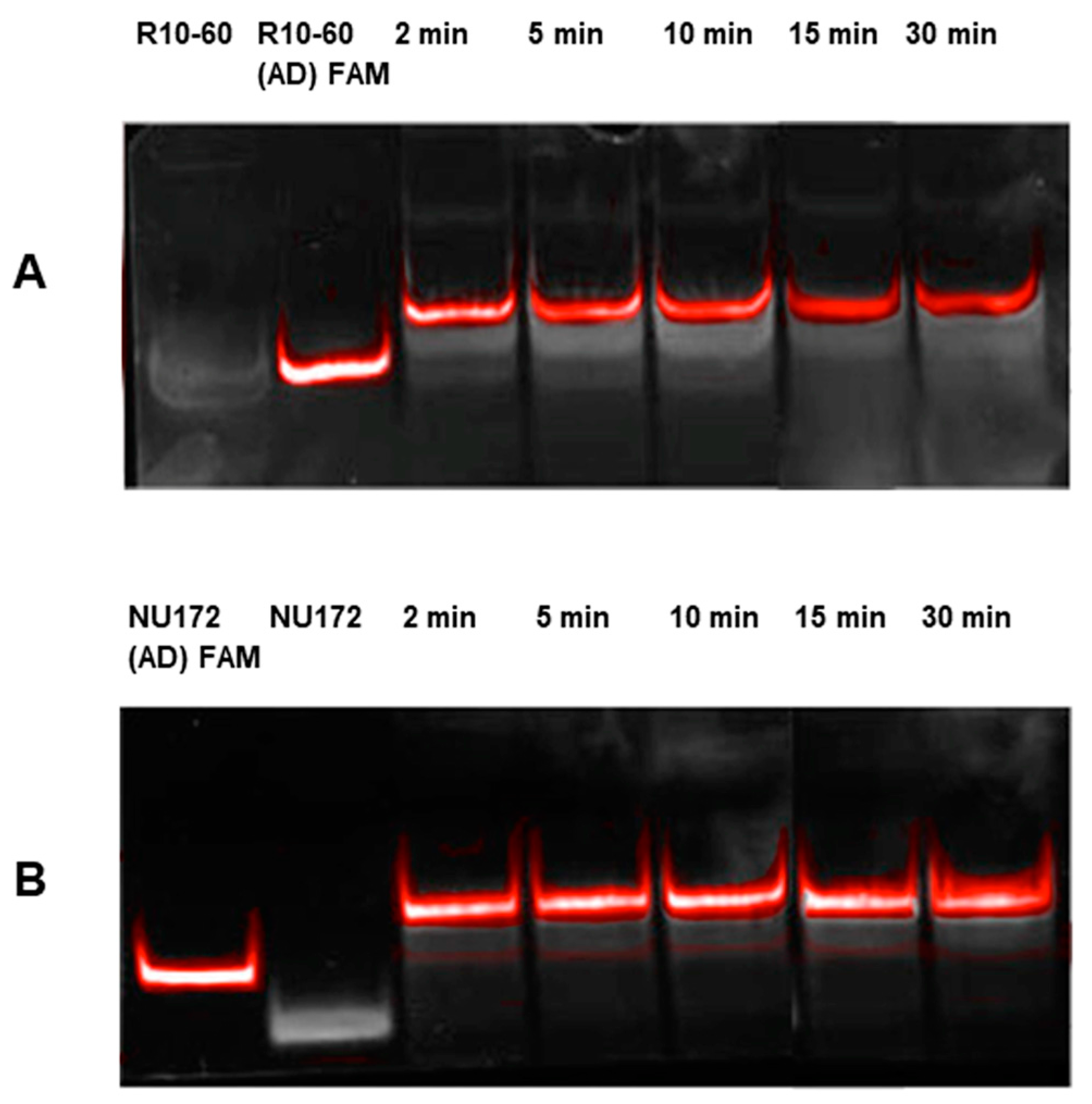
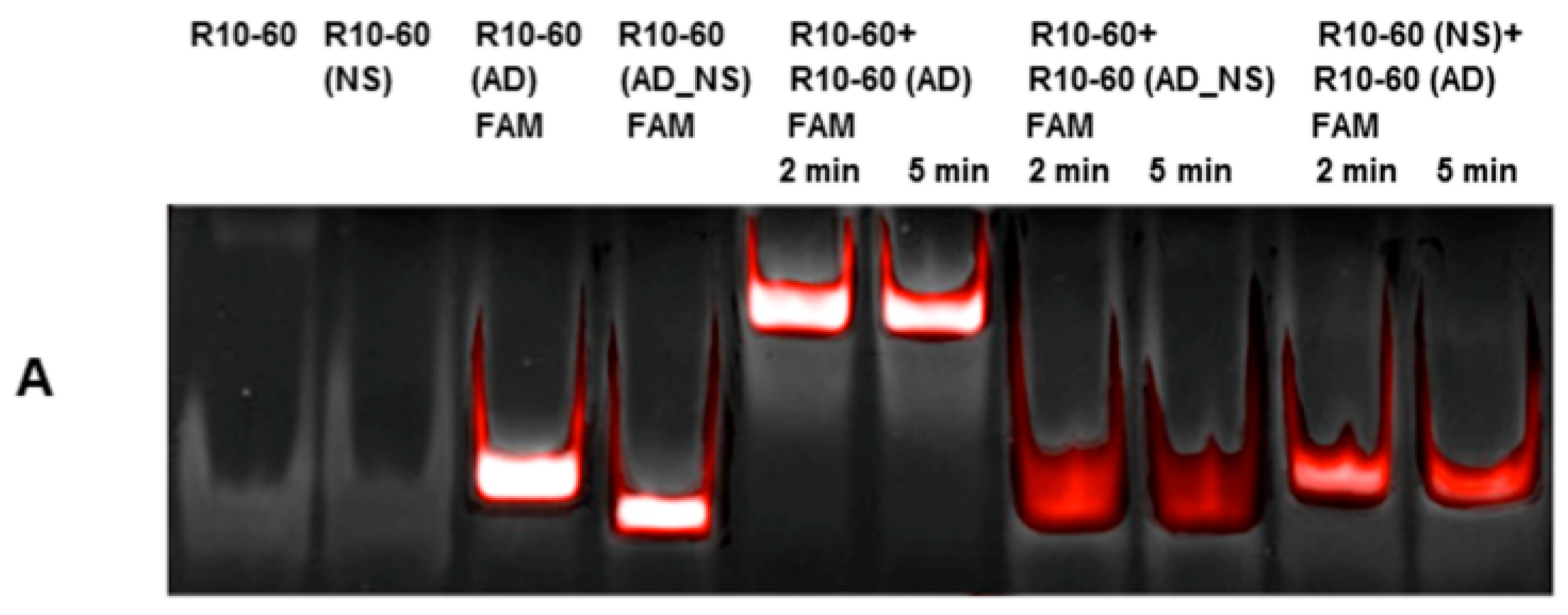
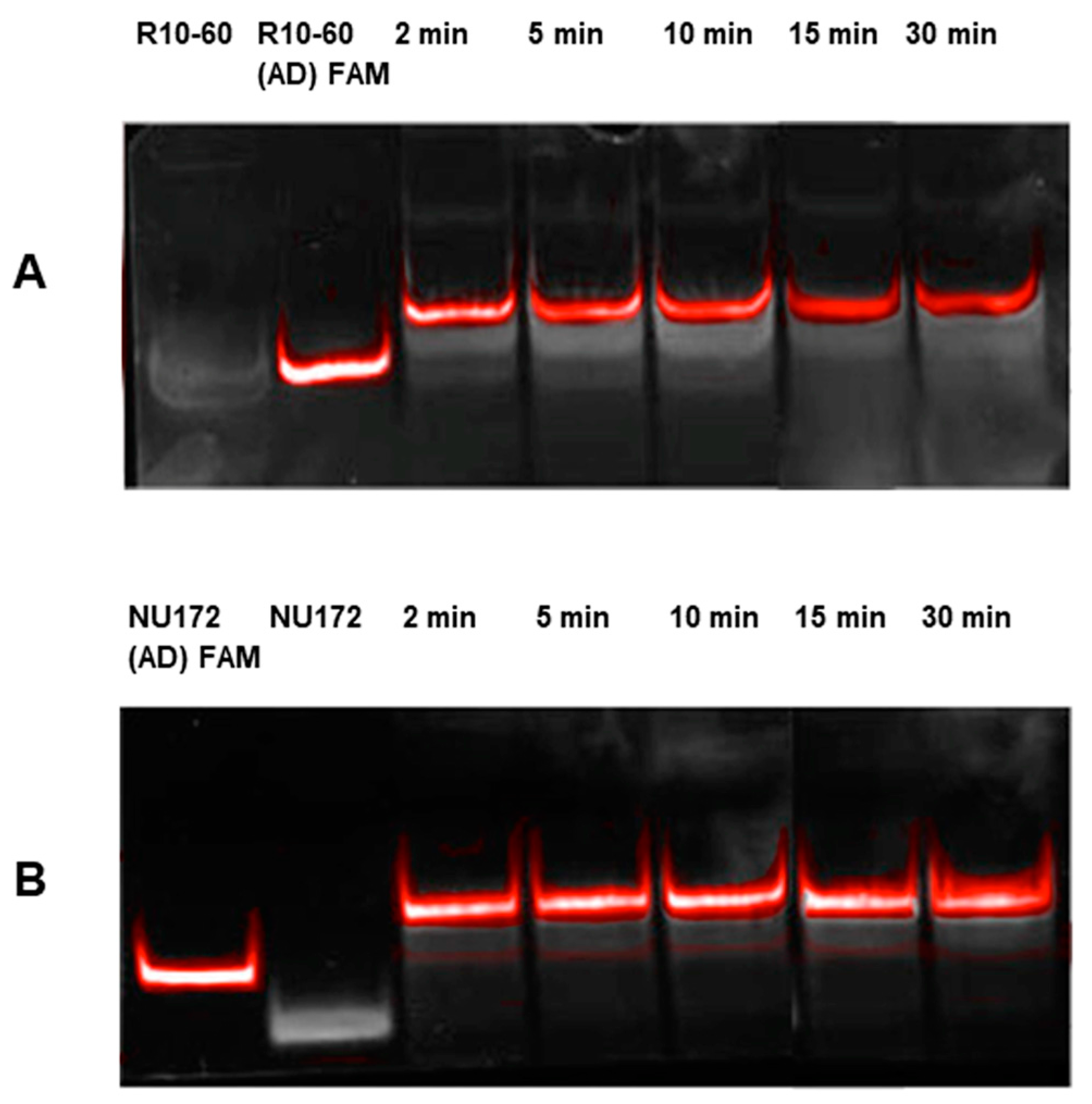
Aptamers, respective ADs, and nonsense ADs or aptamers (10 μM) were denatured at 95 °C and cooled down to 37 °C to induce the three-dimensional folding of aptamers and ADs. The incubation of the aptamers with their respective AD was performed in 200 μL human serum at a molar ratio of 1:1 (1 μg/1 μg) at 37 °C and with shaking at 300 rpm for 2, 5, 10, 15, and 30 min. Nonsense controls were analyzed after 2 and 5 min of incubation. After each incubation time, samples were purified by phenol-chloroform precipitation and concentrated using Amicon Ultra 0.5 mL 10K centrifugal filters (Merck Millipore, Darmstadt, Germany) according to the manufacturer´s instructions. All steps were performed at 37 °C.
The aptamer/AD complexes were visualized using nondenaturing polyacrylamide gel electrophoresis (PAGE). Therefore, 400 ng of aptamers (R10-60, NU172) and their corresponding FAM (fluorescein amidite)-labeled AD, and nonsense controls were loaded onto gels, and 20 μL of concentrated samples after serum incubation was run on 10% nondenaturing polyacrylamide gel. Gels were first visualized on an UV transilluminator to detect the FAM-labeled AD or AD_NS and then stained with GelRed™ (Biotium Inc., Hayward, CA, USA) to detect all oligonucleotides. Subsequently, gels were documented using the gel documentation system (Gel Doc XR, Bio-Rad laboratory GmbH, Munich, Germany). The images were colored and overlaid using Adobe Photoshop Elements 14.0.
