Effect of Repetitive Lysine-Tryptophan Motifs on the Eukaryotic Membrane

Abstract
:1. Introduction
2. Results and Discussion
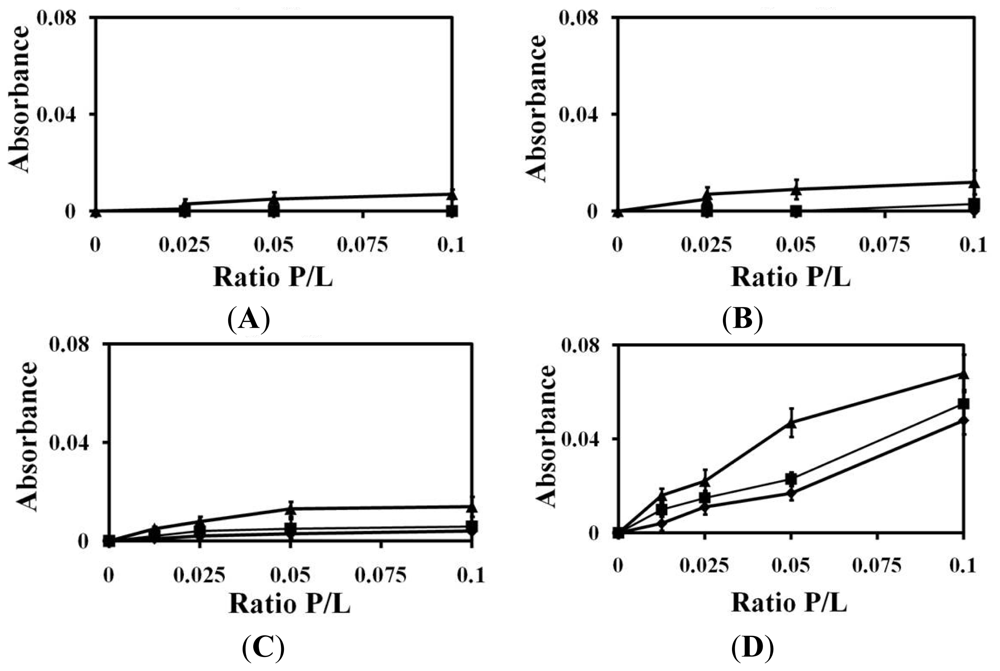
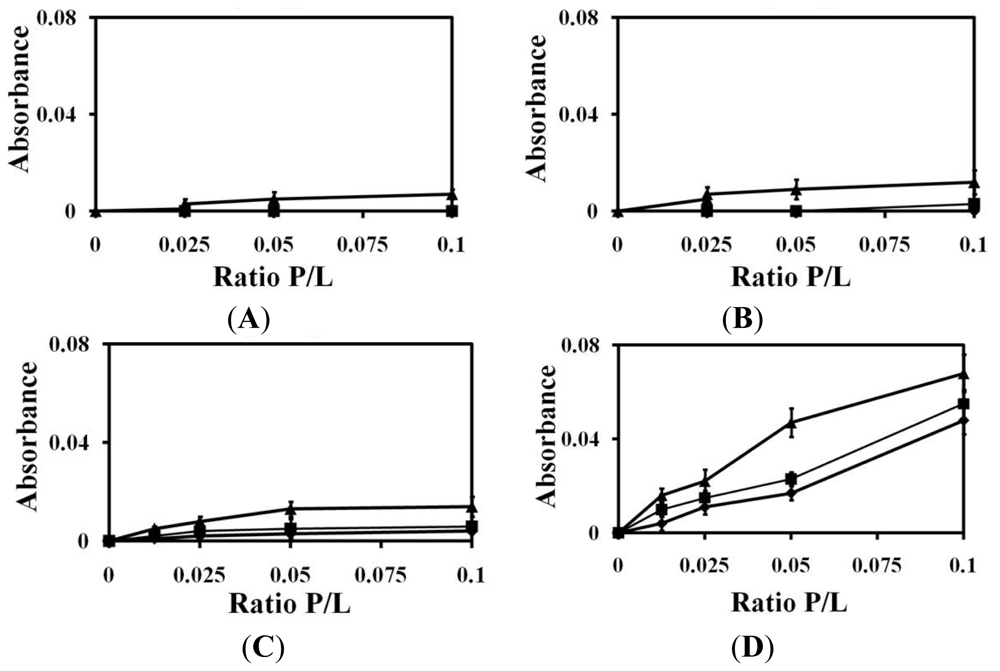
2.1. Liposomes Aggregation
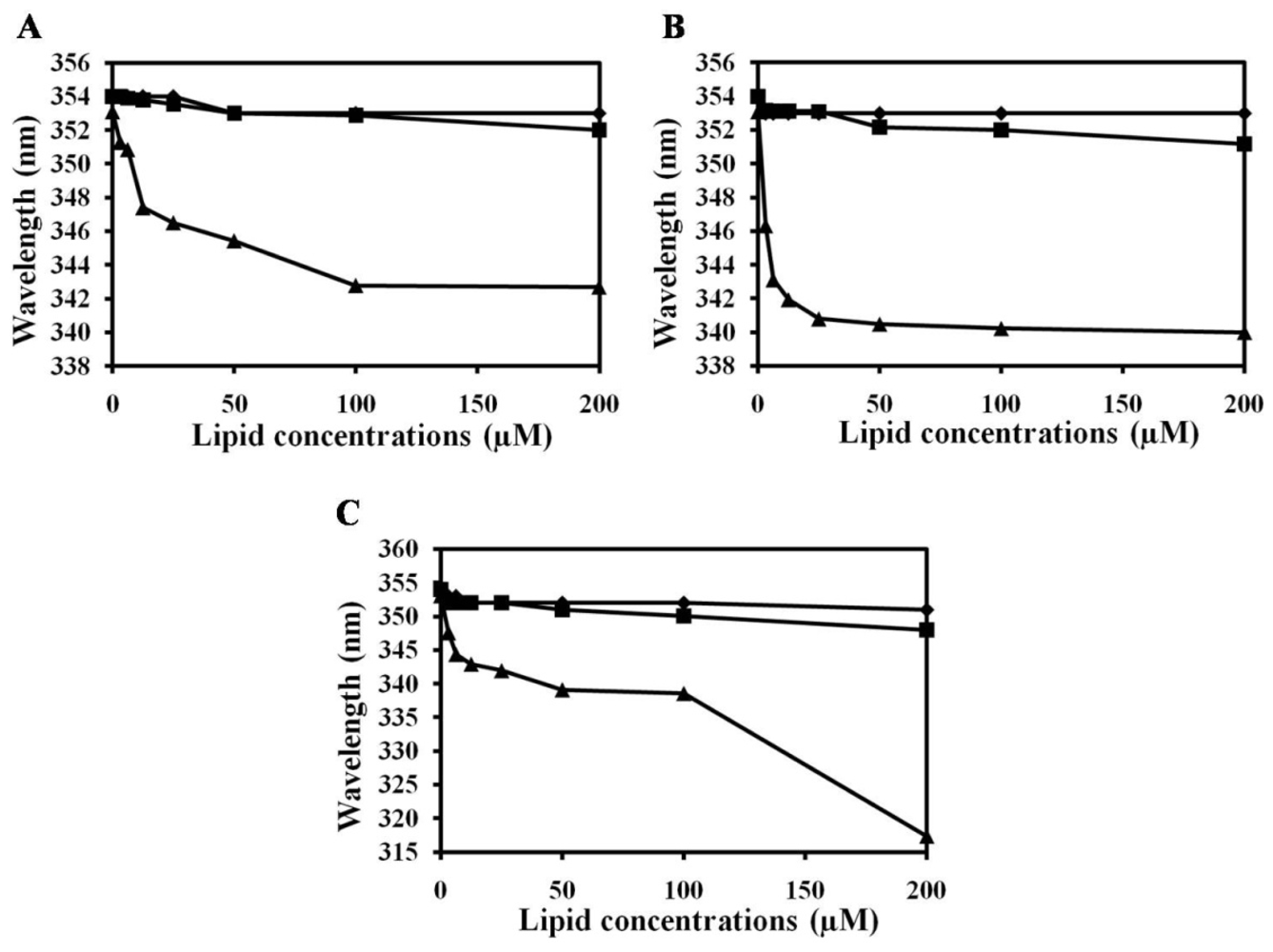
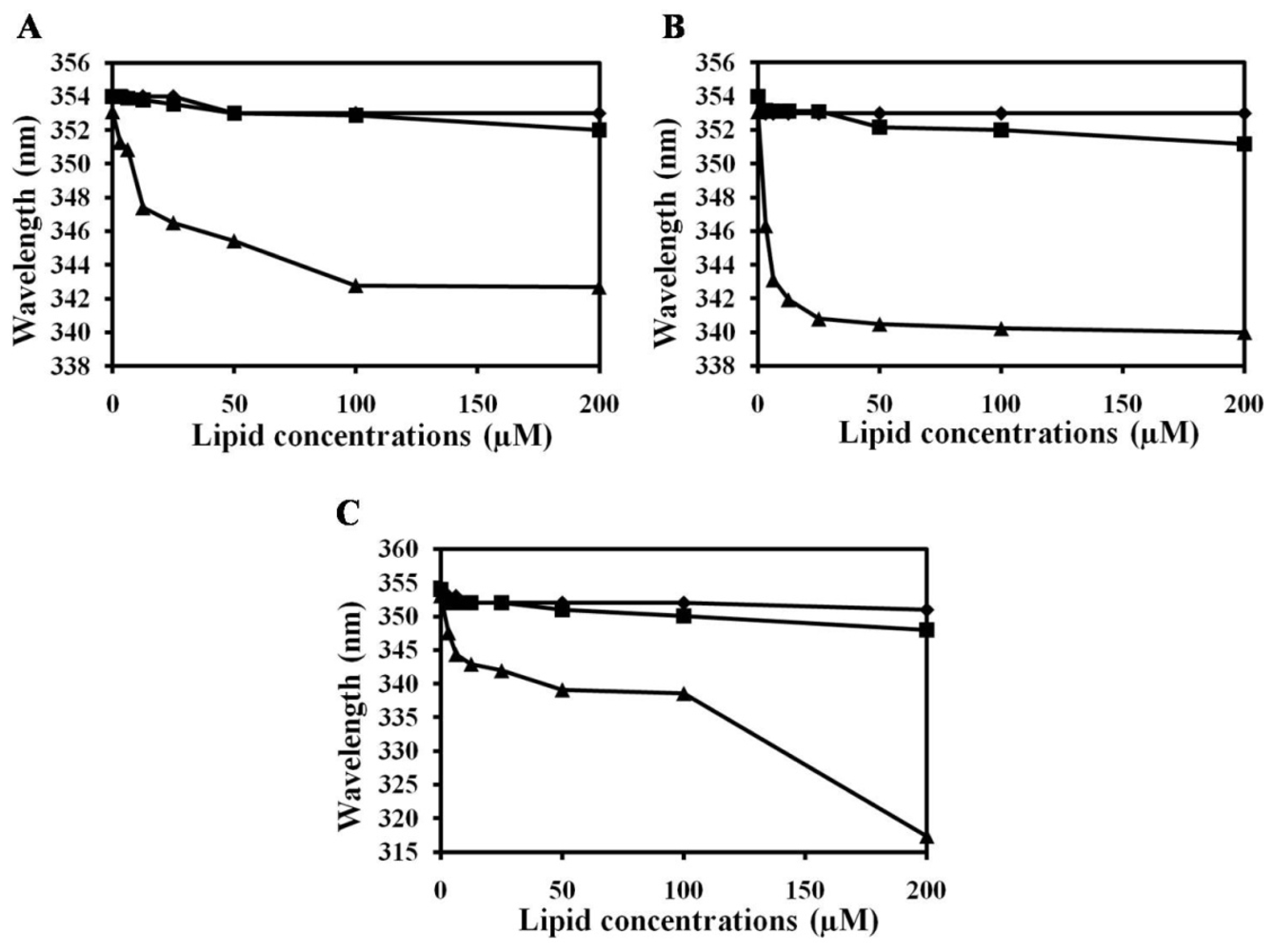
2.2. Characterization of Trp Environment Using Fluorescence Spectroscopy
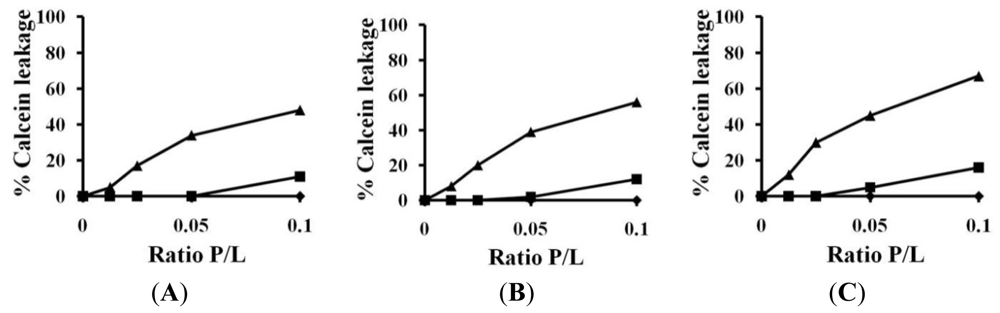
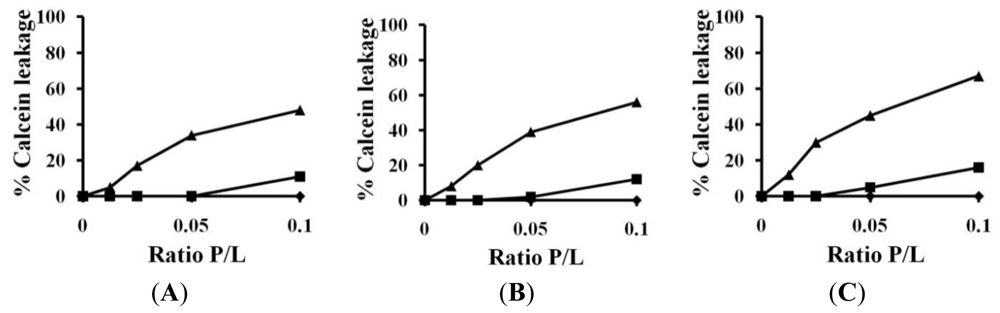
2.3. Calcein Leakage
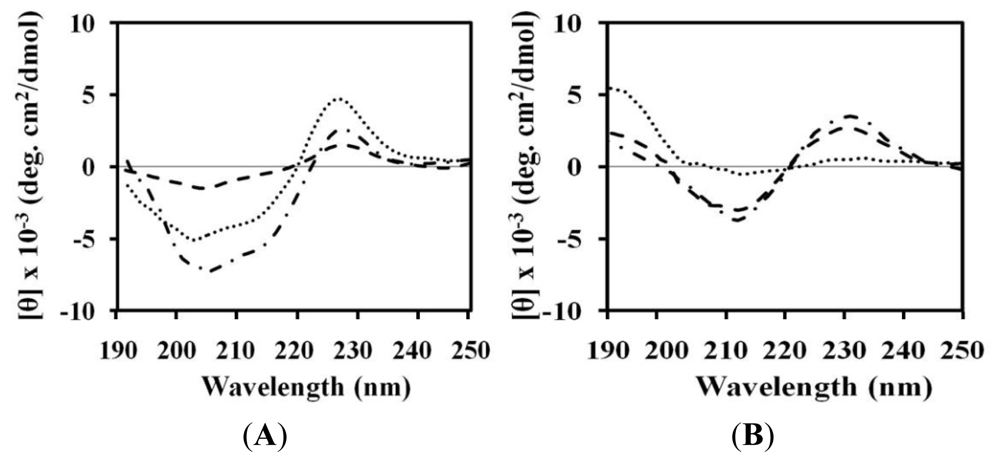
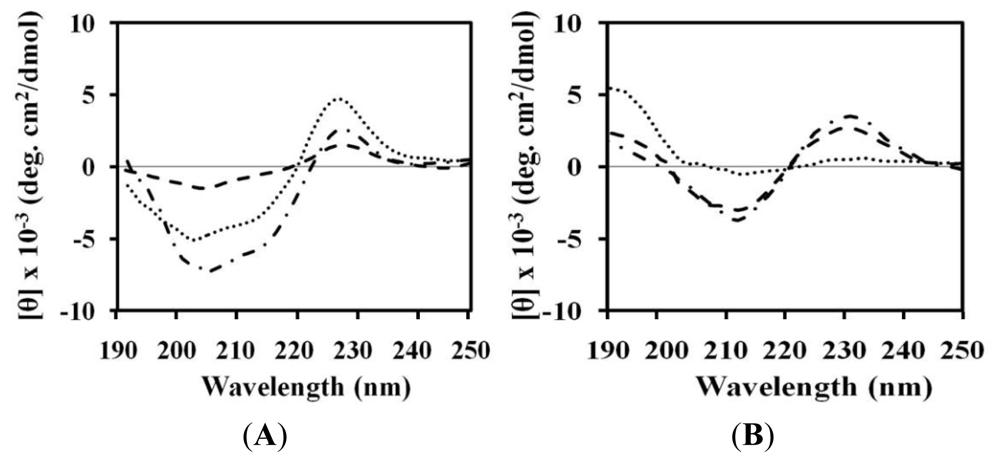
2.4. Circular Dichroism (CD)
3. Experimental Section
3.1. Materials
3.2. Peptide Synthesis and Purification
3.3. Preparation of Large Unilamellar Vesicles (LUVs)
3.4. Liposome Aggregation
3.5. Trp Fluorescence and Acrylamide Quenching Assay
3.6. Calcein Leakage from Liposomes
3.7. Circular Dichroism (CD) Spectroscopy
4. Conclusions
Acknowledgements
References
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar]
- Dürr, U.H.N.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta 2006, 1758, 1499–1512. [Google Scholar]
- Abraham, T.; Lewis, R.N.; Hodges, R.S.; McElhaney, R.N. Isothermal titration calorimetry studies of the binding of the antimicrobial peptide gramicidin S to phospholipid bilayer membranes. Biochemistry 2005, 44, 11279–11285. [Google Scholar]
- Verkleij, A.J.; Zwaal, R.F.; Roelofsen, B.; Comfurius, P.; Kastelijn, D.; van Deenen, L.L. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim. Biophys. Acta 1973, 323, 178–193. [Google Scholar]
