Synthesis and Structure-Activity Relationships of LP1 Derivatives: N-Methyl-N-phenylethylamino Analogues as Novel MOR Agonists

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
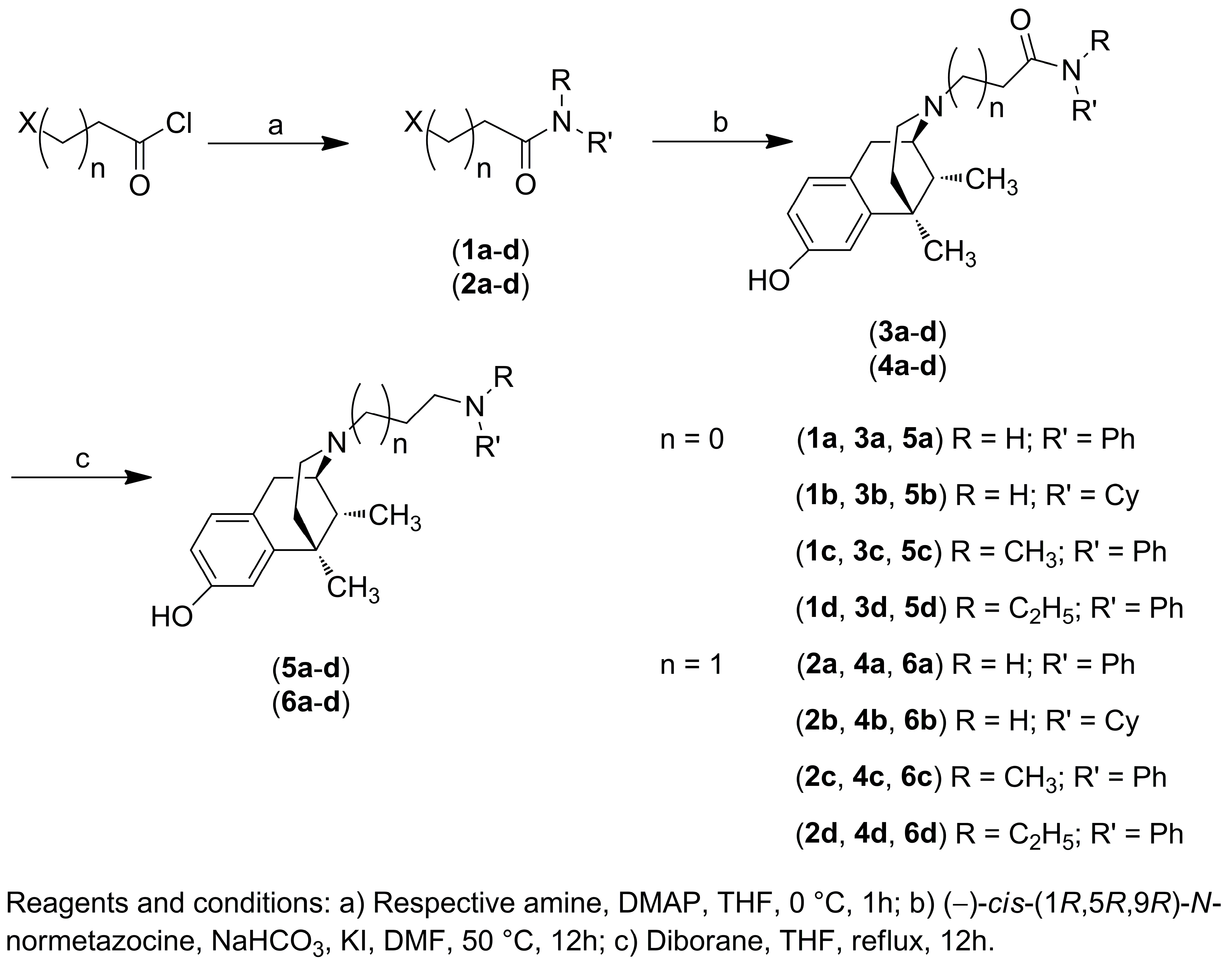
2.1. Chemistry
2.2. Pharmacology
2.1.1. In Vitro Radioligand Binding Assay
2.1.2. Adenylyl Cyclase-Mediated Effects
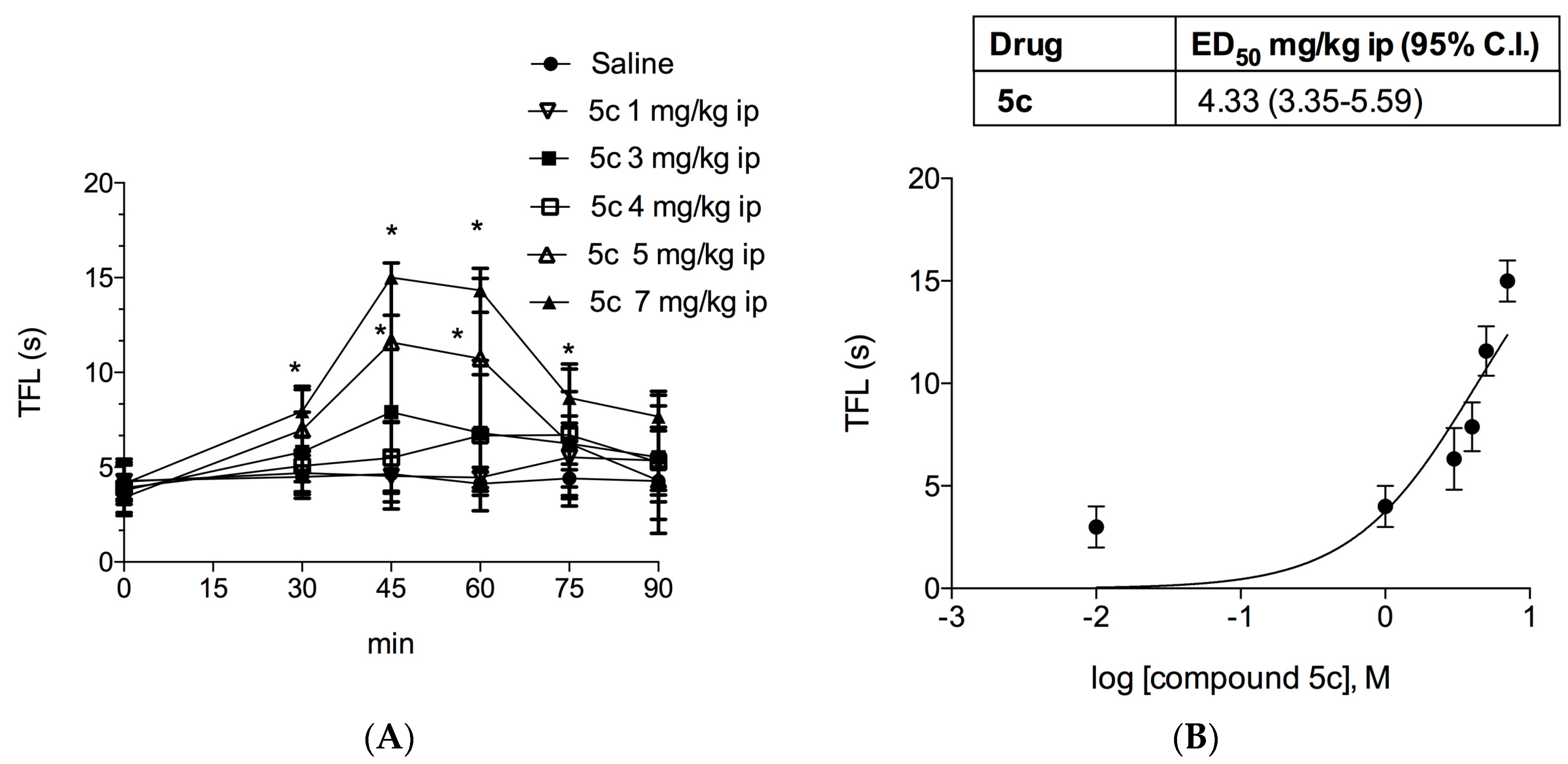
2.1.3. Tail Flick Test
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Chemistry
4.3. Receptor Binding Assays
4.4. cAMP Accumulation Assay
4.5. In Vivo Pharmacology
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Williams, D.A.; Roche, V.F.; Roche, R.A. Central Analgesics. In Foye’s Principles of Medicinal Chemistry, 7nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; pp. 658–699. ISBN 1609133455. [Google Scholar]
- Janecka, A.; Fichna, J.; Janecki, T. Opioid receptors and their ligands. Curr. Top. Med. Chem. 2004, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Marrazzo, A.; Parenti, C.; Pasquinucci, L. Benzomorphan scaffold for opioid analgesics and pharmacological tools development: A comprehensive review. Eur. J. Med. Chem. 2018, 148, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Lipkowski, A.W.; Misicka, A.; Porreca, F.; Davis, P.; Yamamura, H.I.; Stropova, D.; Hruby, V.J. Benzomorphan alkaloids: Natural peptidomimetics of opioid peptide pharmacophores. Lett. Pept. Sci. 1995, 2, 177–181. [Google Scholar] [CrossRef]
- Aceto, M.D.; May, E.L.; Harris, L.S.; Bowman, E.R.; Cook, C.D. Pharmacological studies with a nonpeptidic, delta-opioid (−)-(1R,5R,9R)-5,9-dimethyl-2′-hydroxy-2-(6-hydroxyhexyl)-6,7 benzomorphan hydrochloride ((−)-NIH 11082). Eur. J. Pharm. 2007, 566, 88–93. [Google Scholar] [CrossRef] [PubMed]