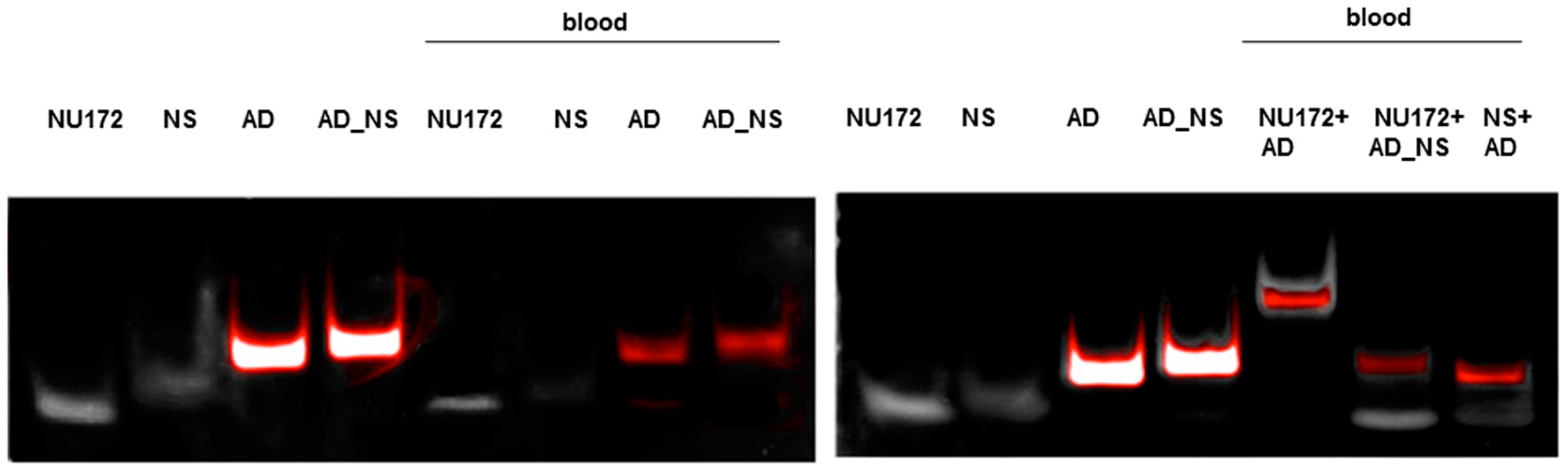
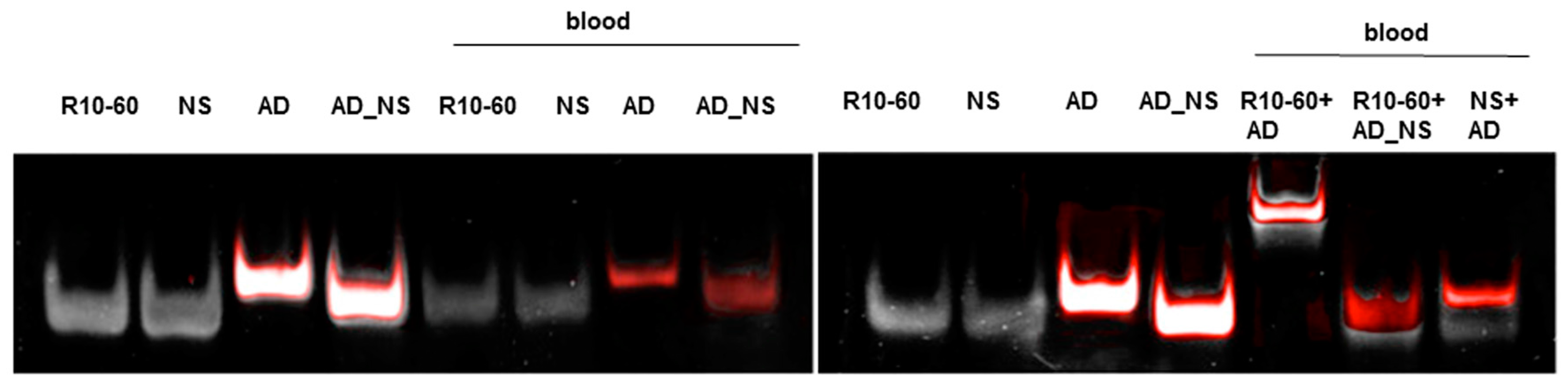
2.7. Generation of Aptamer/AD Complexes in Human Whole Blood
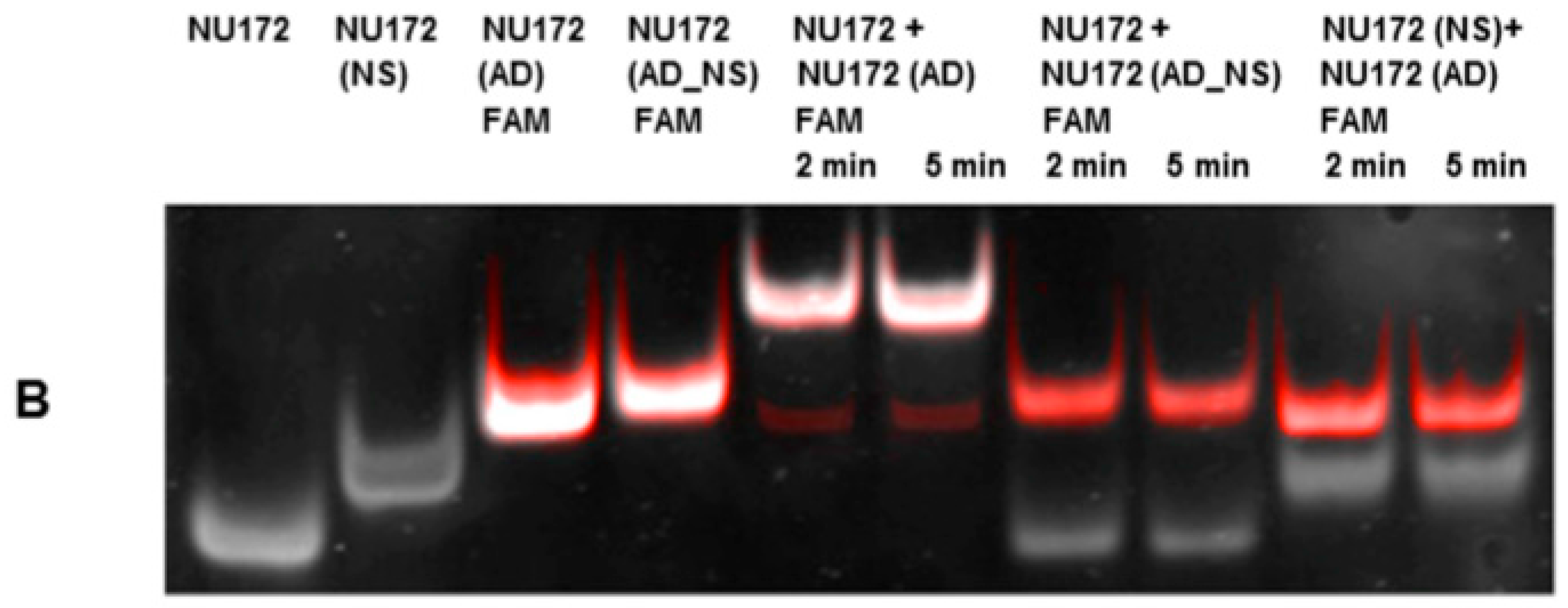
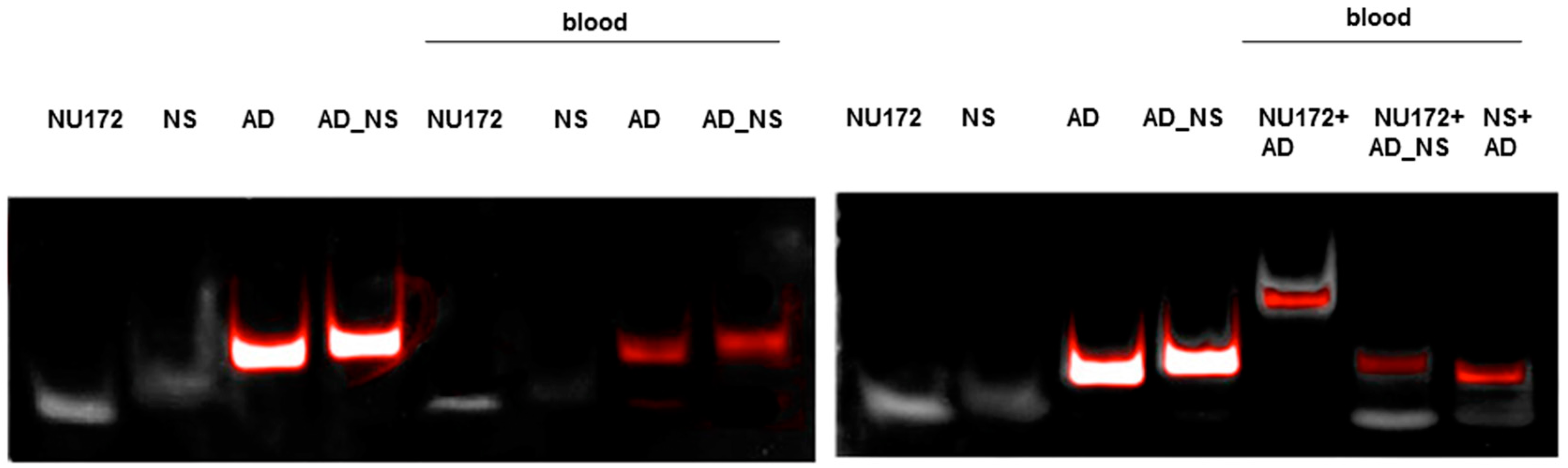
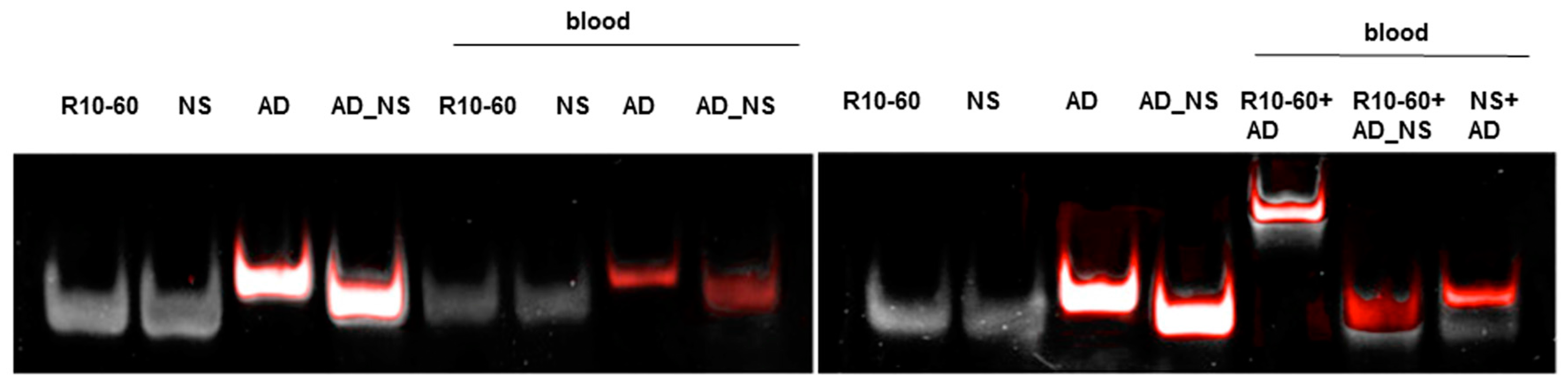
To fold oligonucleotides into their unique three-dimensional structures, aptamers, respective AD, and the controls, AD_NS or aptamer (NS), (10 μM) were denatured at 95 °C and then cooled down to 37 °C. The incubation of aptamers with their respective AD was performed in 1 mL human neutral blood at a molar ratio of 1:1 (5 μg/5 μg) at 37 °C. After the addition of the aptamer or NS aptamer, the blood was incubated with shaking at 300 rpm for 2 min. Thereafter, AD or AD_NS was added to the blood. After 2 min of incubation with shaking at 300 rpm, serum was generated using kaolin-coated granulate (Sarstedt AG & Co., Nümbrecht, Germany) as a clot activator. Samples were purified using phenol-chloroform precipitation and concentrated using Amicon Ultra 0.5 mL 10K centrifugal filters (Merck Millipore, Darmstadt, Germany) according to the manufacturer´s instructions. All steps were performed at 37 °C.
The generated aptamer/AD complexes were visualized using nondenaturing PAGE. For this, 400 ng of the aptamers (R10-60, NU172) and their corresponding FAM-labeled AD, and nonsense controls were loaded onto gels, and 10 μL of concentrated samples was run on 10% nondenaturing polyacrylamide gel. Firstly, FAM-labeled AD or AD_NS in the samples was visualized on an UV transilluminator and then stained with GelRed™ (Biotium Inc., Hayward, CA, USA) to detect all oligonucleotides. Gels were documented using the gel documentation system (Gel Doc XR, Bio-Rad laboratory GmbH, Munich, Germany). The images were colored and overlaid using Adobe Photoshop Elements 14.0.