- Vogel, H.J.; Schibli, D.J.; Jing, W.; Lohmeier-Vogel, E.M.; Epand, R.F.; Epand, R.M. Towards a structure-function analysis of bovine lactoferricin and related tryptophan- and arginine-containing peptides. Biochem. Cell Biol 2002, 80, 49–63. [Google Scholar]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar]
- Lugtenberg, B.; van Alphen, L. Molecular architecture and functioning of the outer membrane of Escherichia coli and other gram-negative bacteria. Biochim. Biophys. Acta 1983, 737, 51–115. [Google Scholar]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem 2005, 280, 33960–33967. [Google Scholar]
- Epand, R.F.; Schmitt, M.A.; Gellman, S.H.; Epand, R.M. Role of membrane lipids in the mechanism of bacterial species selective toxicity by two alpha/beta-antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1343–1350. [Google Scholar]
- Shai, Y.; Fox, J.; Caratsch, C.; Shih, Y.L.; Edwards, C.; Lazarovici, P. Sequencing and synthesis of pardaxin, a polypeptide from the Red Sea Moses sole with ionophore activity. FEBS Lett 1988, 242, 161–166. [Google Scholar]
- Oren, Z.; Shai, Y. Selective lysis of bacteria, but not mammalian cells by diastereomers of melittin: Structure-function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem 1998, 273, 3718–3724. [Google Scholar]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem 2005, 280, 12316–12329. [Google Scholar]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317. [Google Scholar]
- Melo, M.N.; Ferre, R.; Castanho, M.A. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol 2009, 7, 245–250. [Google Scholar]
- Zhu, W.L.; Nan, Y.H.; Hahm, K.S.; Shin, S.Y. Cell selectivity of an antimicrobial peptide melittin diastereomer with D-amino acid in the leucine zipper sequence. J. Biochem. Mol. Biol 2007, 40, 1090–1094. [Google Scholar]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar]