- May, E.L.; Aceto, M.D.; Bowman, E.R.; Bentley, C.; Martin, B.R.; Harris, L.S.; Medzihradsky, F.; Mattson, M.V.; Jacobson, A.E. Antipodal alpha-N-(methyl through decyl)-N-normetazocines (5,9 alpha-dimethyl-2′-hydroxy-6,7-benzomorphans): In vitro and in vivo properties. J. Med. Chem. 1994, 37, 3408–3418. [Google Scholar] [CrossRef] [PubMed]
- May, E.L.; Jacobson, A.E.; Mattson, M.V.; Traynor, J.R.; Woods, J.H.; Harris, L.S.; Bowman, E.R.; Aceto, M.D. Synthesis and in vitro and in vivo activity of (−)-(1R,5R,9R)- and (+)-(1S,5S,9S)-N-alkenyl-, -N-alkynyl-, and -N-cyanoalkyl-5,9-dimethyl-2′-hydroxy-6,7-benzomorphan homologues. J. Med. Chem. 2000, 43, 5030–5036. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, M.D.; Aceto, M.D.; Harris, L.S.; Woods, J.H.; Traynor, J.R.; Coop, A.; May, E.L. The influence of esters and carboxylic acids as the N-substituent of opioids. Part 1: Benzomorphans. Bioorg. Med. Chem. 2008, 16, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Arico, G.; Ronsisvalle, G.; Pasquinucci, L.; Parenti, C. Multitarget Opioid/Non-opioid Ligands: A Potential Approach in Pain Management. Curr. Med. Chem. 2016, 23, 4506–4528. [Google Scholar] [CrossRef] [PubMed]
- Pasquinucci, L.; Prezzavento, O.; Marrazzo, A.; Amata, E.; Ronsisvalle, S.; Georgoussi, Z.; Fourla, D.D.; Scoto, G.M.; Parenti, C.; Aricò, G.; et al. Evaluation of N-substitution in 6,7-benzomorphan compounds. Bioorg. Med. Chem. 2010, 18, 4975–4982. [Google Scholar] [CrossRef] [PubMed]
- Pasquinucci, L.; Parenti, C.; Turnaturi, R.; Aricò, G.; Marrazzo, A.; Prezzavento, O.; Ronsisvalle, S.; Georgoussi, Z.; Fourla, D.D.; Scoto, G.M.; et al. The benzomorphan-based LP1 ligand is a suitable MOR/DOR agonist for chronic pain treatment. Life Sci. 2012, 90, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Parenti, C.; Turnaturi, R.; Aricò, G.; Marrazzo, A.; Prezzavento, O.; Ronsisvalle, S.; Scoto, G.M.; Ronsisvalle, G.; Pasquinucci, L. Antinociceptive