2.8. Blocking of Aptamers by Generation of Aptamer/AD Complexes
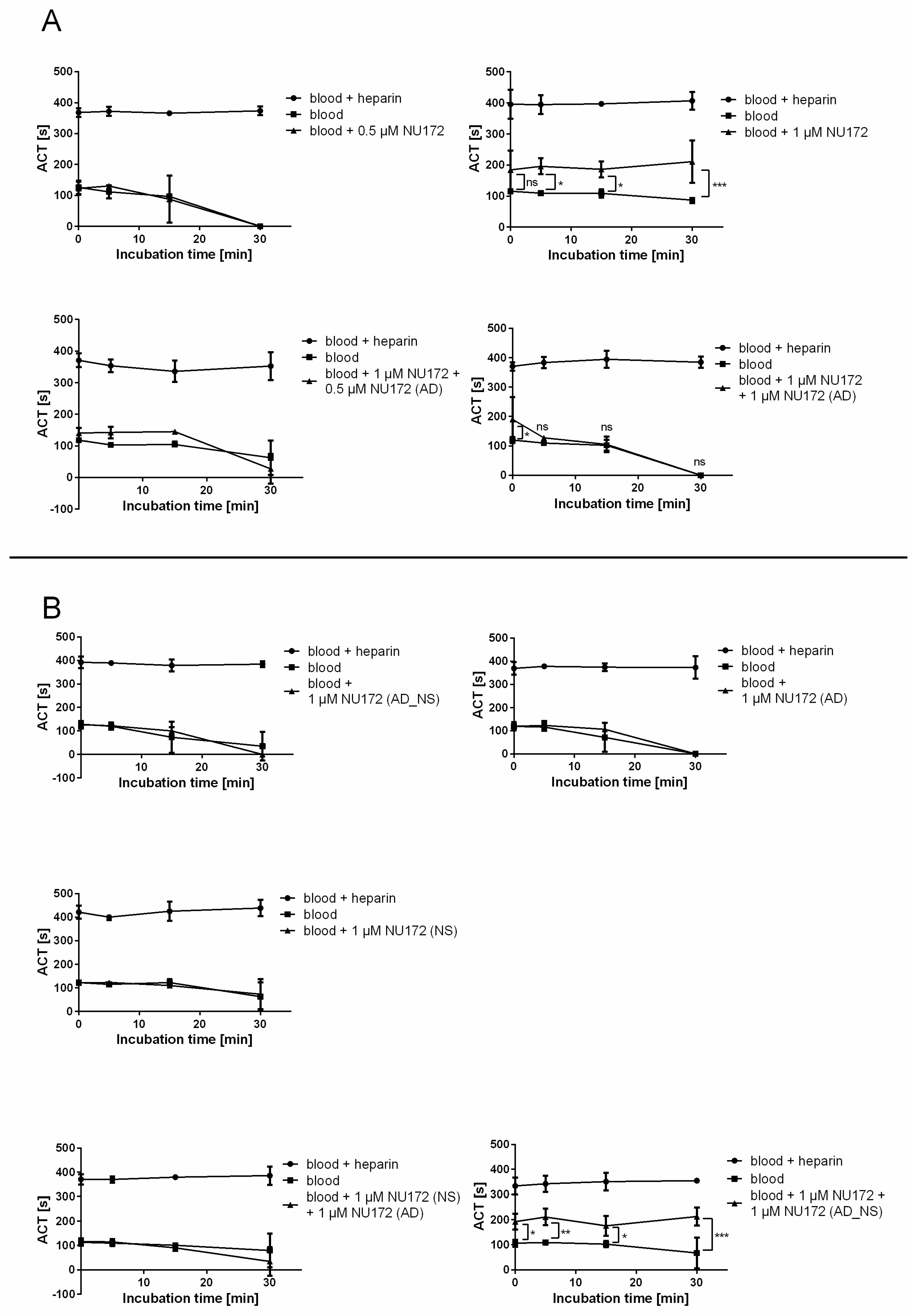
2.8.1. Determination of ACT of Blood after Addition of Thrombin Aptamer AD (NU172 (AD)) to NU172
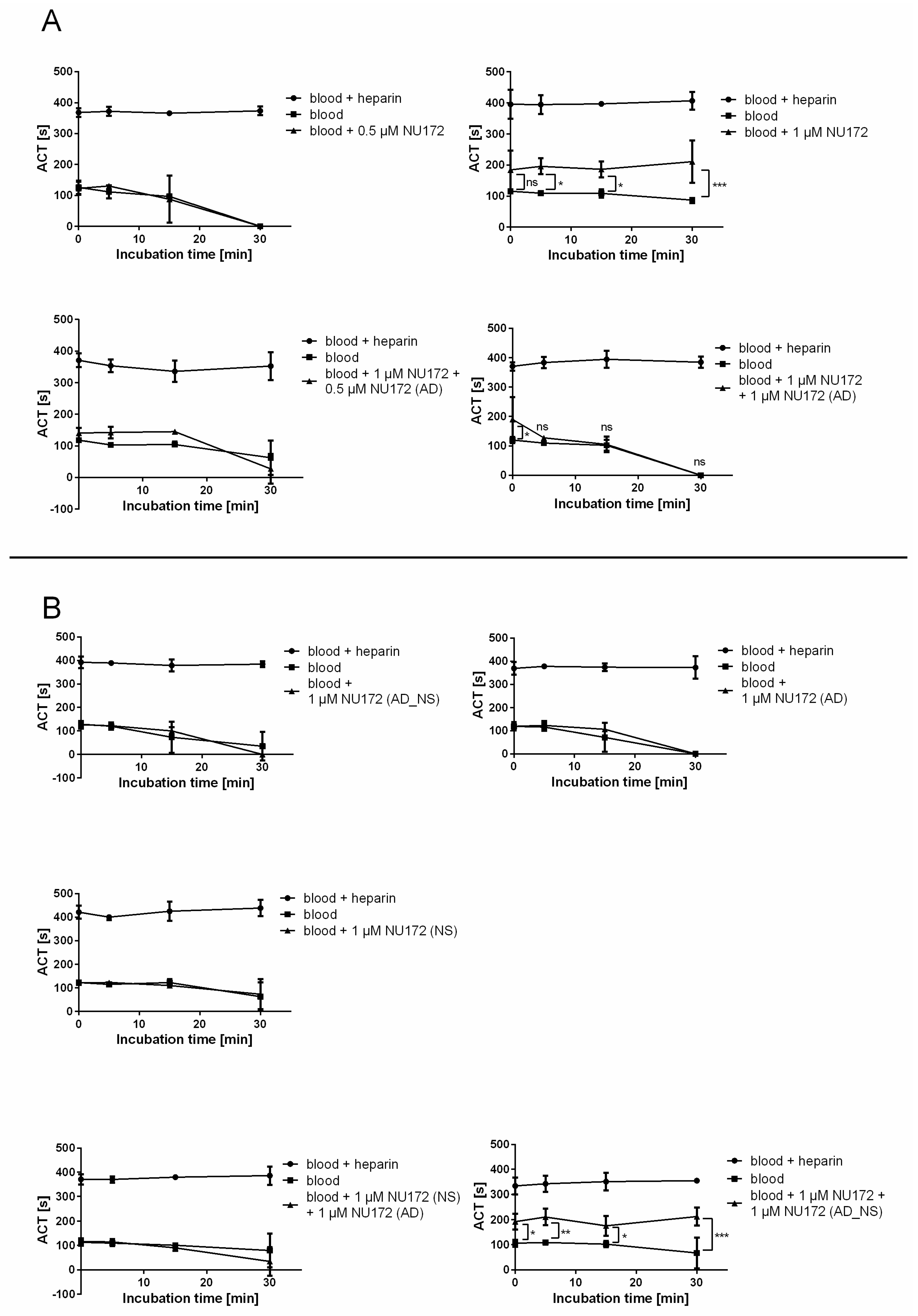
To monitor the functional inhibition time of NU172 by its AD, ACT was determined after addition of 0.5 or 1 μM NU172 aptamer alone, as well as after the addition of 1 or 0.5 μM NU172 (AD) to the 1 μM NU172-containing human blood. The ACT of blood without oligonucleotides was measured as a negative control and blood containing heparin served as a positive control. Additionally, the ACT of blood containing 1 μM NU172 (AD_NS), 1 μM NU172 (NS), 1 μM NU172 (AD), or 1 μM NU172 (NS) and 1 μM NU172 (AD), or 1 μM NU172 and 1 μM NU172 (AD_NS) was analyzed.