- Wang, P.; Nan, Y.H.; Yang, S.T.; Kang, S.W.; Kim, Y.; Park, I.S.; Hahm, K.S.; Shin, S.Y. Cell selectivity and anti-inflammatory activity of a Leu/Lys-rich alpha-helical model antimicrobial peptide and its diastereomeric peptides. Peptides 2010, 31, 1251–1261. [Google Scholar]
- Strom, M.B.; Haug, B.E.; Sker, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The Pharmacophore of short cationic antibacterial peptides. J. Med. Chem 2003, 46, 1567–1570. [Google Scholar]
- Liu, Z.; Brady, A.; Young, A.; Rasimick, B.; Chen, K.; Zhou, C.; Kallenbach, N.R. Length effects in antimicrobial peptides of the (RW)n series. Antimicrob. Agents Chemother 2007, 51, 597–603. [Google Scholar]
- Strom, M.B.; Rekdal, O.; Svendsen, J.S. Antimicrobial activity of short arginine- and tryptophan-rich peptides. J. Pept. Sci 2002, 8, 431–437. [Google Scholar]
- Jing, W.; Hunter, H.N.; Hagel, J.; Vogel, H.J. The structure of the antimicrobial peptide Ac-RRWWRF-NH2 bound to micelles and its interactions with phospholipid bilayers. J. Pept. Res 2003, 61, 219–229. [Google Scholar]
- Dathe, M.; Nikolenko, H.; Klose, J.; Bienert, M. Cyclization increases the antimicrobial activity and selectivity of arginine- and tryptophan- containing hexapeptides. Biochemistry 2004, 43, 9140–9150. [Google Scholar]
- Gopal, R.; Seo, C.H.; Song, P.I.; Park, Y. Effect of repetitive lysine-tryptophan motifs on the bactericidal activity of antimicrobial peptides. Amino Acids 2012. [Epub ahead of print]. [Google Scholar]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother 2007, 51, 1398–1406. [Google Scholar]