profile of LP1, a non-peptide multitarget opioid ligand. Life Sci. 2012, 90, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Parenti, C.; Turnaturi, R.; Aricò, G.; Gramowski-Voss, A.; Schroeder, O.H.; Marrazzo, A.; Prezzavento, O.; Ronsisvalle, S.; Scoto, G.M.; Ronsisvalle, G.; et al. The multitarget opioid ligand LP1’s effects in persistent pain and in primary cell neuronal cultures. Neuropharmacology 2013, 71, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Accolla, M.L.; Turnaturi, R.; Sarpietro, M.G.; Ronsisvalle, S.; Castelli, F.; Pasquinucci, L. Differential scanning calorimetry approach to investigate the transfer of the multitarget opioid analgesic LP1 to biomembrane model. Eur. J. Med. Chem. 2014, 77, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Pasquinucci, L.; Turnaturi, R.; Prezzavento, O.; Arena, E.; Aricò, G.; Georgoussi, Z.; Parenti, R.; Cantarella, G.; Parenti, C. Development of novel LP1-based analogues with enhanced delta opioid receptor profile. Bioorg. Med. Chem. 2017, 25, 4745–4752. [Google Scholar] [CrossRef] [PubMed]
- Pasquinucci, L.; Turnaturi, R.; Aricò, G.; Parenti, C.; Pallaki, P.; Georgoussi, Z.; Ronsisvalle, S. Evaluation of N-substituent structural variations in opioid receptor profile of LP1. Bioorg. Med. Chem. 2016, 24, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Bohotin, C.R.; Asim, M.F.; Stübegger, K.; Schmidhammer, H. In vitro and in vivo pharmacological profile of the 5-benzyl analogue of 14-methoxymetopon, a novel mu opioid analgesic with reduced propensity to alter motor function. Eur. J. Pharm. Sci. 2010, 41, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Prezzavento, O.; Arena, E.; Sánchez-Fernández, C.; Turnaturi, R.; Parenti, C.; Marrazzo, A.; Catalano, R.; Amata, E.; Pasquinucci, L.; Cobos, E.J. (+)-and (−)-Phenazocine enantiomers: Evaluation of their dual opioid agonist/σ(1) antagonist properties and antinociceptive effects. Eur. J. Med. Chem. 2017, 125, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Morou, E.; Georgoussi, Z. Expression of the third intracellular loop of the delta opioid receptor inhibits signalling by opioid receptors and other GPCRs. J. Pharm. Exp. Ther. 2005, 315, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Fourla, D.D.; Papakonstantinou, M.P.; Vrana, S.; Georgoussi, Z. Selective interactions of the C-termini of the δ- and μ-opioid receptor signaling. Cell Signal. 2012, 24, 2315–2328. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, M.P.; Karoussiotis, C.; Georgoussi, Z. RGS2 and RGS4 proteins: New modulators of the κ-opioid receptor signaling. Cell Signal. 2015, 27, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Prezzavento, O.; Arena, E.; Parenti, C.; Pasquinucci, L.; Aricò, G.; Scoto, G.M.; Grancara, S.; Toninello, A.; Ronsisvalle, S. Design and synthesis of new bifunctional sigma-1 selective ligands with antioxidant activity. J. Med. Chem. 2013, 56, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Bliss, C.I. Statistics in Biology; McGraw-Hill: New York, NY, USA, 1967; pp. 558–639. [Google Scholar]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Compound | n | R | R′ | Ki (nM) ± SEM a,b | ||
| MOR | DOR | KOR | ||||
| 5a | 0 | H | Ph | 83 ± 30 | 270 ± 10 | 100 ± 5.0 |
| 5b | 0 | H | C6H11 | 8.3 ± 0.8 | 70 ± 2.0 | 19.8 ±0.9 |
| 5c | 0 | CH3 | Ph | 6.1 ± 0.5 | 147 ± 5.7 | 31 ± 1.3 |
| 5d | 0 | C2H5 | Ph | 160 ± 7.0 | 411 ±14 | 28.7 ± 1.0 |
| 6a | 1 | H | Ph | 7.0 ± 0.6 | 117 ± 5.2 | 71 ± 2.0 |
| 6b | 1 | H | C6H11 | 48 ± 1.4 | >5000 | 126 ± 4.5 |
| 6c | 1 | CH3 | Ph | 68 ±1.6 | >5000 | 94 ± 3.5 |