Measurements were performed using a Hemochron Jr. II (International Technidyne Corporation, Edison, NJ, USA). This microcoagulation system utilizes a mechanical endpoint clotting mechanism which is monitored optically. Thereby, the ACT of whole blood samples can be measured within disposable test cuvettes. Hemochron® Jr. High Range ACT (ACT+) cuvettes were used for the detection of the ACT of blood samples. After warming of the cuvettes in the system, samples were transferred onto the test channel and the clotting time was measured in seconds.
After the withdrawal of venous blood, blood from neutral monovettes was incubated for 2 min with or without oligonucleotides. Heparin containing blood was respectively incubated for 2 min without addition of aptamers. Subsequently, the measurement of the ACT was performed at different times: 0 min (T0), after 5 min (T5), 15 min (T15), and 30 min (T30). Therefore, 50 μL of samples was transferred into the test cuvettes.
To determine the clotting time, neutral blood (without heparin) was spiked with 0.5 and 1 μM NU172 aptamer or 1 μM of each oligonucleotide. After 2 min of incubation, 50 μL of each sample was measured at T0, T5, T15, and T30. Furthermore, the ACT of blood samples was measured after the addition of 0.5 μM and 1 μM NU172 (AD) or after the addition of 1 μM NU172 (AD_NS) to 1 μM NU172 containing neutral blood samples, as well as after the addition of 1 μM NU172 (AD) to 1 μM NU172 (NS)-containing neutral blood, as described above. All experiments were performed at 37 °C with blood samples from 3 donors (n = 3).
2.8.2. Investigation of the Interruption of R10-60 Binding to TLR9 by Antidote Addition
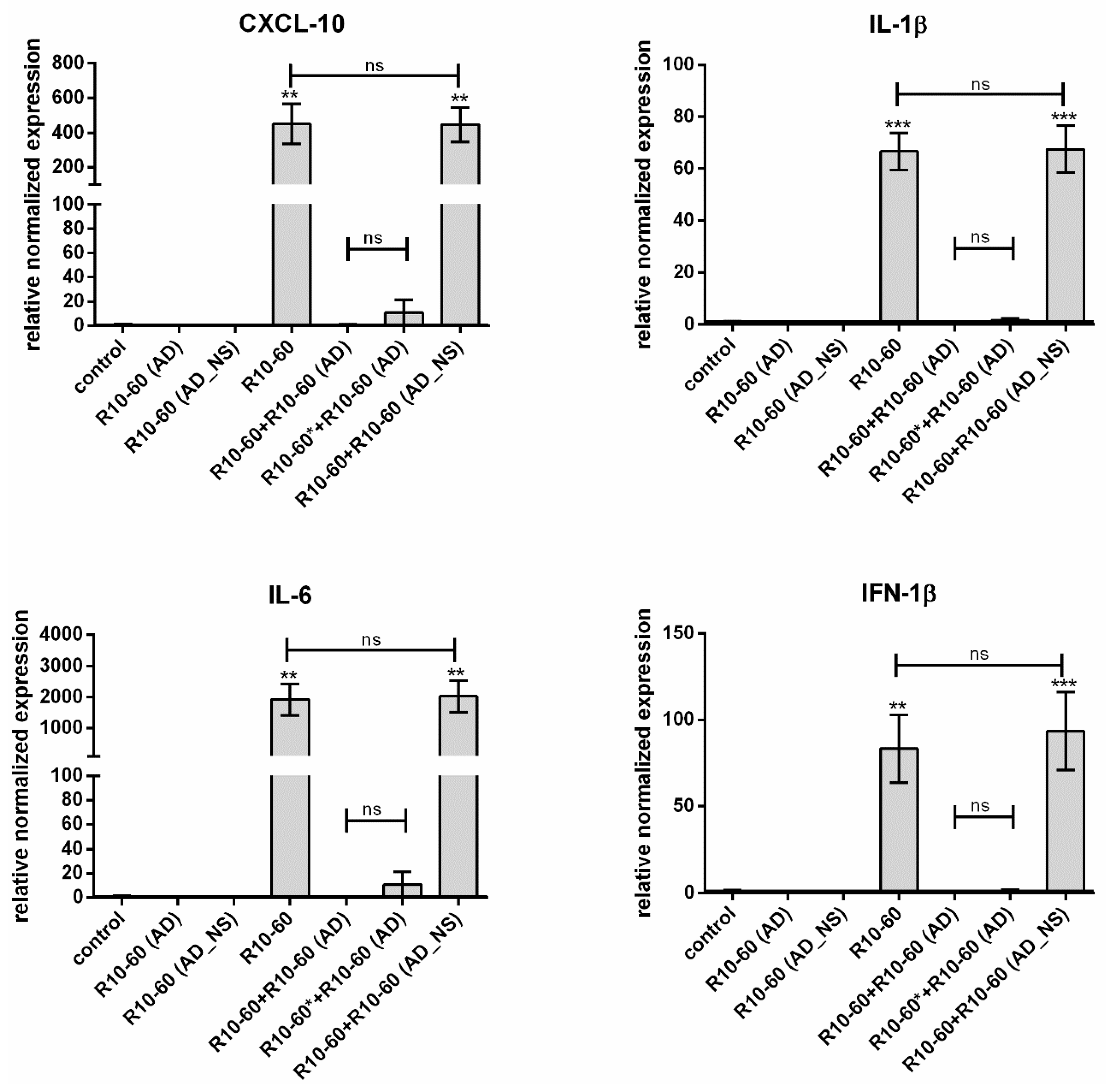