- Ramamoorthy, A.; Lee, D.K.; Narasimhaswamy, T.; Nanga, R.P.R. Cholesterol reduces pardaxin’s dynamics—a barrel—stave mechanism of membrane disruption investigated by solid-state NMR. Biochim. Biophys. Acta 2010, 1798, 223–227. [Google Scholar]
- McHenry, A.J.; Sciacca, M.F.M.; Brender, J.R.; Ramamoorthy, A. Does cholesterol suppress the antimicrobial peptide induced disruption of lipid raft containing membranes? Biochim. Biophys. Acta 2012, 1818, 3019–3024. [Google Scholar]
- Bhattacharjya, S.; Ramamoorthy, A. Multifunctional host defense peptides: Functional and mechanistic insights from NMR structures of potent antimicrobial peptides. FEBS J 2009, 276, 6465–6473. [Google Scholar]
- Ramamoorthy, A. Beyond NMR spectra of antimicrobial peptides: Dynamical images at atomic resolution and functional insights. Solid State Nucl. Magn. Reson 2009, 35, 201–207. [Google Scholar]
- Gottler, L.M.; Ramamoorthy, A. Structure, membrane orientation, mechanism and function of pexiganan—A highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta 2009, 1788, 1680–1686. [Google Scholar]
- March, E.N.; Buer, B.C.; Ramamoorthy, A. Fluorine—A new element in the design of membrane-active peptides. Mol. Biosyst 2009, 5, 1143–1147. [Google Scholar]
- Matsuyama, K.; Natori, S. Mode of action of sapecin, a novel antibacterial protein of Sarcophaga peregrina (flesh fly). J. Biochem 1990, 108, 128–132. [Google Scholar]
- Dhople, V.M.; Nagaraj, R. Generation of analogs having potent antimicrobial and hemolytic activities with minimal changes from an inactive 16-residue peptide corresponding to the helical region of Staphylococcus aureus δ-toxin. Protein Eng. 1995, 8, 315–318. [Google Scholar]