| 6d | 1 | C2H5 | Ph | 86 ± 3.0 | 1060 ± 152 | 56 ±1.7 |
| LP1 | 0.83 ± 0.05 | 29.1 ± 1.0 | 110 ± 6.0 | |||
| DAMGO | 1.16 ± 0.1 | - | - | |||
| U50,488 | - | - | 0.34 ± 0.1 | |||
| Naltrindole | - | 1.13 ± 0.1 | ||||
| Compound | IC50 (nM) ± SD a,b | Imax (%) ± SD c | ||
|---|---|---|---|---|
| MOR | KOR | MOR | KOR | |
| 5a | 55.3 ± 7.0 | 1000 ± 65 | 60 ± 4 | - |
| 5b | 74.0 ± 3.5 | 180 ± 50 | 28 ± 1 | 58 ± 4 |
| 5c | 11.5 ± 2.5 | ND d | 72 ± 5 | - |
| 5d | 66 ± 1.3 | ND | 55 ± 3 | - |
| 6a | 7.4 ± 1.1 | 1400 ± 69 | 50 ± 3 | 53 ± 4 |
| 6b | ND | >5000 | - | 44 ± 3 |
| 6c | 21.61 ± 3.5 | ND | 50 ± 3 | - |
| 6d | 9.51 ± 2.0 | ND | 40 ± 2 | - |
| LP1 | 4.8 ± 0.5 | - | 73 ± 3.8 | - |
| DAMGO | 3.18 ± 0.3 | - | 73 ± 0.3 | - |
| U50,488 | 0.82 ± 0.03 | 68 ± 5 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turnaturi, R.; Parenti, C.; Prezzavento, O.; Marrazzo, A.; Pallaki, P.; Georgoussi, Z.; Amata, E.; Pasquinucci, L. Synthesis and Structure-Activity Relationships of LP1 Derivatives: N-Methyl-N-phenylethylamino Analogues as Novel MOR Agonists. Molecules 2018, 23, 677. https://doi.org/10.3390/molecules23030677
Turnaturi R, Parenti C, Prezzavento O, Marrazzo A, Pallaki P, Georgoussi Z, Amata E, Pasquinucci L. Synthesis and Structure-Activity Relationships of LP1 Derivatives: N-Methyl-N-phenylethylamino Analogues as Novel MOR Agonists. Molecules. 2018; 23(3):677. https://doi.org/10.3390/molecules23030677
Chicago/Turabian StyleTurnaturi, Rita, Carmela Parenti, Orazio Prezzavento, Agostino Marrazzo, Paschalina Pallaki, Zafiroula Georgoussi, Emanuele Amata, and Lorella Pasquinucci. 2018. "Synthesis and Structure-Activity Relationships of LP1 Derivatives: N-Methyl-N-phenylethylamino Analogues as Novel MOR Agonists" Molecules 23, no. 3: 677. https://doi.org/10.3390/molecules23030677