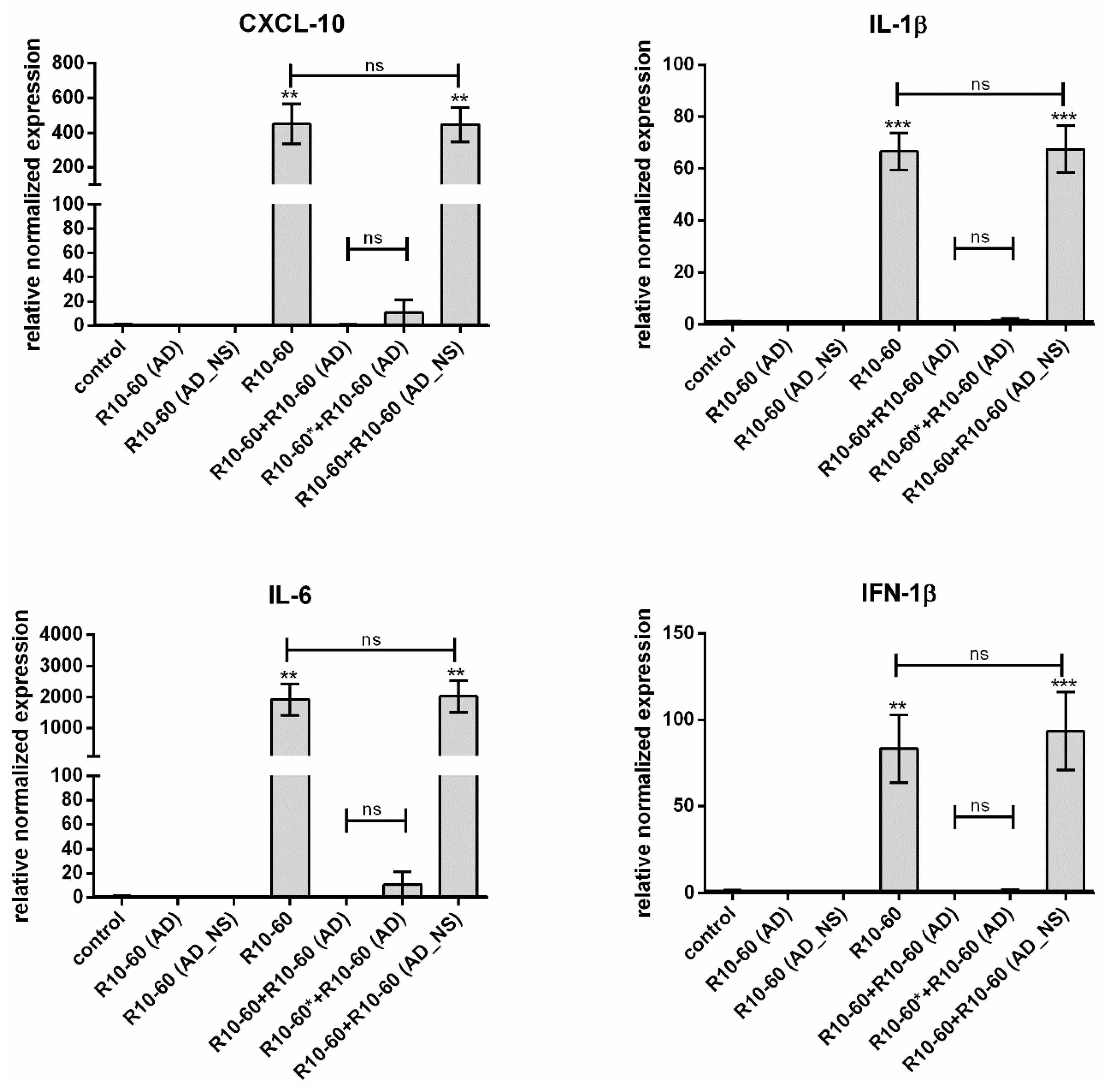
In our previous study, we demonstrated that the recognition of the aptamer R10-60 by TLR9 in PMDC05 cells leads to the expression of pro-inflammatory cytokines [
9], e.g., IFN-1β, IL-6, CXCL-10, and IL-1β. Thus, we determined the neutralization of the R10-60 aptamer binding ability to the TLR9 by the addition of R10-60 (AD) to the PMDC05 cells and subsequently analyzing the expression of IFN-1β, IL-6, CXCL-10, and IL-1β.
2.9. Incubation of PMDC05 Cells with R10-60 Aptamer and R10-60 (AD)
PMDC05 cells were incubated in 48-well suspension plates without and with addition of 10 μM R10-60 aptamer, R10-60 (AD), or R10-60 (AD_NS) alone, or with 10 μM R10-60 aptamer and 10 μM R10-60 (AD) or 10 μM R10-60 (AD_NS), for 4 h at 37 °C. Additionally, cells were incubated for 2 min with 10 μM R10-60 aptamer and then R10-60 (AD) was added to the PMDC05 medium and incubated also for 4 h at 37 °C. A PMDC05 cell suspension of 450 μL containing 1 × 106 cells was used per well of a 48-well plate.
2.10. Isolation of Total RNA from PMDC05 Cells and Synthesis of cDNA
After 4 h of incubation, total RNA was isolated from PMDC05 cells using an Aurum™ Total RNA Mini Kit (BioRad Laboratories, Munich, Germany) according to the manufacturer’s instructions. The RNA concentration was measured using a BioPhotometer (Eppendorf, Hamburg, Germany). To perform real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses, 300 ng RNA of each sample was transcribed using an iScriptTM cDNA synthesis kit (Bio-Rad Laboratories, Munich, Germany) into cDNA. Reverse transcription was performed under the following conditions: 5 min at 25 °C, 30 min at 42 °C, and 5 min at 85 °C. Synthesized cDNA was diluted 1:10 for qRT-PCR.