- Blondelle, S.E.; Lohner, K.; Aguilar, M. Lipid induced conformation and lipid-binding properties of cytolytic and antimicrobial peptides: Determination and biological specificity. Biochim. Biophys. Acta 1999, 1462, 89–108. [Google Scholar]
- Som, A.; Vemparala, S.; Ivanov, I.; Tew, G.N. Synthetic mimics of antimicrobial peptides. Biopolymers 2008, 90, 83–93. [Google Scholar]
- Gopal, R.; Park, S.C.; Ha, K.J.; Cho, S.J.; Kim, S.W.; Song, P.I.; Nah, J.W.; Park, Y.; Hahm, K.S. Effect of leucine and lysine substitution on the antimicrobial activity and evaluation of the mechanism of the HPA3NT3 analog peptide. J. Pept. Sci 2009, 15, 589–594. [Google Scholar]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: The importance of interfacial activity. J. Am. Chem. Soc 2009, 131, 7609–7617. [Google Scholar]
- Zhu, W.L.; Shin, S.Y. Effects of dimerization of the cell-penetrating peptide Tat analog on antimicrobial activity and mechanism of bactericidal action. J. Pept. Sci 2009, 15, 345–352. [Google Scholar]
- Torrent, M.; de la Torre, B.G.; Nogués, V.M.; Andreu, D.; Boix, E. Bactericidal and membrane disruption activities of the eosinophil cationic protein are largely retained in an N-terminal fragment. Biochem. J 2009, 421, 425–434. [Google Scholar]
- Javadpour, M.M.; Barkley, M.D. Self-assembly of designed antimicrobial peptides in solution and micelles. Biochemistry 1997, 36, 9540–9549. [Google Scholar]
- Feder, R.; Dagan, A.; Mor, A. Structure-activity relationship study of antimicrobial dermaseptin S4 showing the consequences of peptide oligomerization on selective cytotoxicity. J. Biol. Chem 2000, 275, 4230–4238. [Google Scholar]
- Chongsiriwatana, N.P.; Barron, A.E. Comparing bacterial membrane interactions of antimicrobial peptides and their mimics. Methods Mol. Biol 2010, 618, 171–182. [Google Scholar]
- Subbalakshmi, C.; Krishnakumari, V.; Sitaram, N.; Nagaraj, R. Interaction of indolicidin, a 13-residue peptide rich in tryptophan and proline and its analogues with model membranes. J. Biosci 1998, 23, 9–13. [Google Scholar]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar]
- Schmidtchen, A.; Pasupuleti, M.; Mörgelin, M.; Davoudi, M.; Alenfall, J.; Chalupka, A.; Malmsten, M. Boosting antimicrobial peptides by hydrophobic oligopeptide end tags. J. Biol. Chem 2009, 284, 17584–17594. [Google Scholar]
- William, C.W. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol 2010, 5, 905–917. [Google Scholar]
- Wimley, W.C.; Hristova, K.; Ladokhin, A.S.; Silvestro, L.; Axelsen, P.H.; White, S.H. Folding of beta-sheet membrane proteins: A hydrophobic hexapeptide model. J. Mol. Biol 1998, 277, 1091–1110. [Google Scholar]
- Ladokhin, A.S.; White, S.H. Folding of amphipathic alpha-helices on membranes: Energetics of helix formation by melittin. J. Mol. Biol 1999, 285, 1363–1369. [Google Scholar]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struc. Biol 1996, 3, 842–848. [Google Scholar]
- Thennarasu, S.; Huang, R.; Lee, D.K.; Yang, P.; Maloy, L.; Chen, Z.; Ramamoorthy, A. Limiting an antimicrobial peptide to the lipid-water interface enhances its bacterial membrane selectivity: A case study of MSI-367. Biochemistry 2010, 49, 10595–10605. [Google Scholar]