2.11. qRT-PCR
The analysis of IFN-1β, IL-6, CXCL-10, and IL-1β mRNA expression in samples was performed using iQ SYBR Green Supermix (BioRad Laboratories, Munich, Germany) according to the manufacturer´s recommendations. All qRT-PCR reactions were performed in triplicate in a CFX Connect™ Real-Time PCR Detection System (BioRad Laboratories, Munich, Germany). Expression of the constitutively expressed gene
GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as an internal control for RNA input. The primers used for the specific amplification of transcripts are listed in
Table 2 and were ordered from Ella Biotech (Martinsried, Germany). PCR cycling conditions consisted of an initial hot start at 95 °C for 3 min to activate the hot-start iTaq DNA polymerase and to denature the template DNA, followed by 40 cycles of denaturing at 95 °C for 15 s, annealing at 63 °C for 30 s and elongation at 72 °C for 10 s. At the end of amplification process, melt-curve analysis was performed consisting of 50 melt cycles, beginning at 70 °C to 95 °C with increments of 0.5 °C per cycle to control the specific length of the amplified product. All qRT-PCR data were collected and analyzed using the Bio-Rad CFX Manager
TM Software Version 3.0 (Bio-Rad Laboratories, Munich, Germany). Levels of mRNA were normalized to GAPDH and the results are shown relative to control mRNA levels in samples without oligonucleotide addition.
2.12. Statistical Analysis
Data are shown as means ± standard deviation (SD) or means ± standard error of the mean (SEM). One-way analysis of variance (ANOVA) for repeated measures followed by the Bonferroni’s multiple comparison test was performed to compare the groups in qRT-PCR measurements. Two-way ANOVA followed by the Bonferroni’s multiple comparison test was used to compare the groups in ACT measurement experiments. All statistical analyses were performed double-tailed using GraphPad Prism version 5.01. Differences of p < 0.05 were considered statistically significant.
4. Discussion
The intake of medicines is also associated with unwanted side effects. Thus, the interest in the development of new drugs with reduced side effects and targeted drug delivery has increased in recent years. Aptamers, having high binding specificity and affinity to specific targets, such as cells or proteins (growth factors, transcription factors, enzymes, immunoglobulins, and receptors), and the ability to modulate the targets’ activities, represent an interesting class of drugs. The high binding affinities and the option to use a complementary oligonucleotide as an AD for the regulation or neutralization of aptamer activity [
4,
10,
11] is a great advantage of these DNA-based drugs. The binding of an AD to the aptamer can disrupt the less stable tertiary structure of the aptamer by the formation of more thermodynamically stable Watson–Crick base-pairing [
12].
In this study, ADs against the thrombin binding aptamer NU172 (ARC2172) [
6] and the TLR9 binding aptamer R10-60 [
7,
8] were used. The ability to abrogate the target binding and the inhibitory and activating effects of the aptamers was analyzed. Furthermore, the required time for the generation of double-stranded complexes was examined by gel electrophoresis analyses.
Already after 2 min, aptamer/AD complexes were generated. Thereby, the ability of the AD to quickly bind to complementary sequences was demonstrated in human serum containing thousands of proteins with varying concentrations, wherein albumin and immunoglobulin G (IgG) account for more than 60% [
13]. Furthermore, the anticoagulant effect of NU172 could be abrogated 5 min after the addition of 1 μM NU172 (AD) to 1 μM NU172 in human whole blood. Additionally, the immune stimulating effect of R10-60 was prevented by the addition of R10-60 (AD) to the PMDC05 cells. Thereby, ADs were able to efficiently bind to the complementary aptamer sequences without the need for denaturation of the oligonucleotides’ three-dimensional structure, for example by heat treatment or addition of denaturing agents. The sequence-specific binding of the ADs was demonstrated by using nonsense ADs or nonsense aptamers. However, it should also be mentioned that the complexation of aptamers in tissues could take a longer time when ADs are delivered intravenously. Nevertheless, to enable the rapid complexation of aptamers, an AD could be delivered site-specifically by local application or targeted delivery.
Oligonucleotides can enter the cells by nonspecific endocytosis as well as via interaction with different cell surface receptors, such as the receptor for advanced glycation end-products (RAGE) [
14], DEC-205 [
15], and macrophage scavenger receptors [
16]. After the delivery of sequestered nucleic acids in endosomes, TLR9 can sense DNA molecules and lead to the production of inflammatory cytokines. In the case of R10-60, the exact mechanism of internalization is unknown, but it is suggested that cell surface receptors are involved [
8]. In this study, we suppose that the R10-60 AD binds outside the cells to the R10-60 aptamers and forms complexes with them. Thereby, the potential interaction of R10-60 with receptors on the cell surface could be prevented, or, in the case of non-specific endocytosis, the endosome-sequestered R10-60 AD complexes could no longer bind and activate TLR9.
The kinetics of the AD binding to the aptamer can differ after the binding of the aptamer to its target. In this study, a rapid complexation of NU172 with NU172 (AD) was demonstrated in serum, which did not contain the coagulation factors. However, a rapid neutralization of the aptamer´s effect could also be shown in human blood containing the target, 5 min after the addition of the AD. In previous studies, Rusconi and colleagues also demonstrated that an AD to the FIXa RNA aptamer was able to neutralize the aptamer-induced anticoagulation rapidly (within 10 min) in plasma [
3,
4].
Furthermore, for the in vivo application of aptamer–AD pairs, especially in human blood, the nucleic acids should possess adequate stability to be effective. In contrast to unmodified RNA aptamers with a half-life of seconds, DNA aptamers are more stable, generally with a half-life of approximately 60 to 120 min [
17,
18]. However, the stability of aptamers can be further increased by the incorporation of modified nucleotides and 5′ end modifications, such as conjugation of polyethylene glycol (PEG) or capping of the 3′ end with an inverted deoxythymidine (dT) residue [
19,
20].
The results of this study demonstrated the rapid and specific binding of ADs to aptamers. Here, we selected two different aptamers with their ADs to demonstrate this interesting feature. Hereby, we aim to highlight that besides target binding, a further important advantage of aptamers is that their effect can be rapidly modulated or interrupted by a corresponding AD.
5. Conclusions
In this study, we demonstrated the quick binding of ADs to their complementary single-stranded DNA molecules (aptamers) in human serum and blood, and in cell culture medium containing 10% FBS. The AD-based neutralization of the aptamer’s tertiary structure and thereby its function, enables the generation of controllable drugs. Furthermore, this phenomenon can also be used for other applications, e.g., to deliver drugs conjugated to aptamers to desired sites in vivo or to modulate the release of bound targets from aptamers. Thus, the application of non-coding nucleic acid pairs is a powerful tool for the modulation of cell and protein activities, delivery of cargos, and site-specific immobilization of desired molecules at defined positions. The findings of this study demonstrate the diverse application possibilities of aptamer-AD pairs.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}