- Andrä, J.; Monreal, D.; Martinez de Tejada, G.; Olak, C.; Brezesinski, G.; Gomez, S.S.; Goldmann, T.; Bartels, R.; Brandenburg, K.; Moriyon, I. Rationale for the design of shortened derivatives of the NK-lysin-derived antimicrobial peptide NK-2 with improved activity against Gram-negative pathogens. J. Biol. Chem 2007, 282, 14719–14728. [Google Scholar]
- Hawrani, A.; Howe, R.A.; Walsh, T.R.; Dempsey, C.E. Origin of low mammalian cell toxicity in a class of highly active antimicrobial amphipathic helical peptides. J. Biol. Chem 2008, 283, 18636–18645. [Google Scholar]
- Zhao, H.; Sood, R.; Jutila, A.; Bose, S.; Fimland, G.; Nissen-Meyer, J.; Kinnunen, P.K. Interaction of the antimicrobial peptide pheromone Plantaricin A with model membranes: Implications for a novel mechanism of action. Biochim. Biophys. Acta 2006, 1758, 1461–1474. [Google Scholar]
- Pandey, B.K.; Ahmad, A.; Asthana, N.; Azmi, S.; Srivastava, R.M.; Srivastava, S.; Verma, R.; Vishwakarma, A.L.; Ghosh, J.K. Cell-selective lysis by novel analogues of melittin against human red blood cells and Escherichia coli. Biochemistry 2010, 49, 7920–7929. [Google Scholar]
- Lee, J.K.; Park, S.C.; Hahm, K.S.; Park, Y. Antimicrobial HPA3NT3 peptide analogs: Placement of aromatic rings and positive charges are key determinants for cell selectivity and mechanism of action. Biochim. Biophys. Acta 2012, 1828, 443–454. [Google Scholar]
- Brender, J.R.; McHenry, A.J.; Ramamoorthy, A. Does cholesterol role in the bacterial selectivity of antimicrobial peptides? Front. Immunol 2012, 3, 195–198. [Google Scholar]
- Steward, J.C. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem 1980, 104, 10–14. [Google Scholar]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J 1999, 341, 501–513. [Google Scholar]
- Mao, D.; Wallace, B.A. Differential light scattering and adsorption flattening optical effects are minimal in the circular dichroism spectra of small unilamellar vesicles. Biochemistry 1984, 23, 2667–2673. [Google Scholar]
- Matsuzaki, K.; Sugishita, K.; Miyajima, K. Interactions of an antimicrobial peptide, magainin 2, with lipopolysaccharide containing liposomes as a model for outer membranes of gram-negative bacteria. FEBS Lett 1999, 449, 221–224. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | KSV (M−1) a | ||
|---|---|---|---|
| PC | PC:SM (2:1,w/w) | PC:CH (2:1 w/w) | |
| (KW)3 | 7.5 | 7.1 | 6.8 |
| (KW)4 | 7.2 | 6.8 | 6.2 |
| (KW)5 | 3.2 | 2.5 | 1.7 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gopal, R.; Lee, J.K.; Lee, J.H.; Kim, Y.G.; Oh, G.C.; Seo, C.H.; Park, Y. Effect of Repetitive Lysine-Tryptophan Motifs on the Eukaryotic Membrane. Int. J. Mol. Sci. 2013, 14, 2190-2202. https://doi.org/10.3390/ijms14012190
Gopal R, Lee JK, Lee JH, Kim YG, Oh GC, Seo CH, Park Y. Effect of Repetitive Lysine-Tryptophan Motifs on the Eukaryotic Membrane. International Journal of Molecular Sciences. 2013; 14(1):2190-2202. https://doi.org/10.3390/ijms14012190
Chicago/Turabian StyleGopal, Ramamourthy, Jong Kook Lee, Jun Ho Lee, Young Gwon Kim, Gwang Chae Oh, Chang Ho Seo, and Yoonkyung Park. 2013. "Effect of Repetitive Lysine-Tryptophan Motifs on the Eukaryotic Membrane" International Journal of Molecular Sciences 14, no. 1: 2190-2202. https://doi.org/10.3390/ijms14012190